

Abstract
Background
Each gene in the chromosome 11q23 cluster of NCAM1, TTC12, ANKK1 and DRD2 is functionally linked to dopamine in brain. Many association studies of DRD2 and substance dependence (SD), including alcohol dependence (AD) and drug dependence (DD) have been reported; the results have been inconsistent. Recent association studies have considered this cluster more comprehensively, examining the association of SD with several risk variants mapped to the other genes in the cluster. Because comorbid AD with DD (AD+DD) is common, we hypothesized that heterogeneity of the SD diagnoses studied might have contributed to the inconsistency of prior results.
Methods
We conducted two separate association studies of AD+DD and AD without DD (AD-only) in 1,090 European-Americans using family-based and case-control designs and 43 single nucleotide polymorphisms mapped to this cluster. We used a generalized linear model and haplotype score tests for the case-control sample, and the family-based association test for the family sample.
Results
For AD+DD, the risk regions centered on TTC12 exon 3 (optimal individual haplotype simulated p (poihs) = 0.000015), and another extended from ANKK1 exon 8 to DRD2^C957T (poihs = 0.0028), in both samples. NCAM1 exon 12 markers showed global significance in both designs, but were significant for specific haplotypes (poihs = 0.0029) only for the family sample. For AD-only, NCAM1 intron 14–18 and the junction of ANKK1 and DRD2 were associated globally. Population stratification was excluded as the basis for these results. Linkage disequilibrium contrast tests supported selection at TTC12 exon 3 and ANKK1 exon 2.
Conclusions
We conclude that variants in TTC12 exon 3, NCAM1 exon 12, and the two 3′ ends of ANKK1 and DRD2 co-regulate risk for comorbid AD and DD.
Keywords: comorbid alcohol and drug dependences, DRD2, ANKK1, TTC12, NCAM1
Introduction
Substance dependence (SD) has a complex etiology, with constituents of risk that include genetic, neurobiological, and environmental factors. The D2 dopamine receptor gene (DRD2), the protein product of which links to the brain mesocortical dopamine reward system, has been the focus of much attention in addiction genetics, and in psychiatric genetics in general. There is a large volume of inconsistent research on the possible association of SD with the putative DRD2 TaqI “A” polymorphism, which has been shown to be located in ANKK1, an adjacent gene (Neville et al., 2004).
The long history of inconsistent reports could reflect more complicated genetic substrates than have heretofore been recognized. In two recent studies, we (Gelernter et al., 2006; Yang et al., 2007) found a broader genomic region on chromosome 11q23, including a gene cluster of NCAM1, TTC12, ANKK1 and DRD2 (“NTAD” cluster), spanning ~542 kb, to be associated with the genetic risk of nicotine dependence (ND) and alcohol dependence (AD), respectively.
We speculated previously that these genes could be functionally related. Within this gene cluster, a haplotype spanning TTC12 and ANKK1 was strongly associated with ND in European- and African-American (EA and AA, respectively) family samples (Gelernter et al., 2006). Haplotypes around TTC12 exon 3, NCAM1 exon 12, and exons 2 and 5 of ANKK1 were also associated with AD in two other independent EA samples, one population-based and one family-based (Yang et al., 2007). These associations were not attributable to linkage disequilibrium (LD) with DRD2 and DRD2 itself showed only very limited association. Among these identified susceptibility regions, the association signals from TTC12 were consistently prominent. The present study considers the latter samples (i.e., from Yang et al, 2007) in the context of additional diagnostic information, with the goal of adding to our understanding of the nature of the associated phenotypes.
While the functions of TTC12 and ANKK1 are not well established, present knowledge suggests that they may be related in part to the development or modulation of dopaminergic neurotransmission. TTC12 encodes a protein called TPARM, which includes one tetratricopeptide repeat domain (TPR) and three armadillo repeat domains (ARMs) (Katoh and Katoh, 2003). Its ARM, included in a family of proteins containing several 42-amino acid repeat domains, is thought to play a variety of roles at the cellular level, including regulation of desmosome assembly, cell adhesion, neurogenesis, signal transduction, and post developmental synaptogenesis. β-catenin, a prototypical ARM-containing protein, was recently identified as a risk locus for bipolar affective disorder (Baum et al., 2007). These findings, and others, suggest that the ARMs encoded by TTC12 might also be involved in the cascade of events in the Wnt signal transduction pathway that occurs in early neuronal development (Castelo-Branco and Arenas, 2006; Castelo-Branco et al., 2004), and could potentially influence the level of dopamine D2 receptors and thereby modulate SD risk.
ANKK1 was also found to associate with the executive attention network in the anterior cingulate gyrus, a dopamine-rich region of the brain (Fossella et al., 2006). A recent study of association between ND and a region spanning ANKK1 and DRD2 reported a putatively functional polymorphism, rs2734849, in an African American sample (Huang et al., 2008); this variant alters the expression level of NF-κB regulated genes (Huang et al., 2008). Further, NF-κB1 gene encoding the transcription factor NF-κB was associated with AD (Edenberg et al, 2008). Another recent extensive analysis of association between AD and a smaller region spanning only ANKK1 and DRD2 from the Collaborative Study on the Genetics of Alcoholism (COGA) showed that the strongest evidence of association is in a linkage disequilibrium (LD) block in the 5′ end of ANKK1 (Dick et al., 2007b). This LD block extends from an upstream intergenic single nucleotide polymorphism (SNP) to exon 2 of ANKK1, which to some degree replicated our finding of an association of AD to ANKK1 (Yang et al., 2007).
We previously reported association of SNPs in this region to two different SD disorders: viz. AD and ND. These results led us to ask whether one of these phenotypes (i.e., AD), or another that is highly comorbid with it (i.e., drug dependence (DD)), is driving the association. Further, some or all of the associations within this cluster that we reported could be attributable to a common risk factor for multiple forms of SD. Since dopamine is a major constituent of reward pathways and is strongly believed to be involved in many forms of SD (Connor et al., 2007; Sasabe et al., 2007; Shahmoradgoli Najafabadi et al., 2005; Volkow et al., 2006; Xu et al., 2004), this seems a likely possibility. One way to answer this question is to subdivide samples characterized by AD, based on the presence or absence of DD comorbidity. We performed this study with the intention to determine whether some, or all, of the risk loci we identified in this gene cluster are specific to AD or comorbid AD+DD.
Presumably, complex biological mechanisms are responsible for SD, and the heterogeneity of SD phenotypes reflects the existence of a spectrum of diseases with different mechanisms. The co-existence of many different SD disorders in an individual is common clinically; in the United States, the odds ratio of comorbid DD with AD, adjusted for demographic characteristics, is as high as 18.7 (Compton et al., 2007). Narrowing down the shared and specific genetic risk factors for different SD disorders would help to understand the biology of this multifaceted set of disorders. To advance such an understanding, in this study, we investigated the association of the NTAD gene cluster with comorbid of AD+DD, or AD alone.
Materials and methods
Subjects
We implemented case control and family study designs. All relevant institutional review boards approved the study protocol and related materials, and all subjects, who were paid for their participation, provided written informed consent. All subjects included in this study were also included in our previous study (Yang et al., 2007) but this sample is completely distinct and nonoverlapping from our previous study of this gene cluster in ND (cf. Gelernter et al 2006, page 3504, Subject recruitment and assessment).
Case-control Sample
A total of 716 EAs were recruited at either the University of Connecticut Health Center or the VA Connecticut Healthcare System-West Haven campus. Among them, 136 were affected with comorbid AD and DD (“AD+DD”), 166 were affected with AD and no comorbid drug dependence (“AD-only”) and 414 were healthy controls. Details of sex and age distributions are shown in Table 1. Each affected subgroup was examined separately in relation to the 414 control subjects for association analyses. AD+DD subjects met lifetime DSM-III-R [APA, 1987] or DSM-IV [APA, 1994] diagnostic criteria for AD and also met diagnostic criteria for cocaine dependence (CD) and/or opioid dependence (OD) (see Table 1). Healthy controls, recruited by advertisement from the general population, were screened to exclude subjects with AD, DD, major Axis I psychiatric disorders such as mood disorders, major anxiety disorders, and psychotic disorders (i.e., schizophrenia or schizophrenia-like disorders). The Structured Clinical Interview for DSM-III-R (SCID-III-R) or DSM-IV (SCID-IV), the Computerized Diagnostic Interview Schedule for DSM-III-R (C-DIS-R), the Schedule for Affective Disorders and Schizophrenia (SADS), or an unstructured interview were used for screening. Many of the subjects were not characterized with respect to ND, so this diagnosis could not be considered.
Table 1.
Demographic and clinical characteristics of the study sample.
|
Case-control sample | |
| AD+DDa (total number) | 136 |
| Number of males (%) | 93 (68.4) |
| Age (years)b | 36.6 ± 7.8 |
| AD-onlyc (total number) | 166 |
| Number of males (%) | 135 (81.3) |
| Age (years)b | 44.7 ± 9.8 |
| Control (total number) | 414 |
| Number of males (%) | 167 (40.3) |
| Age (years)b | 27.9 ± 8.5 |
|
Family sample | |
| Number of subjects (% male) | 374 (50) |
| Number of nuclear families | 112 |
| Number of informative familiesb,d | 42.9 ± 10.8 |
| Family sizeb | 3.3 ± 0.9 |
| Number of parents | 200 |
| Number of siblings (% male) | 174 (53.4) |
| Age (years)b | |
| Parents | 61.8 ± 10.1 |
| All offspring | 34.2 ± 8.1 |
| Number with AD (%) | 163 (43.6) |
| Number with CD (%) | 99 (26.5) |
| Number with OD (%) | 78 (20.9) |
AD+DD indicates alcohol dependence (AD) comorbid with drug dependence (DD).
mean± standard deviation.
AD-only indicates AD without other DD.
number of informative families, defined as those with at least one heterozygous parent, for the 43 studied SNPs.
Family Sample
Four hundred eighty-eight EAs in 143 nuclear families were recruited at the same centers and were assessed using the SCID or the Semi-Structured Assessment for Drug Dependence and Alcoholism (SSADDA), as described elsewhere (Gelernter et al., 2005; Pierucci-Lagha et al., 2007; Pierucci-Lagha et al., 2005). Each member of the family sample was evaluated for comorbidity status. A family classified as AD+DD was one in which not all affected family members were AD-only. Thirty-one and 112 families were identified as AD-only and AD+DD families, respectively. The 31 AD-only families, which included a total of 114 subjects, provided insufficient statistical power for analysis, so association analyses were conducted only for the 374 subjects in the AD+DD families. Details of the demographic characteristic for the samples included here are presented in Table 1. The study sample included all subjects presented elsewhere (Yang et al., 2007), but 16 affected subjects from that analysis who had missing DD status were excluded from the present study.
Marker Selection and Genotyping
Forty-three SNPs in the NTAD gene cluster were selected (cf. Gelernter et al., 2006). These 43 SNPs (Figure 1(A)) were designated in sequence order (from the 5′ end of NCAM1 to the 5′ end of DRD2) as N1-N17 (mapped to NCAM1, 5′ to 3′), N18-N19 and T1-T2 (intergenic between NCAM1 and TTC12), T3-T9 (mapped to TTC12, 5′ to 3′), T10 (intergenic between TTC12 and ANKK1), A1-A7 (mapped to ANKK1, 5′ to 3′), D1-D4 (mapped to DRD2, 3′ to 5′), and D5-D7 (intergenic, 5′ to DRD2; for details refer to online supplemental Table 1). SNPs were genotyped as described previously (Gelernter et al., 2006). In addition, 37 ancestry informative markers (AIMs), as discussed in Yang et al. (2005a, 2005b), including 36 short tandem repeat (STR) markers and one SNP (rs2814778) in the Duffy antigen (FY) gene were genotyped in all cases and controls to infer ancestry. Two markers did not pass the HWE test. They are SNP N4 (p-value = 7.6×10−5) only in the control sample and SNP A4 (p-value = 2.3×10−6) only in the AD-only sample. Since causal variants in the affected sample could cause a failure of the HWE test, SNP A4 in the AD-only sample could be a risk-influencing variant. As for SNP N4, it fails the HWE test only in the control sample, and we kept this marker in the analysis for comparison purpose. Genotyping errors were detected and resolved (details described in Yang et al. (2007).
Figure 1.
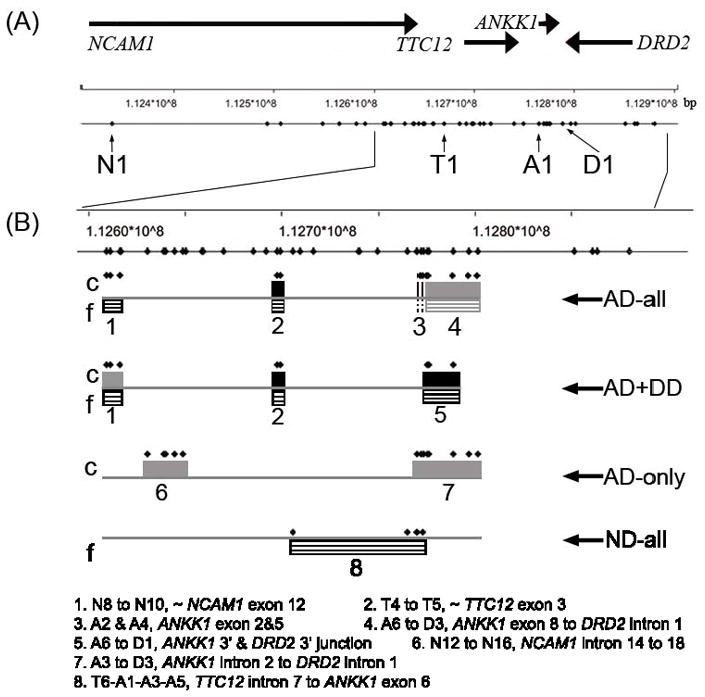
Map of the NCAM1, TTC12, ANKK1 and DRD2 gene region. (A) Relative gene locations, and the distribution of 43 SNPs studied. (B) Associated regions identified in analyzing phenotypes of AD-all, AD+DD, AD-only and ND-all. Solid black or grey indicates a finding in the case-control (“c”) sample, black or grey with white stripes indicates a finding in the family (“f”). A finding in black color indicates significance both in global and individual specific haplotypic association; a finding in grey color indicates significance in global haplotypic association only. Each diamond above the block indicates the position of the corresponding SNP in the associated haplotype. The number underneath each block is annotated for the corresponding genetic region.
Statistical Analysis
Inferences of Linkage Disequilibrium and Haplotype Blocks
LD, haplotype blocks, and allele frequencies were estimated or inferred using the Haploview 3.32 program (Barrett et al., 2005). The LD plots encapsulate the standardized LD coefficients, D’, LD patterns and haplotype blocks (online supplemental Figures 1–4). Haplotype blocks in the LD plots were defined according to the criteria of Gabriel et al (2002).
Association Analysis for the Case-Control Design
A generalized linear model (GLM implemented in the R Project for Statistical Computing) was used to test single marker genotypic additive effects for the relevant traits (AD+DD or AD-only), adjusting for the covariates sex and age. Haplotype-based association analysis was also completed in a GLM framework using the Haplo.stats Package in R (Schaid et al., 2002). The permutation test on global haplotypic effect was implemented by randomly permuting the case vs. control status (null hypothesis of no association), computing and comparing the test statistic with the observed one, replicating the simulation an appropriate number of times, and summarizing with the p-value, which is the probability of the permutated test statistic being greater than the observed one. Only haplotypes with frequency greater than 5.0% were examined for the haplotype-specific tests. The critical level of significance for the haplotype-specific tests within each haplotype set was adjusted by Bonferroni correction, i.e., the conventional α = 0.05 was divided by the number of major haplotypes observed in each case. The LD contrast test (Zaykin et al., 2006) was employed to investigate differences in LD between cases and controls (cf. Yang et al., 2007, for more details), and to infer from the differences evidence of selection of risk variants in the disease group. Population stratification for the case-control sample was tested using the program Structure 2.1. More details regarding the analyses were reported elsewhere (Yang et al., 2007).
Association Analysis for the Family Design
The family-based association test (FBAT)(Horvath et al., 2001; Horvath et al., 2004) was used to test the association of the single SNPs and haplotypes of various SNPs to AD+DD. The FBAT model and parameters used included an additive genetic model under the null hypothesis of no linkage and no association, biallelic mode, minimum number of informative families of 20 for each analysis, and offset of zero. The “hbat” command in FBAT was issued to analyze haplotypic association with an assumption of a biallelic mode of inheritance for haplotype-specific association and a multi-allelic mode for testing all haplotypes as a whole for the global association test. Permutation tests (with 100,000 iterations) were used to calculate all p-values of global associations. The significance level of haplotype-specific tests was set up the same as that in the case-control design.
Results
For AD+DD in the case-control sample, seven nominally significant individual SNPs emerged: T6, T7, T8, A5, A6, D1 and D2 (p = 0.049 to 0.0025), though none was significant after Bonferroni correction, as shown in Table 2. The global haplotype analyses revealed three risk regions: almost the entirety of TTC12, the two merging 3′ regions of DRD2 and ANKK1, and around exon 12 of NCAM1.
Table 2.
Association analyses for AD-only and AD+DD for the case-control sample.
| Starting SNPc | MAFa | p-valuesb | p-valuesb | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Control | AD-Only | AD+DD | AD-onlyd (166 cases) & control | AD+DDe (136 cases) & control | |||||||
| 1SNP | 3SNP-hapf | 4SNP-hap | 5SNP-hap | 1SNP | 3SNP-hap | 4SNP-hap | 5SNP-hap | ||||
| N1 | 0.295 | 0.330 | 0.335 | 0.96 | 0.97 | 0.91 | 0.67 | 0.14 | 0.42 | 0.94 | 0.94 |
| N2 | 0.499 | 0.531 | 0.481 | 0.72 | 0.75 | 0.39 | 0.33 | 0.43 | 0.67 | 0.81 | 0.90 |
| N3 | 0.352 | 0.314 | 0.366 | 0.9 | 0.13 | 0.23 | 0.38 | 0.20 | 0.42 | 0.74 | 0.79 |
| N4 | 0.408 | 0.354 | 0.392 | 0.39 | 0.35 | 0.65 | 0.36 | 0.64 | 0.80 | 0.91 | 0.57 |
| N5 | 0.221 | 0.203 | 0.225 | 0.82 | 0.54 | 0.21 | 0.37 | 0.71 | 0.76 | 0.37 | 0.40 |
| N6 | 0.424 | 0.411 | 0.394 | 0.34 | 0.32 | 0.46 | 0.68 | 0.63 | 0.25 | 0.21 | 0.48 |
| N7 | 0.426 | 0.417 | 0.366 | 0.32 | 0.52 | 0.83 | 0.71 | 0.24 | 0.28 | 0.59 | 0.86 |
| N8 | 0.455 | 0.491 | 0.508 | 0.59 | 0.97 | 0.88 | 0.84 | 0.075 | 0.015 | 0.78 | 0.87 |
| N9 | 0.198 | 0.220 | 0.174 | 0.54 | 0.92 | 0.91 | 0.84 | 0.39 | 0.70 | 0.90 | 0.95 |
| N10 | 0.170 | 0.165 | 0.145 | 0.77 | 0.66 | 0.55 | 0.74 | 0.65 | 0.92 | 0.98 | 0.85 |
| N11 | 0.413 | 0.439 | 0.402 | 0.4 | 0.37 | 0.57 | 0.51 | 0.75 | 1.00 | 0.62 | 0.43 |
| N12 | 0.242 | 0.299 | 0.227 | 0.12 | 0.49 | 0.58 | 0.012 | 0.93 | 0.35 | 0.21 | 0.26 |
| N13 | 0.143 | 0.146 | 0.132 | 0.97 | 0.39 | 0.24 | 0.72 | 0.73 | 0.49 | 0.42 | 0.97 |
| N14 | 0.212 | 0.180 | 0.225 | 0.9 | 0.29 | 0.47 | 0.64 | 0.18 | 0.58 | 0.38 | 0.76 |
| N15 | 0.110 | 0.142 | 0.105 | 0.068 | 0.41 | 0.58 | 0.28 | 0.70 | 0.91 | 0.89 | 0.72 |
| N16 | 0.245 | 0.276 | 0.246 | 0.28 | 0.64 | 0.62 | 0.77 | 0.91 | 0.67 | 0.51 | 0.62 |
| N17 | 0.387 | 0.394 | 0.376 | 0.65 | 0.95 | 0.64 | 0.56 | 0.87 | 0.32 | 0.57 | 0.10 |
| N18 | 0.412 | 0.410 | 0.433 | 0.66 | 0.39 | 0.24 | 0.34 | 0.52 | 0.45 | 0.079 | 0.069 |
| N19 | 0.286 | 0.279 | 0.315 | 0.9 | 0.79 | 0.40 | 0.28 | 0.19 | 0.066 | 0.011 | 0.057 |
| T1 | 0.317 | 0.326 | 0.298 | 0.56 | 0.71 | 0.41 | 0.086 | 0.64 | 0.20 | 0.23 | 0.0041 |
| T2 | 0.335 | 0.372 | 0.417 | 0.55 | 0.34 | 0.18 | 0.21 | 0.055 | 0.18 | 0.021 | 0.019 |
| T3 | 0.221 | 0.244 | 0.260 | 0.41 | 0.12 | 0.26 | 0.36 | 0.59 | 0.022 | 0.017 | 0.013 |
| T4 | 0.350 | 0.369 | 0.420 | 0.7 | 0.27 | 0.37 | 0.53 | 0.080 | 0.00055 | 0.0039 | 0.0011 |
| T5 | 0.494 | 0.479 | 0.463 | 0.35 | 0.71 | 0.92 | 0.32 | 0.14 | 0.011 | 0.0032 | 0.0051 |
| T6 | 0.326 | 0.346 | 0.419 | 0.45 | 0.77 | 0.51 | 0.41 | 0.037 | 0.0032 | 0.019 | 0.011 |
| T7 | 0.141 | 0.161 | 0.184 | 0.29 | 0.83 | 0.22 | 0.29 | 0.0086 | 0.091 | 0.0090 | 0.020 |
| T8 | 0.142 | 0.154 | 0.176 | 0.77 | 0.32 | 0.33 | 0.31 | 0.038 | 0.017 | 0.030 | 0.032 |
| T9 | 0.223 | 0.233 | 0.272 | 0.43 | 0.14 | 0.11 | 0.14 | 0.91 | 0.016 | 0.033 | 0.013 |
| T10 | 0.292 | 0.309 | 0.368 | 0.44 | 0.060 | 0.0730 | 0.054 | 0.088 | 0.023 | 0.010 | 0.011 |
| A1 | 0.306 | 0.321 | 0.375 | 0.98 | 0.24 | 0.14 | 0.18 | 0.17 | 0.27 | 0.054 | 0.056 |
| A2 | 0.309 | 0.299 | 0.365 | 0.52 | 0.22 | 0.23 | 0.34 | 0.52 | 0.39 | 0.11 | 0.054 |
| A3 | 0.315 | 0.306 | 0.351 | 0.39 | 0.031 | 0.18 | 0.11 | 0.76 | 0.018 | 0.020 | 0.0016 |
| A4 | 0.192 | 0.185 | 0.240 | 0.94 | 0.040 | 0.13 | 0.19 | 0.37 | 0.022 | 0.015 | 0.020 |
| A5 | 0.479 | 0.478 | 0.602 | 0.97 | 0.23 | 0.67 | 0.75 | 0.0025 | 0.039 | 0.024 | 0.062 |
| A6 | 0.283 | 0.332 | 0.357 | 0.41 | 0.79 | 0.61 | 0.091 | 0.049 | 0.11 | 0.29 | 0.15 |
| A7 | 0.203 | 0.168 | 0.252 | 0.63 | 0.36 | 0.010 | 0.43 | 0.22 | 0.17 | 0.057 | 0.058 |
| D1 | 0.438 | 0.478 | 0.530 | 0.42 | 0.0037 | 0.17 | 0.50 | 0.013 | 0.065 | 0.13 | 0.21 |
| D2 | 0.359 | 0.412 | 0.472 | 0.39 | 0.58 | 0.72 | 0.36 | 0.0074 | 0.080 | 0.13 | 0.18 |
| D3 | 0.149 | 0.140 | 0.176 | 0.79 | 0.43 | 0.57 | 0.54 | 0.53 | 0.17 | 0.26 | 0.017 |
| D4 | 0.092 | 0.130 | 0.125 | 0.38 | 0.37 | 0.55 | 0.080 | 0.16 | 0.017 | ||
| D5 | 0.369 | 0.325 | 0.309 | 0.2 | 0.47 | 0.057 | 0.019 | ||||
| D6 | 0.446 | 0.395 | 0.416 | 0.42 | 0.16 | ||||||
| D7 | 0.369 | 0.319 | 0.342 | 0.26 | 0.33 | ||||||
MAF: minor allele frequency.
p-values for the haplotype tests were simulated global statistics and the significance level was set at 0.05, while p-values for the single allelic association were from the χ2 test. The grey-marked p-values are significant.
Starting SNP for the haplotype analyses.
AD-only indicates alcohol dependence (AD) without other drug dependence (DD).
AD+DD indicates AD comorbid with DD.
“hap” abbreviates ”haplotype.”
In contrast, the analyses for AD-only (case-control) gave rise to significant global haplotypic association only in A3-A6, D1-D3, A7-D3, and N12-N16 (p = 0.04 to 0.0037; Table 2); however, no specific individual haplotype was significant. The association analysis for AD+DD in the family sample (Table 3) showed that global haplotypes were significantly associated in the three risk or protective regions, NCAM1 exon 12, TTC12 exon 3 and the junction of ANKK1 and DRD2, corroborating those identified in the global association analysis of AD+DD in the case-control sample. There were small differences for these AD+DD association findings between the family and the case-control samples in terms of the statistical significance level, the number of significant haplotypes, and the extent to which the haplotypes extended along the chromosome in the three regions (compare Table 2 with Table 3). These differences could be due to lower statistical power associated with the family study design, a relatively small sample size in that study, and sampling variation, among other possible explanations.
Table 3.
Association analyses for alcohol dependence comorbid with drug dependence (AD+DD) for the family sample.
| Starting SNPa | Minor Allele | MAFb |
p-valuec |
|||
|---|---|---|---|---|---|---|
| Single SNP | 3SNP-hapd | 4SNP-hap | 5SNP-hap | |||
| N1 | T | 0.25 | 0.24 | 0.024 | 0.082 | 0.026 |
| N2 | G | 0.46 | 0.52 | 0.79 | 0.60 | 0.29 |
| N3 | C | 0.32 | 0.73 | 0.51 | 0.32 | 0.26 |
| N4 | C | 0.33 | 0.88 | 0.31 | 0.32 | 0.15 |
| N5 | C | 0.22 | 0.75 | 0.52 | 0.26 | 0.050 |
| N6 | G | 0.44 | 0.37 | 0.068 | 0.026 | 0.0074 |
| N7 | C | 0.44 | 0.37 | 0.039 | 0.011 | 0.0014 |
| N8 | T | 0.47 | 0.12 | 0.016 | 0.0056 | 0.027 |
| N9 | A | 0.18 | 0.010 | 0.0040 | 0.0049 | 0.0035 |
| N10 | C | 0.16 | 0.95 | 0.48 | 0.15 | 0.29 |
| N11 | C | 0.37 | 0.34 | 0.34 | 0.49 | 0.65 |
| N12 | C | 0.21 | 0.46 | 0.96 | 0.99 | 0.96 |
| N13 | G | 0.12 | 0.53 | 0.95 | 1.00 | 0.87 |
| N14 | A | 0.25 | 0.97 | 1.00 | 0.75 | 0.66 |
| N15 | G | 0.13 | 0.77 | 0.94 | 0.56 | 0.53 |
| N16 | T | 0.21 | 0.89 | 0.59 | 0.90 | 0.62 |
| N17 | A | 0.35 | 0.70 | 0.62 | 0.36 | 0.67 |
| N18 | C | 0.40 | 0.85 | 0.17 | 0.74 | 0.89 |
| N19 | A | 0.33 | 0.68 | 0.42 | 0.60 | 0.080 |
| T1 | A | 0.26 | 0.037 | 0.28 | 0.019 | 0.012 |
| T2 | G | 0.39 | 0.15 | 0.088 | 0.016 | 0.15 |
| T3 | G | 0.27 | 0.34 | 0.024 | 0.18 | 0.17 |
| T4 | G | 0.40 | 0.36 | 0.11 | 0.11 | 0.079 |
| T5 | C | 0.47 | 0.073 | 0.27 | 0.18 | 0.39 |
| T6 | G | 0.34 | 0.34 | 0.22 | 0.28 | 0.28 |
| T7 | G | 0.15 | 0.72 | 0.57 | 0.53 | 0.35 |
| T8 | T | 0.15 | 0.40 | 0.70 | 0.34 | 0.17 |
| T9 | G | 0.23 | 0.51 | 0.70 | 0.54 | 0.51 |
| T10 | A | 0.32 | 0.40 | 0.82 | 0.81 | 0.98 |
| A1 | C | 0.33 | 0.38 | 0.71 | 0.83 | 0.83 |
| A2 | A | 0.32 | 0.94 | 0.76 | 0.24 | 0.066 |
| A3 | T | 0.32 | 0.69 | 0.23 | 0.078 | 0.12 |
| A4 | A | 0.16 | 1.00 | 0.17 | 0.15 | 0.25 |
| A5 | C | 0.47 | 0.11 | 0.22 | 0.053 | 0.053 |
| A6 | G | 0.31 | 0.073 | 0.12 | 0.027 | 0.037 |
| A7 | A | 0.16 | 0.60 | 0.042 | 0.015 | 0.026 |
| D1 | C | 0.44 | 0.16 | 0.048 | 0.083 | 0.29 |
| D2 | C | 0.39 | 0.081 | 0.028 | 0.19 | 0.16 |
| D3 | A | 0.14 | 0.28 | 0.069 | 0.11 | 0.0093 |
| D4 | A | 0.09 | 0.033 | 0.42 | 0.11 | |
| D5 | G | 0.40 | 0.19 | 0.17 | ||
| D6 | T | 0.45 | 0.21 | |||
| D7 | T | 0.40 | 0.28 | |||
Starting SNP for the haplotype analyses.
MAF: minor allele frequency.
p-values were simulated and the significance level was set at 0.05. The grey-marked p-values are significant.
”hap” abbreviates ”haplotype.”
As we moved our focus in the association analysis from the significant global haplotypes to the specific individual haplotypic associations, additional interesting results were revealed. Overall in TTC12, the risk haplotypes extended from T1 to T9 and covered the whole gene (individual haplotype simulated p-values (pihs) = 0.0094 ~ 0.000015 for the case-control sample; 0.0094 ~ 0.0034 for the family sample; Table 4). In the family sample, all the risk haplotypes shared the same polymorphic nucleotides at the same risk locus, and no risk haplotypes contained markers downstream of SNP T5, which is in TTC12 exon 3. In the case-control sample, more risk haplotypes were identified, and those with >10% difference in haplotype frequency between cases and controls centered around T4-T5, i.e., TTC12 exon 3 as well, and contained two “yin-yang” haplotypes “G-A” or “T-C”. The difference in haplotype frequency (between cases and controls) decreased as the risk haplotype was more distant from TTC12 exon 3.
Table 4.
Summary results of significant individual haplotypes associated with alcohol dependence comorbid with drug dependence (AD+DD) in the TTC12 region.
| SNP# | T1 | T2 | T3 | T4 | T5 | T6 | T7 | T8 | T9 | # of famb | Hap-freqc | t-stat/Zd | I-hap pe | Global p | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Allelesa | G/A | A/G | A/G | T/G | A/C | A/G | A/G | G/T | A/G | pooled | control | case | diff | |||||
| Case-Control | 3-SNP | G | A | A | --- | 0.18 | 0.16 | 0.26 | 0.10 | 3.43 | 0.00065 | 0.00055 | ||||||
| A | A | G | --- | 0.12 | 0.11 | 0.15 | 0.04 | 3.56 | 0.00040 | 0.011 | ||||||||
| A | G | T | --- | 0.15 | 0.14 | 0.18 | 0.04 | 3.10 | 0.0020 | 0.0032 | ||||||||
| T | C | G | --- | 0.14 | 0.12 | 0.23 | 0.11 | 3.26 | 0.0012 | 0.00055 | ||||||||
| C | G | A | --- | 0.18 | 0.16 | 0.25 | 0.09 | 3.51 | 0.00048 | 0.011 | ||||||||
| G | A | G | --- | 0.35 | 0.33 | 0.41 | 0.08 | 2.72 | 0.0068 | 0.0032 | ||||||||
| 4-SNP | G | A | G | A | --- | 0.11 | 0.09 | 0.16 | 0.07 | 2.83 | 0.0049 | 0.021 | ||||||
| A | G | A | A | --- | 0.11 | 0.08 | 0.17 | 0.09 | 4.27 | 0.000023 | 0.017 | |||||||
| G | G | A | G | --- | 0.11 | 0.09 | 0.17 | 0.08 | 3.08 | 0.0022 | 0.017 | |||||||
| G | A | A | G | --- | 0.10 | 0.08 | 0.14 | 0.06 | 4.24 | 0.000027 | 0.0039 | |||||||
| A | A | G | T | --- | 0.12 | 0.11 | 0.15 | 0.04 | 3.65 | 0.00028 | 0.0032 | |||||||
| A | G | T | A | --- | 0.14 | 0.13 | 0.16 | 0.03 | 2.68 | 0.0076 | 0.019 | |||||||
| A | T | C | G | --- | 0.15 | 0.12 | 0.23 | 0.11 | 3.29 | 0.0011 | 0.017 | |||||||
| T | C | G | A | --- | 0.15 | 0.12 | 0.23 | 0.11 | 3.80 | 0.00016 | 0.0039 | |||||||
| C | G | A | G | --- | 0.18 | 0.16 | 0.25 | 0.09 | 3.81 | 0.00016 | 0.0032 | |||||||
| 5-SNP | G | A | G | A | A | --- | 0.09 | 0.07 | 0.15 | 0.08 | 4.25 | 0.000025 | 0.019 | |||||
| G | G | G | A | G | --- | 0.11 | 0.09 | 0.16 | 0.07 | 3.11 | 0.0019 | 0.019 | ||||||
| A | G | A | A | G | --- | 0.07 | 0.05 | 0.12 | 0.07 | 4.37 | 0.000015 | 0.013 | ||||||
| G | G | A | G | A | --- | 0.11 | 0.09 | 0.16 | 0.07 | 3.58 | 0.00038 | 0.013 | ||||||
| G | A | A | A | G | --- | 0.10 | 0.09 | 0.12 | 0.03 | 2.61 | 0.0094 | 0.0011 | ||||||
| G | A | A | G | T | --- | 0.10 | 0.08 | 0.14 | 0.06 | 3.95 | 0.000087 | 0.0011 | ||||||
| A | A | G | T | A | --- | 0.11 | 0.11 | 0.14 | 0.03 | 3.00 | 0.0028 | 0.0051 | ||||||
| A | A | T | C | G | --- | 0.14 | 0.12 | 0.23 | 0.11 | 3.24 | 0.0013 | 0.019 | ||||||
| A | T | C | G | A | --- | 0.15 | 0.12 | 0.23 | 0.11 | 3.56 | 0.00040 | 0.013 | ||||||
| T | C | G | A | G | --- | 0.15 | 0.12 | 0.23 | 0.11 | 3.71 | 0.00023 | 0.0011 | ||||||
| C | G | A | G | A | --- | 0.10 | 0.08 | 0.16 | 0.08 | 3.81 | 0.00015 | 0.0051 | ||||||
| Family | G | G | A | 33 | 0.26 | --- | --- | --- | 2.80 | 0.0051 | 0.024 | |||||||
| G | G | G | G | 32 | 0.24 | --- | --- | --- | 2.60 | 0.0094 | 0.019 | |||||||
| G | G | G | A | 32 | 0.25 | --- | --- | --- | 2.68 | 0.0075 | 0.016 | |||||||
| G | G | G | G | A | 30 | 0.24 | --- | --- | --- | 2.93 | 0.0034 | 0.012 | ||||||
The allele listed on top is the common allele. Black square with white allele denotes risk haplotypes.
Number of informative families.
Haplotype frequency for the pooled (combined the case and control samples for the case-control design, but representing overall estimates of haplotype frequency for the family design), control, case, and the difference between cases and controls.
“t-stat” is the t-statistic calculated for the case-control sample, and “Z” for the family sample.
Individual haplotype p-value.
Another region important for AD+DD was observed where the 3′ regions of DRD2 and ANKK1 meet (Table 5). The risk variants were the significant individual risk haplotype G-G-G-G-G in A6-D3 (pihs = 0.0093) identified in the family sample, or G-C-G-G-G in A4-D1 (pihs = 0.0087) in the case-control sample. All 11 risk haplotypes (pihs = 0.017 ~ 0.0028) share the same polymorphic nucleotides at the corresponding SNP loci, consistent with all of them identifying the same risk locus, i.e., the locus was tagged with the same set of markers. The various risk haplotypes, between samples from the two designs, overlapped for 3 SNPs (A6-D1). This common region includes the region of DRD2^C957T (rs6277) in exon 7 of DRD2, and SNP-A6 and SNP-A7 (rs4938016 and rs1800497) in exon 8 of ANKK1.
Table 5.
Summary results of significant individual haplotypes associated with alcohol dependence comorbid with drug dependence (AD+DD) in the ANKK1 and DRD2 regions.
| SNP# | A3 | A4 | A5 | A6 | A7 | D1 | D2 | D3 | # of fam b | Hap-freqc | t-stat/Zd | I-hap pe | Global p | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Allelesa | C/T | G/A | C/T | C/G | G/A | A/G | A/G | G/A | pooled | control | case | diff | ||||
| Case-control | G | C | G | --- | 0.30 | 0.28 | 0.35 | 0.07 | 3.01 | 0.0028 | 0.022 | |||||
| C | G | G | --- | 0.29 | 0.27 | 0.34 | 0.07 | 2.99 | 0.0029 | 0.039 | ||||||
| C | G | C | G | --- | 0.13 | 0.12 | 0.16 | 0.04 | 2.81 | 0.0051 | 0.020 | |||||
| G | C | G | G | --- | 0.28 | 0.27 | 0.33 | 0.06 | 2.76 | 0.0060 | 0.015 | |||||
| C | G | G | G | --- | 0.28 | 0.27 | 0.34 | 0.07 | 2.77 | 0.0058 | 0.024 | |||||
| G | C | G | G | G | 0.28 | 0.27 | 0.33 | 0.06 | 2.63 | 0.0087 | 0.020 | |||||
| Family | G | G | G | 34 | 0.24 | --- | --- | --- | 2.40 | 0.017 | 0.042 | |||||
| G | G | G | 34 | 0.24 | --- | --- | --- | 2.76 | 0.0059 | 0.048 | ||||||
| G | G | G | G | 30 | 0.24 | --- | --- | --- | 2.73 | 0.0064 | 0.027 | |||||
| G | G | G | G | 33 | 0.24 | --- | --- | --- | 2.59 | 0.0096 | 0.015 | |||||
| G | G | G | G | G | 29 | 0.24 | --- | --- | --- | 2.60 | 0.0093 | 0.037 | ||||
The allele listed on top is the common allele. Black square with white alleles in a row denotes risk haplotypes.
Number of informative families.
Haplotype frequency for the pooled (combined the case and control samples for the case-control design, but representing overall estimates of haplotype frequency for the family design), control, case, and the difference between cases and controls.
“t-stat” is the t-statistic calculated for the case-control sample, and “Z” for the family sample.
Individual haplotype p-value.
The third susceptibility region for AD+DD occurred at exon 12 of NCAM1 (Table 6); this association was only observed in the family sample. In the case-control sample, we detected a globally significant 3-SNP haplotype (p = 0.015, Table 2), but no specific individual associated haplotype was found. In the family sample, there was evidence for risk (G-A-A) or protective (T-C-A) alleles in the haplotype N8-N10 (Table 6, p = 0.016 ~ 0.0029), located around exon 12 of NCAM1. All of the protective haplotypes contained the same nucleotides at the corresponding SNP loci, consistent with tagging of the same marker locus, and all had the “C” nucleotide in SNP-N9. The risk haplotype N8-N10 of the “G-A-A” allele (p = 0.0036) had a haplotype frequency (12%) that was lower than those of several protective haplotypes (22% ~ 42%) in this family sample.
Table 6.
Summary results of significant individual haplotypes associated with AD+DD in the NCAM1 region.
| SNP# | N6 | N7 | N8 | N9 | N10 | N11 | N12 | N13 | # of famb | Hap-freqc | t-stat/Zd | I-hap pe | Global p | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Allelesa | A/G | T/C | G/T | C/A | A/C | T/C | T/C | T/G | ||||||
| Family | 3-, 4- or 5-SNP haplotypes | G | A | A | 25.1 | 0.12 | 2.91 | 0.0036 | 0.016 | |||||
| T | C | A | 52 | 0.40 | −2.45 | 0.014 | 0.016 | |||||||
| C | A | T | 37 | 0.42 | −2.40 | 0.016 | 0.0040 | |||||||
| T | T | C | A | 45.4 | 0.38 | −2.82 | 0.0048 | 0.011 | ||||||
| T | C | A | T | 30.8 | 0.24 | −2.58 | 0.0098 | 0.0056 | ||||||
| C | A | T | T | 37.8 | 0.37 | −2.43 | 0.015 | 0.0049 | ||||||
| A | T | T | C | A | 44.4 | 0.38 | −2.98 | 0.0029 | 0.0074 | |||||
| T | T | C | A | T | 27.2 | 0.22 | −2.90 | 0.0037 | 0.0014 | |||||
| T | C | A | T | T | 28.3 | 0.23 | −2.55 | 0.011 | 0.027 | |||||
| C | A | T | T | T | 36.8 | 0.36 | −2.90 | 0.0038 | 0.0035 | |||||
The allele listed on top is the common allele. Black square with white alleles in a row denotes risk haplotypes, while white square with black alleles in a row denotes protective haplotypes.
Number of informative families.
Haplotype frequency for the pooled (combined case and control samples for the case-control design, but representing overall estimates of haplotype frequency for the family design), control, case, and the difference between cases and controls.
“t-stat” is the t-statistic calculated for the case-control sample, and “Z” for the family sample.
Individual haplotype p-value.
LD comparisons between cases and controls for AD+DD and AD-only were performed using LD Contrast Test software. Two regions, one around TTC12 exon 3 (SNPs T2 to T5) and another around ANKK1 exon 2 (SNPs T10 to A3), were identified as differing in the standardized composite LD for AD+DD (p = 0.018 and 0.0012, respectively) and AD-only (p = 0.00005 and 0.00013, respectively). The LD difference gradually disappeared as the number of SNPs that were included increased. The differences in pairwise LD matrices are displayed using ellipse plots (Figure 3) in which the shape of an ellipse indicates the magnitude of LD and the direction shows the sign of the disequilibrium. The more circular ellipse reflects a low degree of LD, and the ellipse oriented at 45° suggests a positive value of LD. The two regions, identified by the LD contrast test, are composed of markers in stronger LD among cases than controls. This phenomenon occurs when selection causes transmission of more disease alleles and/or haplotypes in common among cases than controls. The two identified regions of differential LD between the two groups further corroborate the results of haplotype association analysis and provide evidence of harboring disease variants in the regions.
Figure 3.
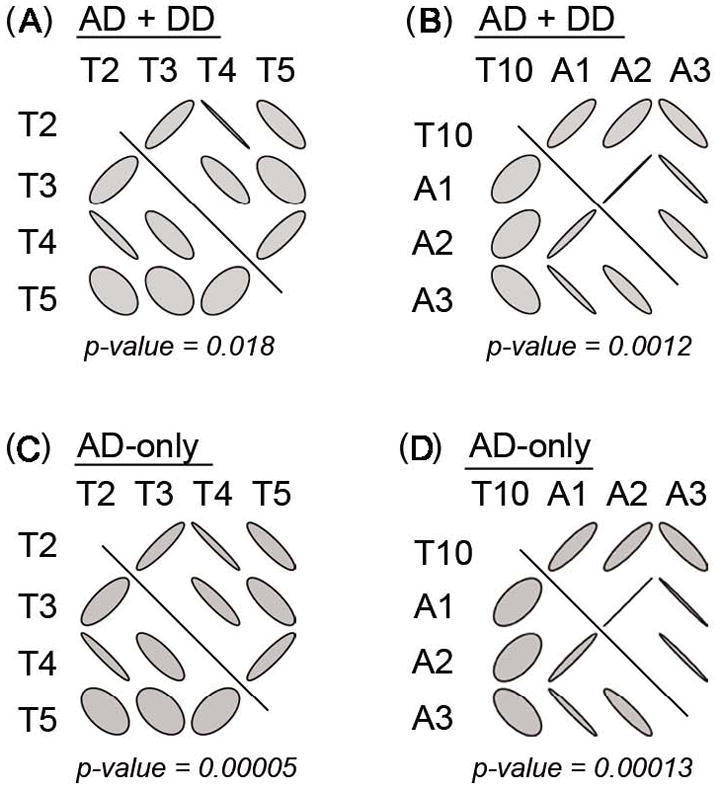
Ellipse plots for the results of the LD contrast tests. For each ellipse plot, the pairwise LD matrices are displayed for the contrast of cases and controls, respectively, above and below the diagonal line. The shape of an ellipse indicates the magnitude of LD and the direction shows the sign of the disequilibrium. The more circular shape of an ellipse reflects a low degree of LD, and 45o-oriented ellipses suggest a positive value of LD.
Population stratification originating from cryptic genetic patterns was examined using a panel of 37 short tandem repeat (STR) markers that we designed to distinguish among major US populations (Yang et al., 2005a; Yang et al., 2005b). There was no evidence of population stratification in these study samples. We conclude that the significant association findings in the case-control sample could not be attributed to population stratification.
In summary, we found evidence that (1) three regions in EAs harbor variants predisposing to AD comorbid with DD: exon 3 in TTC12 (the region for which evidence for association is the strongest and most consistent), the two adjacent 3′ regions of DRD2 and ANKK1 (i.e., between exon 7 of DRD2 and exon 8 of ANKK1), and risk or protective variants close to exon 12 to intron 13 of NCAM1; (2) For AD without DD comorbidity, NCAM1 intron 14 to 18 and the junction of ANKK1 and DRD2 were associated globally; no individual haplotype was significantly associated. We showed in our previous study that overall, risk for AD, irrespective of comorbidity of DD, was attributable in part to variants in four regions: exon 3 of TTC12, exon 12 of NCAM1, and exons 2 and 5 of ANKK1, but not the junction of DRD2 and ANKK1.
Discussion
Findings from this study support the hypothesis that heterogeneity of the SD disorders studied and their high rates of comorbidity are reflected in genetic association findings. The contrast among the findings for the four phenotypes – AD+DD, AD-only, AD irrespective of DD comorbidity (AD-all), and ND irrespective of AD or DD (ND-all) from the current and prior studies, provides important insight not evident when the previous studies are considered individually, as summarized in Table 7 and Figure 1(B). In a previous article (Gelernter et al., 2006), we reported very strong evidence of association of ND (ND-all) to a single haplotype spanning TTC12 and ANKK1. In another recent article (Yang et al., 2007), we reported that risk for AD (AD-all) is attributable in part to variants in four regions within the NTAD gene cluster: exon 3 of TTC12, exon 12/intron13 of NCAM1, and exons 2 and 5 of ANKK1. LD contrasts between cases and controls supported historical selection at TTC12 exon 3 and ANKK1 exon 2, and corroborated the identified risk variants from the association analysis. Further, the risk variants in TTC12 suggested a fundamental neurobiological basis for SD that involves dopaminergic neurotransmission, since the armadillo repeat domain encoded by TTC12 interacts with dopamine through the Wnt signal transduction pathway and may have effects on dopamine neuronal development in the ventral midbrain (Castelo-Branco and Arenas, 2006; Castelo-Branco et al., 2006). In the present article, we examined the effects of DD comorbidity on the observed association, with the goal of evaluating its specificity. To that end, we undertook analyses comparing AD with or without DD comorbidity (AD+DD vs AD-only) using the same 43 SNP markers in the NTAD gene cluster. By doing so, we hoped to learn whether AD or DD was the primary driver of the observed association.
Table 7.
Comparison of the three studies using the same 43 SNPs spanning NCAM1-TTC12-ANKK1-DRD2 gene cluster.
| Study | Gelernter et al, 2006 | Yang et al, 2007 | Current study |
|---|---|---|---|
| Phenotypes | ND-alla | AD-allb | AD+DDc AD-onlyd |
| Study Design | Family | Family, Case-control | Family, Case-control |
| Sample Size | 1615 subjects | 1220 subjects | 1090 subjects (subset of the sample in Yang et al, 2007) |
| Ethnicity | EAe and AAf | EA only | EA only |
| Sample allocation | 319 AA families 313 EA families (independent of the other two studies) |
143 families 318 affecteds 414 controls |
112 families 136 AD+DDs 166 AD-onlys 414 controls |
| Haplotypic association analysis | SNPg selection based on the single SNP association findings and the LDh block structure. Four SNPs were selected for haplotype analysis. | Exhaustive search for haplotypic association by sliding window | Same as Yang et al, 2007 |
| Results: regions with evidence of association |
|
TTC12 exon 3, NCAM1 exon 12, ANKK1 exon 2 & exon 5 |
|
ND-all denotes nicotine dependence (ND) irrespective of other substance dependence (SD).
AD-all denotes alcohol dependence (AD) irrespective of other SD.
AD+DD denotes AD comorbid with DD.
AD-only denotes AD without other DD.
“EA” abbreviates “European American.”
“AA” abbreviates “African American.”
“SNP” abbreviates “single nucleotide polymorphism.”
“LD” abbreviates “linkage disequilibrium.”
The results of these analyses suggest that partitioning the total AD sample reduced genetic heterogeneity. Figure 1 summarizes the location of the four genes in the NTAD gene cluster, the distribution of the 43 SNPs, and the identified susceptibility regions. For AD-all, risk regions were consistent in both samples, with the exception of NCAM1 exon 12 (which was identified as associated only in the family sample). In particular, TTC12 exon 3 was significant in both global and individual haplotypic associations and in both samples. A broader region from ANKK1 exon 2 to DRD2 intron 1 was significant in the global association analyses in both samples, and was narrowed to ANKK1 exons 2 and 5 in the individual specific haplotypes for the case-control sample.
The junction of the 3′ ends of ANKK1 and DRD2 was globally associated with all three phenotypes involving AD (Figure 1), but harbored specific risk variants only for AD+DD. The global significance might be due to a few weak or moderate variants in this region but the individual effect was not strong enough to meet the criterion for statistical significance. A larger sample size for AD-only would be helpful to examine this hypothesis and to detect smaller specific effects from this region. On the other hand, the heterogeneity of AD-all statistically may well have diluted the association signal in this region. Thus, it appears that those strong effects depend on the presence of comorbidity, which might reflect a different neurobiological mechanism or more severe form of substance dependence. This result underscores the importance of the phenotype definition, and possible effects of comorbidity, in studies of complex traits such as substance dependence.
The results for variants mapped to TTC12 for AD+DD were similar to our previously published results in the AD-all analyses, but yielded much greater statistical significance (e.g., with the p-value being reduced from 0.00067 to 0.000023 for the A-G-A-A haplotype at markers T3-T6; see Table 5), and additional specific haplotypes being significant, whereas in the earlier analysis they were not. On the other hand, TTC12 was not observed to harbor risk variants for AD-only, a finding that was probably not due to lack of power, as the sample size was greater for AD-only than for AD+DD.
Interestingly, the LD contrast test supports selection at TTC12 exon 3 for AD-only (p-value = 0.00005), and the LD contrast plot shows the difference between cases and controls (see Figure 3C). “One situation when the LD contrast test is particularly powerful is when dissimilar haplotypes (‘yin yang haplotypes’) carry susceptibility. This could either indicate a true heterogeneity of the mechanism, or that they are ‘proxies’ for an unobserved causative mutation or haplotype.” (personal communication from Dr. Dmitri Zaykin, January 12th, 2007). The haplotypes in TTC12 exon 3 in this sample are exactly “yin-yang haplotypes.” Therefore, it is unknown why there was selection at TTC12 exon 3 for AD-only but no evidence for association emerged. Additional studies with large samples are necessary to validate these findings. It should be noted, however, that in studies from COGA, evidence for an association of GABRA2 and CHRM2 previously observed with AD came only from individuals with comorbid DD, with no evidence of association among individuals with AD but no DD (Agrawal et al., 2006; Dick et al., 2007a). We conclude that TTC12 exon 3 is a risk region for AD+DD, with evidence of association with AD-all due only to the contribution by the comorbid AD+DD part of the sample. This suggests that the analysis of samples with drug dependence in the absence of alcohol dependence is warranted, to ascertain whether the locus predisposes to drug dependence specifically or, as suggested here, to the comorbid phenotype.
The major risk variants identified in the ND study delineate a haplotype spanning TTC12 and ANKK1. This haplotype is composed of four SNPs, T6-A1-A3-A5 (Figure 1), where T6 is very close to the identified risk region in TTC12 exon 3 from the other two studies, and the other 3 SNPs are located in the small ANKK1 region extending from intron 1 to exon 6. This study was analyzed by a different approach, without the “sliding window” exhaustive search for risk variants employed in the other two studies. The identified risk variants were very strongly supported in terms of their statistical significance and were close to the risk regions from the other two studies. In addition, since the ND sample was ascertained for a sib-pair linkage analysis for the primary traits of cocaine and opioid dependence, there is especially high comorbidity with DD in this sample.
We conclude that exon 3 of TTC12, and the region close to exon 12 of NCAM1, both regulate risk for AD+DD. Variants at the two 3′ ends of ANKK1 and DRD2 also regulate risk for AD, with effects depending on comorbidity with DD. The complexity of these relationships, many of which were replicated in our independent samples, may explain prior inconsistent results. Further replication in other samples and functional analysis of this allelic variation are warranted.
Supplementary Material
Online Supplemental Figures:
LD map structure and locations of 43 SNPs in the NTAD gene cluster. The LD maps for the family (AD+DD) and case-control samples (AD+DD, AD-only and control) are shown. The measure of LD (D′) among all possible pairs of SNPs is shown graphically according to the shade of red where white represents very low D′ and dark red represents very high D′.
Supplemental Figure 1. Cases: AD+DD
Supplemental Figure 2. Cases: AD-only
Supplemental Figure 3. Controls
Supplemental Figure 4. AD+DD Family sample
Figure 2.
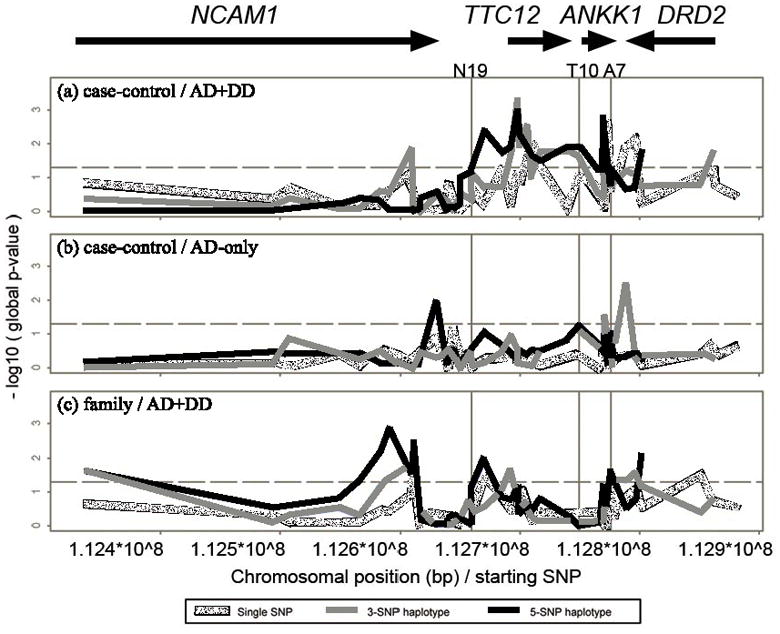
Results of single SNP and haplotype association analyses for the case-control samples of AD+DD and AD-only, and AD+DD of family samples. Global simulated p-values are shown. For haplotype analyses, the value at each SNP location represents the haplotype with SNPs starting from the current location downward from the NCAM1 direction to DRD2. The horizontal dashed lines correspond to type I error rate of 0.05. The three vertical lines from left to right indicate the positions of SNPs N19, T10 and A7, respectively.
Acknowledgments
We thank the volunteer families and individuals who participated in this research study, and Ann Marie Lacobelle, Lisa Naito, and Yakov Lozovatsky for their excellent technical assistance. This work was supported by NIH grants R01 AA11330, P50 AA12870, K08 AA13732, K24 AA13736, R01 AA016015, R01 DA12690, R01 DA12849, K24 DA15105, and M01 RR06192 and the U.S. Department of Veterans Affairs (the VA Connecticut–Massachusetts Mental Illness Research, Education and Clinical Center [MIRECC].
Sources of Support:
This work was supported by NIH grants R01 AA11330, P50 AA12870, P60 AA03510, K08 AA13732, K24 AA13736, R01 AA016015, R01 DA12690, R01 DA12849, K24 DA15105, and M01 RR06192 and the U.S. Department of Veterans Affairs (the VA Connecticut–Massachusetts Mental Illness Research, Education and Clinical Center [MIRECC].
References
- Agrawal A, Edenberg HJ, Foroud T, Bierut LJ, Dunne G, Hinrichs AL, Nurnberger JI, Crowe R, Kuperman S, Schuckit MA, Begleiter H, Porjesz B, Dick DM. Association of GABRA2 with drug dependence in the collaborative study of the genetics of alcoholism sample. Behav Genet. 2006;36(5):640–50. doi: 10.1007/s10519-006-9069-4. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Baum AE, Akula N, Cabanero M, Cardona I, Corona W, Klemens B, Schulze TG, Cichon S, Rietschel M, Nothen MM, Georgi A, Schumacher J, Schwarz M, Abou Jamra R, Hofels S, Propping P, Satagopan J, Detera-Wadleigh SD, Hardy J, McMahon FJ. A genome-wide association study implicates diacylglycerol kinase eta (DGKH) and several other genes in the etiology of bipolar disorder. Mol Psychiatry. 2007 doi: 10.1038/sj.mp.4002012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelo-Branco G, Arenas E. Function of Wnts in dopaminergic neuron development. Neurodegener Dis. 2006;3(1–2):5–11. doi: 10.1159/000092086. [DOI] [PubMed] [Google Scholar]
- Castelo-Branco G, Rawal N, Arenas E. GSK-3beta inhibition/beta-catenin stabilization in ventral midbrain precursors increases differentiation into dopamine neurons. J Cell Sci. 2004;117(Pt 24):5731–7. doi: 10.1242/jcs.01505. [DOI] [PubMed] [Google Scholar]
- Castelo-Branco G, Sousa KM, Bryja V, Pinto L, Wagner J, Arenas E. Ventral midbrain glia express region-specific transcription factors and regulate dopaminergic neurogenesis through Wnt-5a secretion. Mol Cell Neurosci. 2006;31(2):251–62. doi: 10.1016/j.mcn.2005.09.014. [DOI] [PubMed] [Google Scholar]
- Compton WM, Thomas YF, Stinson FS, Grant BF. Prevalence, correlates, disability, and comorbidity of DSM-IV drug abuse and dependence in the United States: results from the national epidemiologic survey on alcohol and related conditions. Arch Gen Psychiatry. 2007;64(5):566–76. doi: 10.1001/archpsyc.64.5.566. [DOI] [PubMed] [Google Scholar]
- Connor JP, Young RM, Lawford BR, Saunders JB, Ritchie TL, Noble EP. Heavy nicotine and alcohol use in alcohol dependence is associated with D2 dopamine receptor (DRD2) polymorphism. Addict Behav. 2007;32(2):310–9. doi: 10.1016/j.addbeh.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Dick DM, Agrawal A, Wang JC, Hinrichs A, Bertelsen S, Bucholz KK, Schuckit M, Kramer J, Nurnberger J, Jr, Tischfield J, Edenberg HJ, Goate A, Bierut LJ. Alcohol dependence with comorbid drug dependence: genetic and phenotypic associations suggest a more severe form of the disorder with stronger genetic contribution to risk. Addiction. 2007a;102(7):1131–9. doi: 10.1111/j.1360-0443.2007.01871.x. [DOI] [PubMed] [Google Scholar]
- Dick DM, Wang JC, Plunkett J, Aliev F, Hinrichs A, Bertelsen S, Budde JP, Goldstein EL, Kaplan D, Edenberg HJ, Nurnberger J, Jr, Hesselbrock V, Schuckit M, Kuperman S, Tischfield J, Porjesz B, Begleiter H, Bierut LJ, Goate A. Family-based association analyses of alcohol dependence phenotypes across DRD2 and neighboring gene ANKK1. Alcohol Clin Exp Res. 2007b;31(10):1645–53. doi: 10.1111/j.1530-0277.2007.00470.x. [DOI] [PubMed] [Google Scholar]
- Edenberg HJ, Xuei X, Wetherill LF, Bierut L, Bucholz K, Dick DM, Hesselbrock V, Kuperman S, Porjesz B, Schuckit MA, Tischfield JA, Almasy LA, Nurnberger JI, Jr, Foroud T. Association of NFKB1, which encodes a subunit of the transcription factor NF-kappaB, with alcohol dependence. Hum Mol Genet. 2008;17(7):963–70. doi: 10.1093/hmg/ddm368. [DOI] [PubMed] [Google Scholar]
- Fossella J, Green AE, Fan J. Evaluation of a structural polymorphism in the ankyrin repeat and kinase domain containing 1 (ANKK1) gene and the activation of executive attention networks. Cogn Affect Behav Neurosci. 2006;6(1):71–8. doi: 10.3758/cabn.6.1.71. [DOI] [PubMed] [Google Scholar]
- Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D. The structure of haplotype blocks in the human genome. Science. 2002;296(5576):2225–9. doi: 10.1126/science.1069424. Epub 2002 May 23. [DOI] [PubMed] [Google Scholar]
- Gelernter J, Panhuysen C, Weiss R, Brady K, Hesselbrock V, Rounsaville B, Poling J, Wilcox M, Farrer L, Kranzler HR. Genomewide linkage scan for cocaine dependence and related traits: significant linkages for a cocaine-related trait and cocaine-induced paranoia. Am J Med Genet B Neuropsychiatr Genet. 2005;136(1):45–52. doi: 10.1002/ajmg.b.30189. [DOI] [PubMed] [Google Scholar]
- Gelernter J, Yu Y, Weiss R, Brady K, Panhuysen C, Yang BZ, Kranzler HR, Farrer L. Haplotype spanning TTC12 and ANKK1, flanked by the DRD2 and NCAM1 loci, is strongly associated to nicotine dependence in two distinct American populations. Hum Mol Genet. 2006;15(24):3498–507. doi: 10.1093/hmg/ddl426. [DOI] [PubMed] [Google Scholar]
- Horvath S, Xu X, Laird NM. The family based association test method: strategies for studying general genotype--phenotype associations. Eur J Hum Genet. 2001;9(4):301–6. doi: 10.1038/sj.ejhg.5200625. [DOI] [PubMed] [Google Scholar]
- Horvath S, Xu X, Lake SL, Silverman EK, Weiss ST, Laird NM. Family-based tests for associating haplotypes with general phenotype data: application to asthma genetics. Genet Epidemiol. 2004;26(1):61–9. doi: 10.1002/gepi.10295. [DOI] [PubMed] [Google Scholar]
- Huang W, Payne TJ, Ma JZ, Beuten J, Dupont RT, Inohara N, Li MD. Significant association of ANKK1 and detection of a functional polymorphism with nicotine dependence in an African-American sample. Neuropsychopharmacology. 2008 Mar 19; doi: 10.1038/npp.2008.37. [DOI] [PubMed] [Google Scholar]
- Katoh M, Katoh M. Identification and characterization of TPARM gene in silico. Int J Oncol. 2003;23(4):1213–7. [PubMed] [Google Scholar]
- Neville MJ, Johnstone EC, Walton RT. Identification and characterization of ANKK1: a novel kinase gene closely linked to DRD2 on chromosome band 11q23.1. Hum Mutat. 2004;23(6):540–5. doi: 10.1002/humu.20039. [DOI] [PubMed] [Google Scholar]
- Pierucci-Lagha A, Gelernter J, Chan G, Arias A, Cubells JF, Farrer L, Kranzler HR. Reliability of DSM-IV diagnostic criteria using the semi-structured assessment for drug dependence and alcoholism (SSADDA) Drug Alcohol Depend. 2007;91(1):85–90. doi: 10.1016/j.drugalcdep.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierucci-Lagha A, Gelernter J, Feinn R, Cubells JF, Pearson D, Pollastri A, Farrer L, Kranzler HR. Diagnostic reliability of the Semi-structured Assessment for Drug Dependence and Alcoholism (SSADDA) Drug Alcohol Depend. 2005;80(3):303–12. doi: 10.1016/j.drugalcdep.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Sasabe T, Furukawa A, Matsusita S, Higuchi S, Ishiura S. Association analysis of the dopamine receptor D2 (DRD2) SNP rs1076560 in alcoholic patients. Neurosci Lett. 2007;412(2):139–42. doi: 10.1016/j.neulet.2006.10.064. [DOI] [PubMed] [Google Scholar]
- Schaid DJ, Rowland CM, Tines DE, Jacobson RM, Poland GA. Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am J Hum Genet. 2002;70(2):425–34. doi: 10.1086/338688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahmoradgoli Najafabadi M, Ohadi M, Joghataie MT, Valaie F, Riazalhosseini Y, Mostafavi H, Mohammadbeigi F, Najmabadi H. Association between the DRD2 A1 allele and opium addiction in the Iranian population. Am J Med Genet B Neuropsychiatr Genet. 2005;134(1):39–41. doi: 10.1002/ajmg.b.30117. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Begleiter H, Porjesz B, Fowler JS, Telang F, Wong C, Ma Y, Logan J, Goldstein R, Alexoff D, Thanos PK. High levels of dopamine D2 receptors in unaffected members of alcoholic families: possible protective factors. Arch Gen Psychiatry. 2006;63(9):999–1008. doi: 10.1001/archpsyc.63.9.999. [DOI] [PubMed] [Google Scholar]
- Xu K, Lichtermann D, Lipsky RH, Franke P, Liu X, Hu Y, Cao L, Schwab SG, Wildenauer DB, Bau CH, Ferro E, Astor W, Finch T, Terry J, Taubman J, Maier W, Goldman D. Association of specific haplotypes of D2 dopamine receptor gene with vulnerability to heroin dependence in 2 distinct populations. Arch Gen Psychiatry. 2004;61(6):597–606. doi: 10.1001/archpsyc.61.6.597. [DOI] [PubMed] [Google Scholar]
- Yang BZ, Kranzler HR, Zhao H, Gruen JR, Luo X, Gelernter J. Association of haplotypic variants in DRD2, ANKK1, TTC12 and NCAM1 to alcohol dependence in independent case control and family samples. Hum Mol Genet. 2007;16(23):2844–53. doi: 10.1093/hmg/ddm240. [DOI] [PubMed] [Google Scholar]
- Yang BZ, Zhao H, Kranzler HR, Gelernter J. Characterization of a likelihood based method and effects of markers informativeness in evaluation of admixture and population group assignment. BMC Genet. 2005a;6:50. doi: 10.1186/1471-2156-6-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang BZ, Zhao H, Kranzler HR, Gelernter J. Practical population group assignment with selected informative markers: characteristics and properties of Bayesian clustering via STRUCTURE. Genet Epidemiol. 2005b;28(4):302–12. doi: 10.1002/gepi.20070. [DOI] [PubMed] [Google Scholar]
- Zaykin DV, Meng Z, Ehm MG. Contrasting linkage-disequilibrium patterns between cases and controls as a novel association-mapping method. Am J Hum Genet. 2006;78(5):737–46. doi: 10.1086/503710. Epub 2006 Mar 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Online Supplemental Figures:
LD map structure and locations of 43 SNPs in the NTAD gene cluster. The LD maps for the family (AD+DD) and case-control samples (AD+DD, AD-only and control) are shown. The measure of LD (D′) among all possible pairs of SNPs is shown graphically according to the shade of red where white represents very low D′ and dark red represents very high D′.
Supplemental Figure 1. Cases: AD+DD
Supplemental Figure 2. Cases: AD-only
Supplemental Figure 3. Controls
Supplemental Figure 4. AD+DD Family sample