

Abstract
The hypocretin (HCRT) neurons are located only in the perifornical area of the lateral hypothalamus and heavily innervate the cholinergic neurons in the basal forebrain (BF), histamine neurons in the tuberomammillary nucleus (TMN), and the noradrenergic locus ceruleus (LC) neurons, three neuronal populations that have traditionally been implicated in regulating arousal. Based on the innervation, HCRT neurons may regulate arousal by driving these downstream arousal neurons. Here, we directly test this hypothesis by a simultaneous triple lesion of these neurons using three saporin-conjugated neurotoxins. Three weeks after lesion, the daily levels of wake were not changed in rats with double or triple lesions, although rats with triple lesions were asleep more during the light-to-dark transition period. The double- and triple-lesioned rats also had more stable sleep architecture compared with nonlesioned saline rats. These results suggest that the cholinergic BF, TMN, and LC neurons jointly modulate arousal at a specific circadian time, but they are not essential links in the circuitry responsible for daily levels of wake, as traditionally hypothesized.
Keywords: 192-IgG-saporin, acetylcholine, Ach, histamine, norepinephrine, noradrenaline, sleep, waking
Introduction
A distributed network of neurons in the hypothalamus and brainstem are hypothesized to be responsible for arousal. Chief among these are neurons containing the neuropeptide hypocretin (HCRT), also known as orexin, located in the perifornical region of the lateral hypothalamus (de Lecea et al., 1998; Peyron et al., 1998). Loss of these neurons has been linked to the sleep disorder narcolepsy (Peyron et al., 2000; Thannickal et al., 2000). Consistent with such a neuronal degeneration, narcoleptic patients have reduced CSF levels of HCRT (Nishino et al., 2001). Mice (Chemelli et al., 1999; Hara et al., 2001) and rats (Gerashchenko et al., 2001a, 2003; Beuckmann et al., 2004) with either loss of HCRT or of the HCRT neurons exhibit symptoms of narcolepsy. Canines with narcolepsy possess a mutation in the hypocretin-2 receptor (Lin et al., 1999) and mice with a deletion of this receptor (Willie et al., 2003) also show symptoms of narcolepsy.
The HCRT neurons project to virtually the entire brain and spinal cord but the innervation of the neuronal populations traditionally implicated in arousal is especially heavy (de Lecea et al., 1998; Peyron et al., 1998). Based on this innervation, and the finding that loss of HCRT leads to a profound sleep disturbance, HCRT may drive arousal by acting on these downstream arousal neurons (Saper et al., 2005). Thus, lesioning of the downstream arousal neurons should decrease wake and increase sleep, including narcoleptic behavior. Here, we test this hypothesis by using three saporin-based neurotoxins to lesion the basal forebrain (BF) cholinergic neurons, the locus ceruleus (LC), and the tuberomammillary nucleus (TMN), and then determining whether rats with such a triple lesion sleep more. These three neuronal populations have been implicated in arousal based on pharmacological studies (Jones, 2005) and on the firing patterns of their respective neurons (Chu and Bloom, 1973; John et al., 2004; Lee et al., 2005). HCRT receptors are present on their neurons (Greco and Shiromani, 2001; Marcus et al., 2001), and application of HCRT in the BF (Blanco-Centurion et al., 2006b), TMN (Huang et al., 2001), or LC (Bourgin et al., 2000) has a potent arousing effect. Individual lesions of the BF cholinergic neurons (Blanco-Centurion et al., 2006a), TMN (Gerashchenko et al., 2004), or LC (Blanco-Centurion et al., 2004) does not change daily amounts of sleep–wake, but this might be because the other nonlesioned arousal groups compensate and stabilize the sleep–wake network. However, a combined triple lesion of the BF cholinergic neurons, TMN, and LC would for the first time directly test whether they are important links in the circuit responsible for wake, as has been traditionally hypothesized.
Materials and Methods
Subjects and surgical procedure.
Forty-four adult male Sprague Dawley rats (400–620 g; 4–6 months; Charles River Laboratories, Wilmington, MA) were deeply anesthetized (with a mixture of 10 mg/kg xylazine and 80 mg/kg ketamine, i.m.) and placed in a stereotaxic apparatus (Kopf, Tujunga, CA). Using a glass micropipette (20 μm tip diameter; FHC, Bowdoinham, ME) coupled to a pressure injector (Picospritzer; General Valve, Fairfield, NJ), the experimental rats (n = 38) were given anti-dopamine-β-hydroxylase saporin (anti-DBH-SAP) to the LC (1 μg/μl in 0.25 μl each side; Chemicon International, Temecula, CA; MAB394) [anterior (A), −0.8; lateral (L), ±1.6; ventral (V), +3.0 mm; coordinates based on interaural line], and hypocretin2-saporin to the TMN (Hcrt2-SAP; also termed orexin-SAP) (250 ng/μl in 0.25 μl; Advanced Targeting Systems, San Diego, CA; IT-20; A, −4.2; L, ±0.7; V, −9.3 mm below the dura; coordinates based on bregma). In these rats, 192-IgG-saporin (2 μg/μl; total volume injected, 3 μl; Chemicon International; MAB390) was placed into the lateral ventricle via the gravity method using a stainless-steel cannula. The cannula and the glass micropipettes were left in place for 5 min and then withdrawn slowly. Control rats (n = 6) were given pyrogen-free saline (Sigma-Aldrich, St. Louis, MO) in lieu of the three different neurotoxins.
At the same time, sleep recording electrodes were implanted as described previously (Gerashchenko et al., 2001a). Briefly, two screws (Plastics One, Roanoke, VA) were inserted 2 mm on either side of the midline suture and 3 mm anterior to bregma (frontal cortex); the other two screws were located 3 mm on either side of the midline and 6 mm posterior to bregma (occipital cortex). The electroencephalogram (EEG) was recorded from two contralateral (frontal-occipital) screw electrodes, whereas the electromyogram (EMG) signal was recorded from two multistranded wires inserted bilaterally into the nuchal muscles. All experimental procedures performed on live animals were approved and complied with the guidelines issued by the local animal care committee.
Polysomnographic recordings and sleep data analysis.
Immediately after surgery, rats were housed individually with food and water available ad libitum, and sleep–wake states were continuously recorded for 3 weeks. The EEG recordings were made in an isolated room at 20–23°C, with a 12 h light/dark cycle schedule (7:00 A.M. lights on; 200 lux). Contralateral frontal-occipital and nuchal muscles voltages were amplified, filtered (0.3–70 Hz for EEG, 0.1–1 kHz for EMG; model 12; Grass, West Warwick, RI), digitized (sampling frequency, 128 Hz; National Instruments, Austin, TX), and stored in a computer using the ICELUS data acquisition software (Mark Opp, University of Michigan, Ann Arbor, MI). Data from 19th and 20th (or 20 and 21 d) postinjection days were analyzed by a technician (E. Winston) blind to the treatment. The data from two consecutive 24 h periods were combined to generate a single value representative of the third postinjection week.
The EEG/EMG data were analyzed in 12 s epochs for wake, non-rapid eye movement (non-REM) sleep, and REM sleep using the ICELUS software. Waking EEG was identified by the presence of desynchronized EEG activity, high EMG integrated activity coupled with low delta power relative to non-REM sleep. Non-REM sleep was characterized by high amplitude–slow wave EEG activity coupled to low EMG activity and high delta power compared with waking. REM sleep was distinguished by hypersynchronized theta EEG activity, low to absent EMG signal relative to non-REM sleep and high theta–low delta power spectra.
Immunohistochemistry.
After sleep recordings were completed, rats were killed with an overdose of Nembutal, perfused with 0.9% saline solution (100 ml) followed by 10% phosphate-buffered formalin (400 ml). The brains were removed, formalin-fixed overnight, and then equilibrated in 30% sucrose–PBS at 4°C. The brains were cut (frozen sections; 40 μm; coronal) in a one-in-five serial order. Free-floating sections in each series were processed separately for detection of adenosine deaminase (ADA) (rabbit anti-ADA; AB176; 1:15K; Chemicon International) to detect the histamine neurons in the TMN, choline acetyltransferase (goat anti-ChAT; AB144P; 1:2K; Chemicon International), DBH (rabbit anti-DBH; AB1585; 1:50K; Chemicon International), hypocretin-1 (goat anti-Orexin A; SC 8070; 1:10K; Santa Cruz Biotechnology, Santa Cruz, CA), and NeuN (mouse anti-NeuN; MAB 377; 1:1000; Chemicon International). Tissue sections stained with NeuN identified the extent of the lesioned area in rats given the HCRT2-SAP to the TMN. Brainstem sections were processed separately to identify the DBH-IR neurons in the LC.
The tissue sections were incubated overnight at room temperature in the primary antibody. The following day, the sections were incubated in biotin-coupled secondary antibody for 1 h (1:500; Chemicon International) followed by incubation in the peroxidase–biotin–avidin complex for 1 h (Vectastain Elite ABC standard kit; Vector Laboratories, Burlingame, CA). The DAB-nickel method was used to visualize the reaction product (DAB kit; Vector Laboratories). For each antibody, the tissue from all of the rats was processed in one batch.
Cell counts.
The tissue sections were processed for visualization of only one phenotype per well (one-in-five series). All positive neurons in each region were counted using a semiautomatic stereology image analysis software (MCiD Elite Workstation; Imaging Research, St. Catherines, Ontario, Canada). Briefly, digitized automatic tiled images were taken at 25× objective (40× for LC). With the aid of the rat brain atlas (Paxinos and Watson, 2006), the region of interest was outlined on the scanned images. For each neuronal phenotype (for example, ChAT), an optical density threshold (scale from 0 to 2.4 with 0 = white) was adjusted to detect all relevant somata of that phenotype (for example, ChAT-IR somata). This threshold was the same for all rats (control and lesioned). Once the threshold detected all of the relevant somata, the images were closely examined for false positives (dirt and debris), and overlapping cell bodies were identified and separated. In the end, clearly identified neuronal perikarya were counted. Because antibodies with a very high degree of specificity were used and the immunoreactive neurons were localized to a specific brain region, the labeled neurons were easily identified (see Fig. 1).
Figure 1.
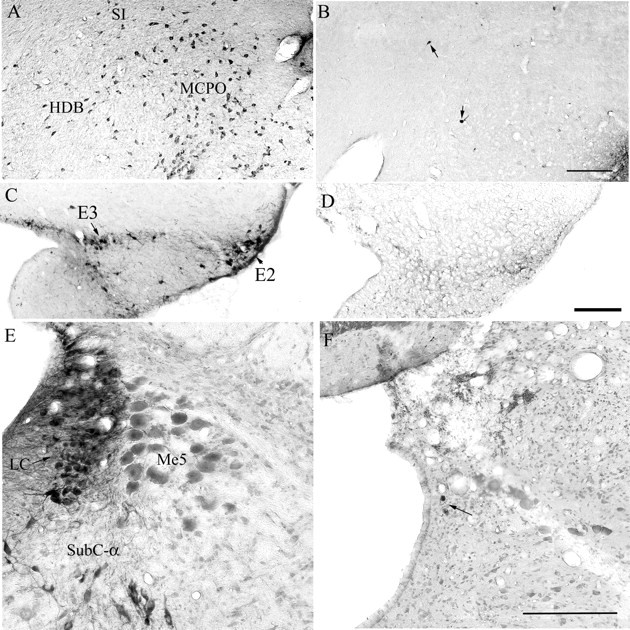
The loss of neurons in three arousal populations after administration of three saporin-conjugated neurotoxins. Photomicrographs A, C, and E depict the cholinergic neurons in the BF, the histamine neurons of the TMN, and the noradrenergic neurons of the LC in saline-treated rats. The cholinergic BF neurons were destroyed with intracerebroventricular injection of 192-IgG-saporin (photo B). The TMN neurons were destroyed with hypocretin-2-saporin (photo D) and the noradrenergic LC neurons were destroyed with anti-dopamine-β-hydroxylase-saporin (photo F). Scale bars: B, D, F, 200 μm. The arrows point to few surviving neurons. E2–E3, Histaminergic clusters within the TMN according to Inagaki et al. (1990); HDB, horizontal limb of the diagonal band of Broca; MCPO, magnocellular preoptic nucleus; Me5, mesencephalic trigeminal nucleus; SI, substantia innominata; SubC-α, subceruleus α.
Somata that were ChAT-IR, DBH-IR, ADA-IR, or HCRT-IR were counted in a specific number of coronal tissue sections depending on the distribution of the phenotype. ChAT-IR neurons in 11 sections taken from planes A +1.0 to −1.3 mm from bregma (Paxinos and Watson, 2006) were counted in the medial septum (MS), horizontal and vertical limbs of the diagonal bands of Broca (HDB; VDB), the magnocellular preoptic nucleus (MCPO), and the substantia innominata (SI). DBH-IR somata in the locus ceruleus were counted in five sections from A −9.3 to −10.2 mm. ADA-IR in the TMN histaminergic neurons (E1–E5 clusters) (Inagaki et al., 1990) were counted in seven sections from A −3.6 to −4.7 mm and HCRT-IR neurons in the perifornical area were counted in six sections from A −2.3 to −3.6 mm.
Statistical analysis.
The data were analyzed using SigmaStat (version 3.5; Systat Software, Point Richmond, CA). One-way ANOVA with post hoc test (Student–Newman–Keul) compared group means (saline vs double vs triple, except in Table 1 in which there are four groups). Pearson product-moment correlations established relationships between sleep and number of neurons lost.
Table 1.
Average (±SEM) number of neurons in three arousal populations after administration of three saporin-conjugated toxins
| Phenotype |
ChAT-IR (BF) |
ADA-IR (TMN) |
DBH-IR (LC) |
HCRT-IR (LH) |
|||
|---|---|---|---|---|---|---|---|
| Group | Cell no. | % Lesion | Cell no. | % Lesion | Cell no. | % Lesion | Cell no. |
| Saline (n = 6) | 4084.8 (85.9) | 0 | 707.8 (62.6) | 0 | 462.3 (21.6) | 0 | 1070.2 (35.1) |
| BF+TMN+LC (n = 6) | 443.5*** (95.2) | 89.2 (2.3) | 174.3*** (30.0) | 75.4 (4.2) | 31.2*** (9.3) | 93.3 (2.0) | 988.3 (115.3) |
| BF+LC (n = 7) | 159.9*** (29.6) | 96.1 (0.7) | 355.9*** (21.5) | 49.7* (3.0) | 26.1*** (5.8) | 94.3 (1.2) | 1020.3 (90.2) |
| TMN+LC (n = 4) | 2589.3*** (359.0) | 36.6** (8.8) | 175.8*** (30.6) | 75.2 (4.3) | 16.5*** (6.1) | 96.4 (1.3) | 1104.8 (115.4) |
In each rat, identified neurons in a specific number of tissue sections corresponding to each arousal population (for details, see Materials and Methods) were counted. The cholinergic neurons in the basal forebrain were lesioned with 192-IgG-saporin and identified with ChAT, the acetylcholine-synthesizing enzyme. To lesion hypocretin receptor-bearing neurons that are present on the histamine neurons in the TMN, hypocretin-2-saporin was administered locally. In the TMN, an antibody against ADA identified the histamine neurons. The LC noradrenergic neurons were lesioned with anti-DBH-saporin.
*p < 0.05 versus BF+TMN+LC or TMN+LC; **p < 0.01 versus BF+TMN+LC or BF+LC; ***p < 0.0001 versus saline. A one-way ANOVA was used to test the means in each column followed by post hoc tests.
Results
Neurotoxin-induced neuronal loss
Figure 1 depicts the loss of the cholinergic, adenosine deaminase-IR and DBH-IR neurons in the BF, TMN, and LC, respectively. With the administration of HCRT2-SAP to the TMN, the outer edges (determined by NeuN staining) reached the caudal portions of the perifornical area. However, because most of the HCRT neurons are located rostrally, there was no significant HCRT neuronal loss (Table 1, Fig. 2). The rats administered the three saporin conjugates were divided into two groups based on the degree of neuronal loss in each of the three arousal populations (Table 1). One group was classified as a triple lesion group (BF+TMN+LC; n = 6) based on the extent of neuronal loss in the three arousal populations (89.2% of ChAT-BF, 75.4% of ADA-TMN, and 93.3% of DBH-LC). A second group was classified as a double lesion (BF+LC lesion; n = 7), and rats in this group had significant lesions of the ChAT-BF (96.1%) and the DBH-LC (94.3%) neurons, but only partial loss of ADA-IR neurons in the TMN (49.7%). The third group consisted of rats given saline (n = 6) against which the double (BF+LC) and triple (BF+TMN+LC) lesion groups were compared. Four other rats had extensive lesions of the ADA-TMN (75.2%) and DBH-LC (96.4%) but partial loss of the ChAT-BF neurons (36.6%). Because of the small number of rats in this group (TMN+LC lesion), these four rats were not compared against the saline rats, but the sleep data from the individual rats were used to determine the correlations between neuronal loss and sleep changes.
Figure 2.
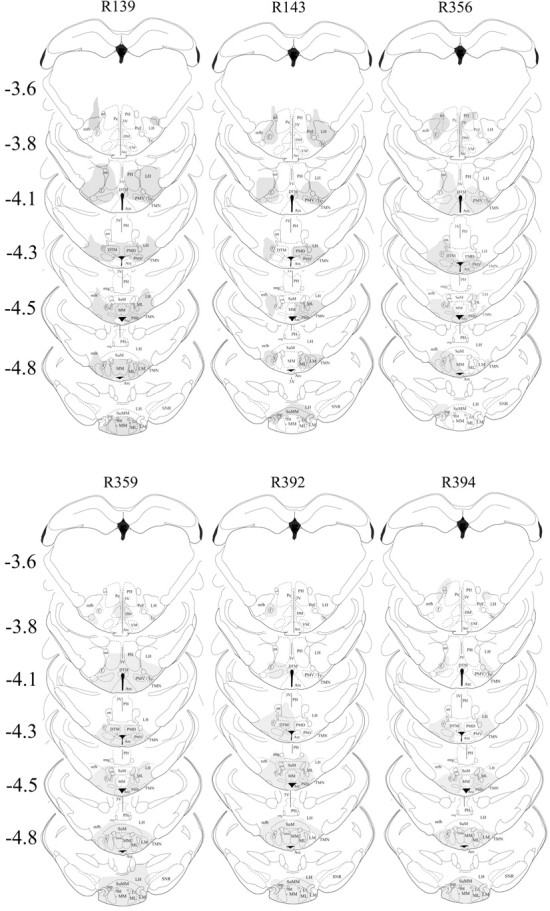
Extent of neuronal loss because of local injection of HCRT2-SAP (250 ng/μl in 0.25 μl each side) into the TMN. HCRT2-SAP kills neurons that possess the HCRT-2 receptor, which is on the histaminergic TMN neurons but also on other adjacent neurons. Thus, the neuronal marker NeuN was used to assess the extent of nonspecific neuronal loss, and those regions devoid of neurons were then drawn onto the corresponding figures taken from the rat brain atlas (Paxinos and Watson, 2006). Each column represents an individual rat (marked by number at the top of each column) in the triple-lesion group (BF+TMN+LC). The numbers to the left indicate anteroposterior distance from bregma in millimeters. Arc, Arcuate n; DM, dorsomedial hypothalamus; DTM, dorsal tuberomammillary n; MM, medial mammillary n; MMn, median mammillary n; ML, medial mammillary n lateral; LH, lateral hypothalamus; LM, lateral mammillary n; PeF, perifornical area; PH, posterior hypothalamus; PMD, premammillary n dorsal; SuM, supramammillary n; SNR, substantia nigra reticular; VM, ventromedial hypothalamus; Te, Terete hypothalamic n; 3V, third ventricle; f, fornix; mfb, medial forebrain bundle; mt, mammilothalamic tract; mtg, mammilotegmental tract; mp, mammillary peduncle.
Effects on sleep–wake
Over the 24 h period (Table 2), the double (BF+LC)- and triple (BF+TMN+LC)-lesioned rats had no significant change in the amount of wake or non-REM sleep, but REM sleep was significantly reduced (double, −18.6%; triple, −25.8%; post hoc test, p < 0.05) compared with saline rats (one-way ANOVA; F(2,16) = 5.18; p < 0.013). During the 12 h light cycle, when nocturnal rodents are primarily asleep, REM sleep was significantly reduced in both the double (−32.2%; p < 0.03)- and triple (−44.4%; p < 0.02)-lesioned rats, compared with saline rats (F(2,16) = 5.84; p < 0.012), but wake or non-REM sleep were not changed (Fig. 3). During the 12 h light-off period when the rats are primarily awake, there were no significant changes in sleep–wake states in the lesioned rats.
Table 2.
Average (±SEM) percent wake, non-REM and REM sleep over the 24 h
| Wake | Non-REM | REM sleep | |
|---|---|---|---|
| Saline (n = 6) | 54.18 (0.99) | 35.68 (0.89) | 10.14 (0.25) |
| BF+LC (n = 7) | 52.27 (6.28) | 39.48 (6.82) | 8.25 (2.01)* |
| BF+TMN+LC (n = 6) | 50.89 (5.52) | 41.59 (5.81) | 7.52 (1.06)* |
The daily average was derived from a 48 h continuous recording of the rat's sleep that was made 3 weeks after delivery of saline or the saporin-conjugated neurotoxins.
*p < 0.05 (post hoc test after one-way ANOVA) versus saline REM sleep.
Figure 3.

Percentage (±SEM) of wake, non-REM, and REM sleep in lesioned and nonlesioned rats. The line graphs summarize the average in 4 h blocks over the 24 h period with the data double-plotted to illustrate the diurnal rhythm of the sleep–wake cycle. The crescent moon symbols represent the 12 h dark cycle. The bar graphs on the right summarize the data during the 12 h day and night cycles. *p < 0.01 versus triple-lesioned rats (post hoc significance after one-way ANOVA); **p < 0.05 versus double- and triple-lesioned rats (post hoc after one-way ANOVA);  p < 0.05 versus double lesion (post hoc after one-way ANOVA).
p < 0.05 versus double lesion (post hoc after one-way ANOVA).
We partitioned the data in 4 h blocks (Fig. 3) to determine whether there were changes in sleep–wake during the light–dark transition periods because in previous studies (Blanco-Centurion et al., 2004; Gerashchenko et al., 2004) in which the LC and TMN were lesioned changes in sleep were noted only during these periods. During the first 4 h of the light-off period (Fig. 3) when the rats normally wake up, the triple-lesioned rats had twice the amount of non-REM sleep (triple, +106.6%) (p < 0.02; post hoc test) and were significantly less awake (−22.9%) (p < 0.01; post hoc test) compared with saline. The rats with the double lesion (BF+LC) were intermediate and not significant from saline (Fig. 3). Thus, the triple lesion had an additive effect on wake and non-REM sleep during the first four hours of the light-off period.
During the light cycle, the levels of wake or non-REM sleep in the two lesioned groups were not different from saline rats (Fig. 3). During the first 4 h of the light cycle, REM sleep was significantly less in both the double (p < 0.04; post hoc test)- and triple (p < 0.001; post hoc test)-lesioned rats (Fig. 3) compared with saline (one-way ANOVA; F(2,16) = 8.86; p < 0.003). During this time period, the triple-lesioned rats also had significantly less REM sleep compared with the double-lesioned rats (p < 0.05; post hoc test), indicating an additive effect of the lesion. In the middle third of the light cycle, REM sleep levels were lower in the lesioned rats but not statistically significant. In the last third of the light cycle, REM sleep was significantly less in the double (p < 0.04)- and triple (p < 0.05)-lesioned rats compared with saline (F(2,16) = 4.05; p < 0.04).
To identify whether the lesions changed the architecture of sleep, the number and length of bouts of wake, non-REM and REM sleep were analyzed (Fig. 4). Normally, rats have many brief (<1 min) bouts of wake, non-REM and REM sleep, but in the lesioned rats these decreased significantly during both the day and night periods (Fig. 4). The lesioned rats had fewer numbers of non-REM sleep bouts that were 1–3 min long (Fig. 4), but the number of non-REM sleep bouts that were 5–10 min long increased significantly (p < 0.02) during both the day (F(2,16) = 7.01; p < 0.007) and night (F(2,16) = 21.17; p < 0.001). The triple-lesioned rats had a decrease in number of REM sleep bouts that were 2–4 min long during the day, but an increase in the length of these bouts at night. Overall, during the 12 h day period, the lesioned rats had significantly fewer numbers of bouts of wake (F(2,16) = 46.10; p < 0.001), non-REM (F(2,16) = 47.6; p < 0.001) and REM sleep (F(2,16) = 20.36; p < 0.001) (Table 3) but significantly longer bouts of wake (F(2,16) = 7.91; p < 0.004) and non-REM sleep (F(2,16) = 15.01; p < 0.001) (Table 3). Thus, the lesioned rats had fewer but longer bouts of wake and non-REM sleep, which produced no net change in these states during the light cycle. However, the number of REM sleep bouts decreased sharply, and because there was no change in the length of the remaining REM sleep bouts there was an overall decrease in this sleep state during the day cycle.
Figure 4.

The number and length of bouts of wake, non-REM, and REM, sleep in saline versus lesioned rats. Each bout of wake, non-REM, and REM sleep was placed into a bin corresponding to a specific length (in minutes) of the bout, and the figure represents the average (±SEM) number of bouts of a specific length for the lesion and saline groups. *p < 0.02 saline versus double and triple; **p < 0.002 saline versus double- and triple-lesioned rats; +p < 0.05 saline versus triple-lesioned rats. Significance represents post hoc test after one-way ANOVA.
Table 3.
Average (±SEM) number and length (in minutes) of bouts of wake, non-REM and REM sleep, and transitions between the sleep–wake states during the 12 h day and night periods
| Group | Number of bouts |
Length of bouts |
||||||
|---|---|---|---|---|---|---|---|---|
| No lesion | BF+LC | BF+TMN+LC | Correlation: number of neurons vs number of bouts | No lesion | BF+LC | BF+TMN+LC | Correlation: number of neurons vs length of bouts | |
| Day | ||||||||
| Wake | 142.3 (5.9) | 90.3 (4.3)* | 82.5 (3.7)* | 0.910*** | 1.6 (0.2) | 2.5 (0.2)** | 2.9 (0.3)* | −0.635# |
| Non-REM | 141.6 (5.9) | 94.8 (2.7)* | 84.2 (4.3)* | 0.905*** | 2.8 (0.1) | 4.4 (0.4)* | 4.9 (0.2)* | −0.779# |
| REM sleep | 77.8 (4.5) | 45.1 (4.6)* | 37.0 (4.9)* | 0.814*** | 1.5 (0.05) | 1.7 (0.1) | 1.7 (0.2) | −0.366 |
| Transitions | 361.7 (14.6) | 230.1 (8.9)* | 203.7 (8.0)* | 0.918*** | ||||
| Night | ||||||||
| Wake | 62.9 (9.0) | 43.9 (6.2) | 40.6 (6.7) | 0.495 | 10.4 (1.9) | 13.8 (2.3) | 16.6 (5.6) | −0.297 |
| Non-REM | 62.2 (8.9) | 45.7 (6.3) | 43.8 (7.3) | 0.424 | 2.1 (0.1) | 3.5 (0.2)* | 4.3 (0.3)*0 | −0.835*** |
| REM sleep | 30.7 (3.7) | 24.1 (4.4) | 22.6 (3.9) | 0.334 | 0.9 (0.1) | 1.7 (0.1)* | 1.9 (0.2)* | −0.756*** |
| Transitions | 155.8 (21.3) | 113.8 (16.5) | 107.1 (17.3) | 0.444 | ||||
The correlation coefficient represents the relationship between individual sleep–wake states and the number of neurons that are cholinergic (in BF), adenosine deaminase-IR (marker of histamine neurons in the TMN), and dopamine β-hydroxylase-IR (marker of noradrenaline neurons in the LC). Correlations were derived from data from all rats (n = 23) in Table 1, and not from group data.
*p < 0.01 versus nonlesioned; **p < 0.05 versus nonlesioned; ***p < 0.0001, Pearson product-moment correlation; #p < 0.01, Pearson product-moment correlation; 0p < 0.05 versus BF+LC. A one-way ANOVA was used to test the means of the three groups for each state followed by post hoc tests.
At night, the lesioned rats had significantly longer bouts of non-REM (F(2,16) = 39.0; p < 0.001) and REM sleep (F(2,16) = 12.7; p < 0.001) (Table 3). However, because there was a sharp decrease in the number of short bouts of these sleep states, the overall nighttime levels of sleep were unchanged in the lesioned rats. The number and length of bouts were significantly correlated with the number of ChAT-, ADA-, and DBH-immunoreactive neurons (Table 3). During the day, the number of bouts of wake, non-REM and REM sleep increased in relationship with the number of cholinergic BF, histamine, and LC neurons (correlations cited in Table 3). However, as the number of these neurons increased, the length of wake and non-REM sleep decreased (correlations cited in Table 3). At night, the length of non-REM and REM sleep bouts were inversely correlated with the number of the arousal neurons.
Because of the changes in the number and length of bouts the lesioned rats had significantly fewer transitions between the sleep–wake states indicating a less fragmented, more stable sleep–wake architecture during the day, the rat's normal sleep period (F(2,16) = 28.4; p < 0.001). Indeed, during the day, there was a direct relationship between the number of cholinergic BF, histamine, and LC neurons and number of transitions (correlations cited in Table 3).
The lesioned rats did not display narcoleptic behavior such as cataplexy or sleep onset REM sleep behavior. The behavior of these rats was not videotaped but a sudden drop in muscle tone signifying cataplexy would have been detected in the EEG/EMG recordings (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). In our previous studies (Gerashchenko et al., 2001a, 2003; Zhang et al., 2007), such episodes were easily identified.
Discussion
The primary hypothesis that the cholinergic BF, histamine TMN, and noradrenergic LC neurons represent key links in the circuit regulating arousal was not supported because rats with lesions of all three of these neuronal phenotypes had normal levels of wake. We expected an additive effect on wake as a result of the combined lesion of three arousal populations, but to our surprise the overall daily levels of wake were not significantly different compared with saline rats. The three arousal populations lesioned in this study have been central to theories of sleep–wake generation for a number of years (Lin et al., 1988; Szymusiak, 1995; Jones, 2005). When HCRT was linked to narcolepsy and given that HCRT neurons project heavily to these major arousal neurons, it raised the possibility that daily levels of sleep–wake are regulated by the action of HCRT onto downstream arousal neurons (Saper et al., 2005). This is the first study in which three of the downstream targets of the HCRT neurons have all been lesioned simultaneously, thereby directly testing their contribution to arousal.
Lesions were made using saporin-based neurotoxins that selectively kill specific neurotransmitter-containing neurons. 192-IgG-SAP, which binds to the p75 NGF receptor present on the BF cholinergic neurons, was used to lesion these neurons. The neurotoxin kills these neurons, resulting in 80–90% selective ChAT denervation of rat neocortex and hippocampus (Heckers et al., 1994). In the ventral pallidum and sublenticular substantia innominata (caudal portions of Ch4), some of the cholinergic neurons [7.1% by one estimate (Woolf et al., 1989)] do not contain the p75 NGF receptor and are spared by the toxin (Heckers et al., 1994). These ChAT+/p75− neurons project to the amygdala (Hecker and Mesulam, 1994) and are unlikely to regulate waking, because in humans, adenosine, a potential regulator of sleep homeostasis, does not increase in the amygdala with 40 h waking (Zeitzer et al., 2006). The LC neurons were lesioned with anti-DBH-SAP, which targets the norepinephrine-synthesizing enzyme, dopamine β-hydroxylase, but does not lesion other adjacent neuronal phenotypes (Blanco-Centurion et al., 2004). The TMN neurons were lesioned with HCRT2-SAP, which kills HCRT receptor-bearing neurons (Gerashchenko et al., 2001a, 2004). The HCRT2-SAP kills the histamine TMN neurons and other nonhistamine neurons that possess the HCRT receptor within the injection area. In the triple-lesion group, the saporin-conjugated toxins destroyed 89% of the BF cholinergic neurons, 75% of the histaminergic TMN neurons, and 93% of the noradrenergic neurons in the LC.
In previous studies, individual lesions of the BF cholinergic neurons (Blanco-Centurion et al., 2006a), the noradrenergic LC (Blanco-Centurion et al., 2004), or the histaminergic TMN neurons (Gerashchenko et al., 2004) were made. Deletion of the BF cholinergic neurons (−95%) produced a diminution in theta activity during REM sleep (Gerashchenko et al., 2001b), but did not change the daily levels of wake, non-REM sleep, or REM sleep, nor did they change the homeostatic response to sleep loss (Blanco-Centurion et al., 2006a). The histamine neurons, which are located exclusively in the TMN, project heavily to the sleep-active preoptic neurons (Sherin et al., 1996), but their lesion (−82%) also did not change the daily levels of sleep (Gerashchenko et al., 2004). The lesion data are supported by data in knock-out mice. For instance, mice deficient in histamine (histidine decarboxylase-null mice) display sleep abnormalities only during the light–dark transition period (Parmentier et al., 2002). The daily amount of sleep–wake is not changed in DBH-null mice (Hunsley and Palmiter, 2003) or in rats with lesions of the LC with DBH-SAP (Blanco-Centurion et al., 2004).
The lack of a more robust overall effect on sleep after individual lesions of the BF, TMN, and LC, or of the pons might be attributable to compensation from nonlesioned arousal neurons that might stabilize the sleep–wake network. Therefore, for the first time, all three were lesioned yet the daily levels of wake did not decrease. These findings do not support current models (Saper et al., 2005) that waking is driven by the action of HCRT on these downstream targets.
Significant changes in the level of sleep–wake were noted only during the light-to-dark transition period with the triple-lesioned rats being asleep when they should be awake. The selective changes in sleep during the light–dark transition period suggest that the lesioned neurons modulate waking levels only at certain circadian times. We suggest that, in these nocturnal rodents, the cholinergic BF, histamine, and LC neurons combine to wake up the animal when the lights turn off. During this time period, the HCRT levels are low and the HCRT influence has waned (Fujiki et al., 2001; Yoshida et al., 2001; Desarnaud et al., 2004), but the rats have to wake up. We suggest that, during this time period, the combined action of the cholinergic BF, histamine, and LC neurons offsets the faded HCRT signal, and arouses the animal. This is in contrast to the existing model (Saper et al., 2005), which suggests that HCRT stimulates the downstream arousal neurons. Similarly, at the end of the wake–active period (night cycle), when the HCRT signal is at its peak (Fujiki et al., 2001; Yoshida et al., 2001; Desarnaud et al., 2004), the animals have to fall asleep, and at this time the cholinergic BF, histamine, and LC neurons withdraw their waking influence, thereby balancing the strong HCRT signal.
These results suggest that the relationship between HCRT and the three downstream arousal neurons is not one way, with the HCRT neurons driving the monoaminergic neurons, as proposed (Saper et al., 2005). Instead, we suggest that the monoamines and HCRT balance each other to modulate waking; when one is low the other is high and vice versa. If this is the case, then the double- and triple-lesioned rats would be expected to have increased HCRT drive. We believe this to be the case because in the lesioned rats there is a significant decrease in REM sleep over the 24 h and during the day cycle. It is unlikely that the daytime reduction in REM sleep in the lesioned rats is attributable to loss of the TMN, because when only the TMN is lesioned (Gerashchenko et al., 2004) there is no reduction in REM sleep. Similarly, mice deficient in histamine (histidine decarboxylase-null mice) do not have a day cycle reduction in REM sleep (Parmentier et al., 2002).
The second finding of this study is that the loss of the three arousal neurons did not produce a more fragmented sleep–wake pattern as would be expected in the current model (Saper et al., 2005). Deletions of both HCRT and DBH in mice (combined knock-out) also does not lead to more fragmented sleep versus single gene deletions (Hunsley et al., 2006). In the present study, the lesioned rats had fewer short (<1 min) bouts of wake, non-REM and REM sleep, and more long bouts (5–10 min long) of non-REM sleep. This resulted in fewer transitions between the three states. The lengthening of non-REM sleep bouts may be attributable to the loss of the inhibition of the sleep-active preoptic neurons.
Could the remaining unlesioned BF, TMN, and LC neurons provide near-normal function? Lesions rarely destroy all of the neurons, but a behavioral abnormality should be evident when large portions of key neuronal populations are destroyed. This is the case in neurodegenerative diseases including narcolepsy in which all of the underlying HCRT neurons need not be destroyed for a sleep abnormality to be clearly evident (Thannickal et al., 2000; Gerashchenko et al., 2001). The evidence against the BF, TMN, and LC as conduits of a HCRT-wake-promoting effect also comes from knock-out mice (Parmentier et al., 2002; Hunsley and Palmiter, 2003), including double knock-outs (Hunsley et al., 2006). In the present study, the saporin toxins killed specific neurons. The 192-IgG-SAP and DBH-SAP killed the BF cholinergic and LC noradrenergic neurons, respectively. The HCRT2-SAP kills neurons containing the HCRT receptor, and in the TMN in which there is a very dense collection of neurons containing the HCRT receptor 2 (Marcus et al., 2001), the histamine neurons, as well as other neurons that possess the receptor, were killed. Thus, the neurons in three arousal regions that receive HCRT input were killed, and yet daily levels of wake remained unchanged.
Without the cholinergic BF, TMN, and LC neurons, what circuit(s) might maintain the daily levels of wake in the triple-lesioned rats? We suggest that HCRT neurons, which were mostly spared in this study, might exert their wake-promoting actions on the remaining arousal neurons residing in the ascending reticular activating system (Peyron et al., 1998) including the serotoninergic raphe neurons (Brown et al., 2001; Matsuzaki et al., 2002) and the noncholinergic neurons of the BF. The latter possibility is suggested by the finding that, in rats with lesions of the cholinergic BF neurons, HCRT-1 has a potent arousing effect (Blanco-Centurion et al., 2006b). Additionally, other neurons in the pons such as neuropeptide S (Xu et al., 2004), dopamine (Lu et al., 2006), glutamatergic and the pontine cholinergic neurons (Jones, 2005) might network with HCRT. The serotonin neurons could be lesioned with 5–7-dihydroxytryptamine, and the pontine cholinergic neurons could be targeted with a neurotoxin that binds to the urotensin 2 receptors that are only on the cholinergic neurons (Clark et al., 2007). Alternatively, multiple genes could be deleted in mice and the additive effects of such deletions on sleep could be investigated. We suggest that such multiple lesions are the most direct method of determining which brain chemicals regulate sleep–wake states. It is necessary to isolate key regions regulating sleep–wake states, because then one can target these structures with methodologies to reverse the defect.
These results are clinically relevant because it shows that a massive loss of neurons encompassing three arousal populations innervating the entire brain does not cause coma or loss of consciousness. The cholinergic neurons of the BF represent the rostral-most extension of the ascending reticular activating system, innervating the entire cortex and hippocampus. The TMN contains the only group of histamine neurons in the brain, and the LC is one of the principal sources of norepinephrine in the brain. Neurons in all three neuronal populations are active only at a time when the cortex is electrophysiologically awake, which led to the hypothesis that these neurons are critically involved in maintaining wake. However, here we have demonstrated that their loss does not change overall levels of wakefulness.
Footnotes
This work was supported by National Institutes of Health Grants NS30140, NS52287, and MH55772, and Medical Research Service of the Department of Veterans Affairs. We thank Elizabeth Winston for analyzing the sleep data and Dr. Errol Baker for help with statistics.
References
- Beuckmann CT, Sinton CM, Williams SC, Richardson JA, Hammer RE, Sakurai T, Yanagisawa M. Expression of a poly-glutamine-ataxin-3 transgene in orexin neurons induces narcolepsy-cataplexy in the rat. J Neurosci. 2004;24:4469–4477. doi: 10.1523/JNEUROSCI.5560-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Centurion C, Gerashchenko D, Salin-Pascual RJ, Shiromani PJ. Effects of hypocretin2-saporin and antidopamine-beta-hydroxylase-saporin neurotoxic lesions of the dorsolateral pons on sleep and muscle tone. Eur J Neurosci. 2004;19:2741–2752. doi: 10.1111/j.1460-9568.2004.03366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Centurion C, Xu M, Murillo-Rodriguez E, Gerashchenko D, Shiromani AM, Salin-Pascual RJ, Hof PR, Shiromani PJ. Adenosine and sleep homeostasis in the basal forebrain. J Neurosci. 2006a;26:8092–8100. doi: 10.1523/JNEUROSCI.2181-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Centurion CA, Shiromani A, Winston E, Shiromani PJ. Effects of hypocretin-1 in 192-IgG-saporin-lesioned rats. Eur J Neurosci. 2006b;24:2084–2088. doi: 10.1111/j.1460-9568.2006.05074.x. [DOI] [PubMed] [Google Scholar]
- Bourgin P, Huitron-Resendiz S, Spier AD, Fabre V, Morte B, Criado JR, Sutcliffe JG, Henriksen SJ, de Lecea L. Hypocretin-1 modulates rapid eye movement sleep through activation of locus coeruleus neurons. J Neurosci. 2000;20:7760–7765. doi: 10.1523/JNEUROSCI.20-20-07760.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RE, Sergeeva O, Eriksson KS, Haas HL. Orexin A excites serotonergic neurons in the dorsal raphe nucleus of the rat. Neuropharmacology. 2001;40:457–459. doi: 10.1016/s0028-3908(00)00178-7. [DOI] [PubMed] [Google Scholar]
- Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, Richardson JA, Williams SC, Xiong Y, Kisanuki Y, Fitch TE, Nakazato M, Hammer RE, Saper CB, Yanagisawa M. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–451. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- Chu NS, Bloom FE. Norepinephrine containing neurons: changes in spontaneous discharge patterns during sleeping and waking. Science. 1973;179:908–910. doi: 10.1126/science.179.4076.908. [DOI] [PubMed] [Google Scholar]
- Clark SD, Alderson HL, Winn P, Latimer MP, Nothacker HP, Civelli O. Fusion of diphtheria toxin and urotensin II produces a neurotoxin selective for cholinergic neurons in the rat mesopontine tegmentum. J Neurochem. 2007;102:112–120. doi: 10.1111/j.1471-4159.2007.04529.x. [DOI] [PubMed] [Google Scholar]
- de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FS, Frankel WN, van den Pol AN, Bloom FE, Gautvik KM, Sutcliffe JG. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci USA. 1998;95:322–327. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desarnaud F, Murillo-Rodriguez E, Lin L, Xu M, Gerashchenko D, Shiromani SN, Nishino S, Mignot E, Shiromani PJ. The diurnal rhythm of hypocretin in young and old F344 rats. Sleep. 2004;27:851–856. doi: 10.1093/sleep/27.5.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiki N, Yoshida Y, Ripley B, Honda K, Mignot E, Nishino S. Changes in CSF hypocretin-1 (orexin A) levels in rats across 24 hours and in response to food deprivation. NeuroReport. 2001;12:993–997. doi: 10.1097/00001756-200104170-00026. [DOI] [PubMed] [Google Scholar]
- Gerashchenko D, Kohls MD, Greco M, Waleh NS, Salin-Pascual R, Kilduff TS, Lappi DA, Shiromani PJ. Hypocretin-2-saporin lesions of the lateral hypothalamus produce narcoleptic-like sleep behavior in the rat. J Neurosci. 2001a;21:7273–7283. doi: 10.1523/JNEUROSCI.21-18-07273.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerashchenko D, Salin-Pascual R, Shiromani PJ. Effects of hypocretin-saporin injections into the medial septum on sleep and hippocampal theta. Brain Res. 2001b;913:106–115. doi: 10.1016/s0006-8993(01)02792-5. [DOI] [PubMed] [Google Scholar]
- Gerashchenko D, Blanco-Centurion C, Greco MA, Shiromani PJ. Effects of lateral hypothalamic lesion with the neurotoxin hypocretin-2-saporin on sleep in Long-Evans rats. Neuroscience. 2003;116:223–235. doi: 10.1016/s0306-4522(02)00575-4. [DOI] [PubMed] [Google Scholar]
- Gerashchenko D, Chou TC, Blanco-Centurion CA, Saper CB, Shiromani PJ. Effects of lesions of the histaminergic tuberomammillary nucleus on spontaneous sleep in rats. Sleep. 2004;27:1275–1281. doi: 10.1093/sleep/27.7.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco MA, Shiromani PJ. Hypocretin receptor protein and mRNA expression in the dorsolateral pons of rats. Brain Res Mol Brain Res. 2001;88:176–182. doi: 10.1016/s0169-328x(01)00039-0. [DOI] [PubMed] [Google Scholar]
- Hara J, Beuckmann CT, Nambu T, Willie JT, Chemelli RM, Sinton CM, Sugiyama F, Yagami K, Goto K, Yanagisawa M, Sakurai T. Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron. 2001;30:345–354. doi: 10.1016/s0896-6273(01)00293-8. [DOI] [PubMed] [Google Scholar]
- Hecker S, Mesulam MM. Two types of cholinergic projections to the rat amygdala. Neuroscience. 1994;60:383–397. doi: 10.1016/0306-4522(94)90252-6. [DOI] [PubMed] [Google Scholar]
- Heckers S, Ohtake T, Wiley RG, Lappi DA, Geula C, Mesulam MM. Complete and selective cholinergic denervation of rat neocortex and hippocampus but not amygdala by an immunotoxin against the p75 NGF receptor. J Neurosci. 1994;14:1271–1289. doi: 10.1523/JNEUROSCI.14-03-01271.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZL, Qu WM, Li WD, Mochizuki T, Eguchi N, Watanabe T, Urade Y, Hayaishi O. Arousal effect of orexin A depends on activation of the histaminergic system. Proc Natl Acad Sci USA. 2001;98:9965–9970. doi: 10.1073/pnas.181330998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunsley MS, Palmiter RD. Norepinephrine-deficient mice exhibit normal sleep-wake states but have shorter sleep latency after mild stress and low doses of amphetamine. Sleep. 2003;26:521–526. [PubMed] [Google Scholar]
- Hunsley MS, Curtis WR, Palmiter RD. Behavioral and sleep/wake characteristics of mice lacking norepinephrine and hypocretin. Genes Brain Behav. 2006;5:451–457. doi: 10.1111/j.1601-183X.2005.00179.x. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Toda K, Taniuchi I, Panula P, Yamatodani A, Tohyama M, Watanabe T, Wada H. An analysis of histaminergic efferents of the tuberomammillary nucleus to the medial preoptic area and inferior colliculus of the rat. Exp Brain Res. 1990;80:374–380. doi: 10.1007/BF00228164. [DOI] [PubMed] [Google Scholar]
- John J, Wu MF, Boehmer LN, Siegel JM. Cataplexy-active neurons in the hypothalamus: implications for the role of histamine in sleep and waking behavior. Neuron. 2004;42:619–634. doi: 10.1016/s0896-6273(04)00247-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BE. From waking to sleeping: neuronal and chemical substrates. Trends Pharmacol Sci. 2005;26:578–586. doi: 10.1016/j.tips.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Lee MG, Hassani OK, Alonso A, Jones BE. Cholinergic basal forebrain neurons burst with theta during waking and paradoxical sleep. J Neurosci. 2005;25:4365–4369. doi: 10.1523/JNEUROSCI.0178-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JS, Sakai K, Jouvet M. Evidence for histaminergic arousal mechanisms in the hypothalamus of cat. Neuropharmacology. 1988;27:111–122. doi: 10.1016/0028-3908(88)90159-1. [DOI] [PubMed] [Google Scholar]
- Lin L, Faraco J, Li R, Kadotani H, Rogers W, Lin X, Qiu X, de Jong PJ, Nishino S, Mignot E. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98:365–376. doi: 10.1016/s0092-8674(00)81965-0. [DOI] [PubMed] [Google Scholar]
- Lu J, Jhou TC, Saper CB. Identification of wake-active dopaminergic neurons in the ventral periaqueductal gray matter. J Neurosci. 2006;26:193–202. doi: 10.1523/JNEUROSCI.2244-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus JN, Aschkenasi CJ, Lee CE, Chemelli RM, Saper CB, Yanagisawa M, Elmquist JK. Differential expression of orexin receptors 1 and 2 in the rat brain. J Comp Neurol. 2001;435:6–25. doi: 10.1002/cne.1190. [DOI] [PubMed] [Google Scholar]
- Matsuzaki I, Sakurai T, Kunii K, Nakamura T, Yanagisawa M, Goto K. Involvement of the serotonergic system in orexin-induced behavioral alterations in rats. Regul Pept. 2002;104:119–123. doi: 10.1016/s0167-0115(01)00355-x. [DOI] [PubMed] [Google Scholar]
- Nishino S, Ripley B, Overeem S, Nevsimalova S, Lammers GJ, Vankova J, Okun M, Rogers W, Brooks S, Mignot E. Low cerebrospinal fluid hypocretin (Orexin) and altered energy homeostasis in human narcolepsy. Ann Neurol. 2001;50:381–388. doi: 10.1002/ana.1130. [DOI] [PubMed] [Google Scholar]
- Parmentier R, Ohtsu H, Djebbara-Hannas Z, Valatx JL, Watanabe T, Lin JS. Anatomical, physiological, and pharmacological characteristics of histidine decarboxylase knock-out mice: evidence for the role of brain histamine in behavioral and sleep-wake control. J Neurosci. 2002;22:7695–7711. doi: 10.1523/JNEUROSCI.22-17-07695.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. San Diego: Academic; 2006. [DOI] [PubMed] [Google Scholar]
- Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 1998;18:9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyron C, Faraco J, Rogers W, Ripley B, Overeem S, Charnay Y, Nevsimalova S, Aldrich M, Reynolds D, Albin R, Li R, Hungs M, Pedrazzoli M, Padigaru M, Kucherlapati M, Fan J, Maki R, Lammers GJ, Bouras C, Kucherlapati R, et al. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med. 2000;6:991–997. doi: 10.1038/79690. [DOI] [PubMed] [Google Scholar]
- Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–1263. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- Sherin JE, Shiromani PJ, McCarley RW, Saper CB. Activation of ventrolateral preoptic neurons during sleep. Science. 1996;271:216–219. doi: 10.1126/science.271.5246.216. [DOI] [PubMed] [Google Scholar]
- Szymusiak R. Magnocellular nuclei of the basal forebrain: substrates of sleep and arousal regulation. Sleep. 1995;18:478–500. doi: 10.1093/sleep/18.6.478. [DOI] [PubMed] [Google Scholar]
- Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, Cornford M, Siegel JM. Reduced number of hypocretin neurons in human narcolepsy. Neuron. 2000;27:469–474. doi: 10.1016/s0896-6273(00)00058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willie JT, Chemelli RM, Sinton CM, Tokita S, Williams SC, Kisanuki YY, Marcus JN, Lee C, Elmquist JK, Kohlmeier KA, Leonard CS, Richardson JA, Hammer RE, Yanagisawa M. Distinct narcolepsy syndromes in orexin receptor-2 and orexin null mice. Molecular genetic dissection of non-REM and REM sleep regulatory processes. Neuron. 2003;38:715–730. doi: 10.1016/s0896-6273(03)00330-1. [DOI] [PubMed] [Google Scholar]
- Woolf NJ, Gould E, Butcher LL. Nerve growth factor receptor is associated with cholinergic neurons of the basal forebrain but not the pontomesencephalon. Neuroscience. 1989;30:143–152. doi: 10.1016/0306-4522(89)90360-6. [DOI] [PubMed] [Google Scholar]
- Xu YL, Reinscheid RK, Huitron-Resendiz S, Clark SD, Wang Z, Lin SH, Brucher FA, Zeng J, Ly NK, Henriksen SJ, de Lecea L, Civelli O. Neuropeptide S: a neuropeptide promoting arousal and anxiolytic-like effects. Neuron. 2004;43:487–497. doi: 10.1016/j.neuron.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Fujiki N, Nakajima T, Ripley B, Matsumura H, Yoneda H, Mignot E, Nishino S. Fluctuation of extracellular hypocretin-1 (orexin A) levels in the rat in relation to the light-dark cycle and sleep-wake activities. Eur J Neurosci. 2001;14:1075–1081. doi: 10.1046/j.0953-816x.2001.01725.x. [DOI] [PubMed] [Google Scholar]
- Zeitzer JM, Morales-Villagran A, Maidment NT, Behnke EJ, Ackerson LC, Lopez-Rodriguez F, Fried I, Engel J, Jr, Wilson CL. Extracellular adenosine in the human brain during sleep and sleep deprivation: an in vivo microdialysis study. Sleep. 2006;29:455–461. doi: 10.1093/sleep/29.4.455. [DOI] [PubMed] [Google Scholar]
- Zhang S, Lin L, Kaur S, Thankachan S, Blanco-Centurion C, Yanagisawa M, Mignot E, Shiromani PJ. The development of hypocretin (OREXIN) deficiency in hypocretin/ataxin-3 transgenic rats. Neuroscience. 2007;148:34–43. doi: 10.1016/j.neuroscience.2007.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]