

Abstract
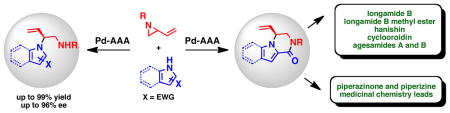
We report that nitrogen heterocycles can serve as competent nucleophiles in the palladium-catalyzed dynamic kinetic asymmetric alkylation of vinyl aziridines. The resulting alkylated products were obtained with high regio-, chemo-, and enantioselectivity. Both substituted 1H-pyrroles and 1H-indoles were successfully employed to give exclusively the branched N-alkylated products. The synthetic utility of this process was demonstrated by applying this method to the preparation of several medicinal chemistry lead compounds and bromopyrrole alkaloids including longamide B, longamide B methyl ester, hanishin, agesamides A and B, and cyclooroidin.
Introduction
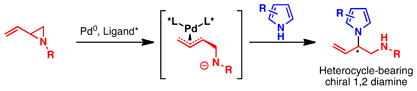
The Pd-catalyzed asymmetric allylic alkylation (Pd-AAA) has become a powerful tool for the construction of stereocenters and has engendered a number of versatile methods for the total synthesis of natural products and other biologically-active targets.1 Unlike other asymmetric reactions, the Pd-AAA is capable of constructing several different types of bonds in an enantioselective fashion, including C–C, C–N, C–O, and C–S bonds. Furthermore, this transformation is highly chemoselective and can be performed in the presence of a large number of functional groups. Despite its versatility, the Pd-AAA typically requires the use of soft stabilized nucleophiles such as malonates or imides and has been limited in its ability to accommodate hard nucleophiles. Our group has extended the scope of this reaction to include hard nucleophiles such as alkoxides, lithium enolates, and benzylic anions employing a family of diphenylphosphino benzoic acid-derived ligands (Figure 1).2 However, hard nitrogen nucleophiles, specifically, nitrogen heterocycles, largely remain unexplored in this transformation (eq 1).3 Likewise, the scope of electrophiles used in the Pd-AAA has been limited to allylic ester derivatives.1 Other, unstabilized leaving groups, such as ethers and amines, are difficult to employ. By taking advantage of ring strain to facilitate ring opening, vinyl epoxides have been successfully employed in the Pd-AAA.4 To date, the analogous vinyl aziridines have remained a challenge in this transformation despite their potential to construct chiral 1,2 diamines (eq 1).
Figure 1.

Diphenylphosphino benzoic acid-based ligands
 |
(1) |
Heterocycle-bearing chiral 1,2 diamines, (see eq 1) are present in a large number of natural products and small molecule pharmaceuticals.5 One important class of natural products containing this structural motif, the bromopyrrole alkaloids, present a significant synthetic challenge due to the presence of numerous contiguous stereocenters, nitrogen-rich polar functional groups, and sensitive pyrrole nuclei (Figure 2). Due to their intriguing structures and biological activities, the bromopyrrole alkaloids have been the target of numerous total syntheses.6 The agelastatins contain a fused tetracyclic structure and possess a variety of interesting biological activities including nanomolar activities against several cancer cell lines.7 Palau’amine possesses a strained hexacylic skeleton, flanked by eight contiguous stereocenters and nine nitrogen atoms and has shown antibiotic, antifungal, as well as immunosuppressant activity.8 Other bromopyrrole alkaloids, including longamide B and its methyl ester, hanishin, agesamides A and B, and cyclooroidin contain pyrrole-fused piperazinones, and possess significant antibiotic and cytotoxic activities.9
Figure 2.

Representative bromopyrrole alkaloids
Like the piperazinones found in pyrrole alkaloids, indole-fused piperazinones and piperazines (Figure 3) have also shown a broad array of biological activities.10 Indole-fused piperazinones are known for their ability to act as conformationally rigid peptidomimetics and have been shown to act as antagonists of the histamine 3 (H3) receptor, which is expressed in the central nervous system and controls histamine levels in the brain.10 These indole-fused heterocycles are present in a large number of drug candidates as well as medicinal chemistry lead compounds.11 Recently, these compounds have been patented for their H3 antagonist activity, which can be used to treat obesity, cardiovascular disorders, Alzheimer’s disease, and other neurological disorders.10
Figure 3.

Representative medicinal chemistry lead compounds
Due to the abundance of biologically active compounds that contain a heterocycle-bearing 1,2 diamine (see eq 1 and Figures 2 and 3), the development of catalytic asymmetric methods to generate these moieties is an important challenge. While many asymmetric alkylation methods exist for functionalizing heterocycles at the carbon atom,12 relatively few examples are known for the direct asymmetric alkylation at nitrogen. Chen and coworkers reported an organocatalytic intermolecular asymmetric N-alkylation of various substituted indoles with Morita Baylis-Hillman carbonates.13 Likewise, Bandini et al. have disclosed that cinchona alkaloid-derived catalysts can facilitate enantioselective N-alkylations of indoles in an intramolecular fashion.14 Most recently, Hartwig et al. reported an elegant Ir-catalyzed chemo-, regio- and enantioselective allylation of indoles with linear allyl carbonates.15 Our group has utilized heterocycles as nitrogen nucleophiles in the Pd-AAA in the context of natural product total syntheses.3 However, no general method has yet been disclosed utilizing these molecules as nucleophiles in the Pd-AAA.
The Pd-AAA can operate through a variety of mechanisms to induce chirality. Unlike many other asymmetric transformations, the Pd-AAA can convert racemic starting materials to enantioenriched products through a dynamic kinetic asymmetric transformation (DYKAT).1e In contrast to a kinetic resolution, which is limited to a maximum theoretical yield of 50% and involves consumption of only one enantiomer of the starting material, a DYKAT has a theoretical yield of 100%. In this transformation, both enantiomers of the starting material are converted into a single, optically active, product. This type of process is desirable, as it minimizes waste and increases the overall synthetic efficiency and atom economy of a reaction by employing all of the starting material.16
While racemic vinyl epoxides have been shown to undergo a Pd-catalyzed DYKAT with a number of carbon and heteroatom nucleophiles, examples with vinyl aziridines remain scant.17 Typically, vinyl aziridines require a strong electron-withdrawing group on the nitrogen atom to increase the electrophilicity of the adjacent carbon atoms.18 The necessity of such a group imposes a limitation on the system, and ideally both electron-rich and electron-deficient vinyl aziridines could be used. Our group has studied these compounds in the Pd-AAA but required the use of isocyanates to activate the vinyl aziridine.17a Thus the utility with these useful, nitrogen-containing electrophiles has been limited, and nucleophiles such as nitrogen heterocycles have remained understudied. The reactions of vinyl aziridines with nitrogen heterocycles, such as pyrrole and indole, would provide access to useful, enantioenriched, heterocycle-bearing diamine products, which comprise the core of several biologically relevant compounds.
Results and Discussion
Reaction Optimization
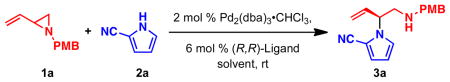
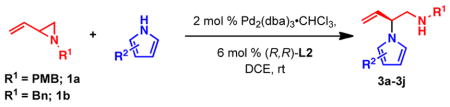
To determine conditions under which nitrogen heterocycles could serve as competent nucleophiles in the Pd-AAA, we studied the reaction of 1H-pyrrole-2-carbonitrile (2a) with vinyl aziridine 1a17a (Table 1). Treatment of this system with 2 mol % Pd2(dba)3•CHCl3, and 6 mol % of ligand L1 in 1,2 dichloroethane (DCE) provided the desired N-alkylated product 3a in a promising 73% yield and 61% ee (entry 1). Previous work with vinyl aziridines showed that changing the ligand had a profound impact on both reactivity and selectivity of the transformation.17a Further screening (entries 2–4) revealed that ligand L2, containing a naphthyl moiety, provided the desired product 3a in 99% yield and 89% ee. Additionally, the choice of solvent (Table 1, entries 5–8) was found to have a dramatic effect on the course of this transformation: conducting the reaction in aromatic solvents (entries 5 and 6) or polar solvents (entries 7–9) had a detrimental effect on the reactivity while showing little impact on the levels of enantioselectivity. The combination of ligand L2 in DCE was found to be optimal for this transformation. Under all the conditions that were evaluated, the branched, N-alkylated product was observed exclusively, demonstrating the profound chemo- and regioselectivity of the transformation.
Table 1.
Selected optimization conditionsa
 | ||||
|---|---|---|---|---|
| entry | ligand | solvent | % yieldb | % eec |
| 1 | L1 | DCE | 73 | 61 |
| 2 | L2 | DCE | 99 | 89 |
| 3 | L3 | DCE | 77 | 72 |
| 4 | L4 | DCE | <5 | 9 |
| 5 | L2 | PhMe | 50 | 89 |
| 6 | L2 | PhCF3 | 52 | 88 |
| 7 | L2 | THF | 52 | 89 |
| 8 | L2 | DME | 44 | 89 |
| 9 | L2 | Dioxane | 60 | 93 |
All reactions were performed with 1.0 equiv of 2a and 1.1 equiv of 1a at ambient temperature at 0.25M in the designated solvent.
Isolated yield.
% ee determined by chiral HPLC.
Substrate Scope
With optimized conditions in hand, we next studied the scope of this transformation using other substituted pyrroles and vinyl aziridines (Table 2). Vinyl aziridines substituted at R1 with either PMB (1a) or Bn (1b) groups reacted smoothly, and typically resulted in similar levels of enantioselectivity. A variety of functional groups were tolerated on the pyrrole nucleus at both the C2- and C3-positions and afforded the branched N-alkylated products in high yield and % ee. The presence of an electron-withdrawing group on the pyrrole ring was necessary for the reaction to proceed, and only starting materials were recovered when an electron-rich or unsubstituted pyrrole was used as the nucleophile (3i,j). To investigate whether C-alkylation could occur, N-methyl pyrrole was subjected to the optimized reaction conditions but resulted in only slight decomposition of the starting materials. The reaction between methyl 1H-pyrrole-2-carboxylate with vinyl aziridines 1a and 1b afforded homoallyl amines 3c and 3d, respectively, as acyclic products which could be stored at −20 °C for several weeks without appreciable cyclization to the piperazinone. The cyclic pyrrole-fused piperazinones could be accessed by heating the acyclic products to 40 °C for 16 h. In cases where lower yields were obtained, such as 3b and 3c, only unreacted pyrrole was observed, with little to no decomposition. On the other hand, unreacted vinyl aziridine was rarely observed at the end of the reaction. When 3-nitro-1H-pyrrole was used, the addition of 1.1 equiv of Cs2CO3 was needed to facilitate the reaction (3h). However, no base was required in other cases, and the amide anion in the π–allyl Pd intermediate (see eq 1) was sufficiently basic to deprotonate the pyrrole N–H and facilitate the transformation.
Table 2.
Reaction scope of pyrroles with vinyl aziridinesa
 | ||
|---|---|---|
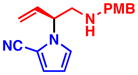 3a 99% yield, 89% ee |
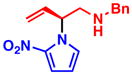 3b 77% yield, 89% ee |
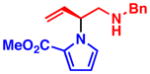 3c 76% yield, 96% ee |
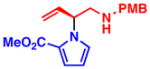 3d 86% yield, 94% ee |
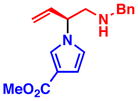 3e 86% yield, 94% ee |
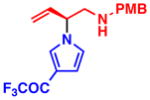 3f 92% yield, 93% ee |
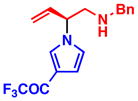 3g 95% yield, 93% ee |
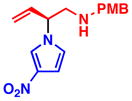 3h 85% yield, 90% eeb |
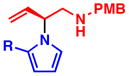 3i, R = Et: NR 3j, R = H: NR |
All reactions were performed with 1.0 equiv of pyrrole nucleophile and 1.1 equiv of 1 at ambient temperature. Isolated yield. % ee was determined by chiral HPLC.
1.1 equiv of Cs2CO3 was added.
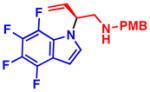
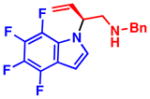
To further expand the generality of this process, indoles were employed as nucleophiles in the transformation. Like pyrroles, electron-deficient indoles reacted smoothly under the optimized reaction conditions providing the desired products in high yield and % ee (Table 3). Although indoles have a high propensity to undergo substitution at C3 in the presence of Pd source and an allylic electrophile, only the N-alkylated, branched products were observed.19 We postulated that N-alkylation could be suppressed in favor of C3 alkylation in the absence of an electron-withdrawing group, or in the presence of an electron-donating group at C2. However, using 2-methyl-1H-indole or unsubstituted indole (4l) led to only unreacted starting materials. Substitution on either the azole, or benzene portion of the indole moiety, was sufficient to facilitate the reaction. However, a strong electron-withdrawing group was necessary to obtain high levels of reactivity and enantioselectivity. When weak electron-withdrawing groups were employed, such as Cl or Br, both the yield and % ee of the product decreased(4j,k); 2-phenyl-1H-indole only afforded trace quantities of the desired product (4m). As for the cases with pyrroles, nitrile (4e,f), ester (4i), halide (4j,k), ketone (4h), and nitro (4c,d,g) functional groups were all tolerated in the transformation and little to no decomposition of the indole nucleophile was observed, demonstrating the high functional group compatibility of this transformation.
Table 3.
Reaction scope of indoles with vinyl aziridinesa
 | ||
|---|---|---|
 4a 89% yield, 90% eeb |
 4b 96% yield, 92% ee |
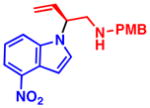 4c 99% yield, 86% ee |
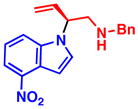 4d 86% yield, 88% ee |
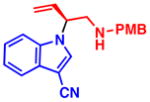 4e 92% yield, 89% ee |
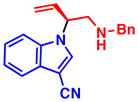 4f 88% yield, 90% ee |
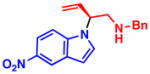 4g 81% yield, 83% ee |
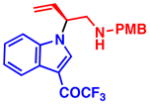 4h 99% yield, 81% eeb |
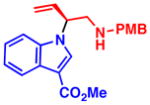 4i 91% yield, 93% ee |
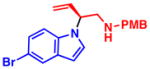 4j 57% yield, 73% eeb |
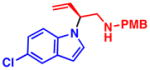 4k 60% yield, 73% eeb |
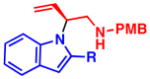 4l, R = H: NR 4m, R = Ph: NR |
All reactions were performed with 1.0 equiv of indole nucleophile and 1.1 equiv of 1 at ambient temperature. Isolated yield. % ee was determined by chiral HPLC.
1.1 equiv of Cs2CO3 was added.
Piperazinone Functionalization and Synthesis of Medicinal Chemistry Leads
When heterocycles containing certain acyl substituents at the 2–position were employed in the transformation, a cyclization occurred, providing access to enantioenriched piperazinones (Table 4). For instance, pyrrole-fused piperazinone 5a was obtained in 97% yield and 90% ee from vinyl aziridine 1a and pyrrole containing a trifluoromethyl ketone at the 2-position. Switching R2 from a trifluoromethyl ketone to a methyl ester also afforded the desired cycloadduct 5b. Like pyrroles, indoles could also be employed as nucleophiles, to afford indole-fused piperazinones utilizing either 1a or 1b as the vinyl aziridine. Piperazinones containing a pyrrole moiety like the one found in 5b, represent the core structure of a number of bromopyrrole alkaloids including longamide B, agesamides A and B, hanishin and cyclooroidin.7 Piperazinones containing indole moieties such as 5c and 5d have also found broad use in medicinal chemistry and show significant biological activities.10
Table 4.
Reactions of nitrogen heterocycles with vinyl aziridines to access piperazinonesa
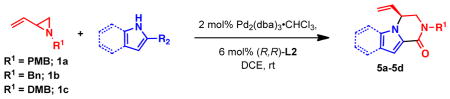 | |||
|---|---|---|---|
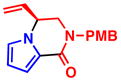 5a, R2 = COCF3 97% yield, 90% eeb |
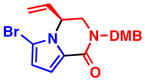 5b, R2 = CO2Me 72% yield, 95% eec |
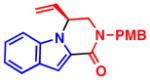 5c, R2 = CO2Me 72% yield, 93% ee |
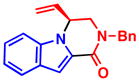 5d, R2 = CO2Me 97% yield, 96% ee |
All reactions were performed with 1.0 equiv of heterocyclic nucleophile and 1.1 equiv of 1 at ambient temperature. Isolated yield. % ee was determined by chiral HPLC.
1.1 equiv of Cs2CO3 was added.
Performed using 2.5 mol % [Pd(C3H5)Cl]2 7.5 mol % (R,R)-L1, in CH2Cl2 at rt.
The indole products isolated from this cyclization were readily transformed into the core of several patented piperazinones (Scheme 1).10 Hydroboration of compound 5d followed by oxidation afforded primary alcohol 6 as the only product, in 85% yield. Oxidation of the alcohol 6 with PhI(OAc)2 in the presence of catalytic TEMPO provided the known carboxylic acid 7 in 81% yield. The sign and magnitude of the optical rotation of this acid was in agreement with previous reports, confirming the absolute configuration from the Pd-AAA.10, 14
Scheme 1.

Synthesis of medicinal chemistry lead compounds.
Compound 5d was easily reduced upon treatment with LiAlH4 to give the corresponding piperazine 8 in 85% yield. Indole-fused piperazines of this general structure are also found in a large number of medicinal chemistry leads, and have been shown to be useful in treating central nervous system disorders, gastrointestinal disorders, diabetes, obesity, and sleep apnea.10 Hydroboration of piperazine 8, followed by oxidation, provided the known alcohol 9 in 93% yield. Although we only show the elaboration of a simple indole into the core of biologically active piperazinone 7 and piperazine 8, utilizing substituted indoles should provide access to more complex members of this substrate class.
A Concise Synthesis of Bromopyrrole Alkaloids
Bromopyrrole alkaloids are an important class of natural products with a range of biological activities.6 Many of these alkaloids contain a piperazinone core that can be accessed through the Pd-AAA of a vinyl aziridine with a bromopyrrole nucleophile. To demonstrate the versatility of the method described herein, the syntheses of several bromopyrrole alkaloids were pursued (Scheme 2). Piperazinone 5b served as a scaffold that was elaborated to a common intermediate from which many bromopyrrole alkaloids were synthesized. Hydroboration of 5b with 9-BBN, followed by oxidation, provided the desired primary alcohol 10 in 82% yield. Cleavage of the dimethoxybenzyl (DMB) group with tetrahydrothiophene followed by bromination with NBS furnished alcohol 11 in 96% over two steps. Alcohol 11 was readily oxidized to longamide B (12) with PhI(OAc)2 in the presence of catalytic TEMPO. Longamide B (12) could then be converted to three additional natural products. Esterification of the free acid in longamide B (12) under known literature conditions20 with either a methyl or ethyl group affords longamide B methyl ester (13), and hanishin (14) respectively. Two additional bromopyrrole alkaloids, agesamides A and B, were readily accessed from alcohol 11. Oxidation of 11 with Dess-Martin periodinate, followed by treatment with potassium cyanide and ammonium carbonate, lead to both agesamides A (15) and B (16). Likewise, installation of the amino-imidazole moiety onto the free acid, affords cyclooroidin (17).21
Scheme 2.

Concise synthesis of bromopyrrole alkaloids
Mechanistic Discussion and Rationalization of Asymmetric Induction
We propose the following mechanistic rationale for the observed stereochemistry based on the “wall and flap” model previously reported.1d, 22 Initial ionization of the two enantiomers of the vinyl aziridine 1 (Scheme 3) with the (R,R)-enantiomer of the Pd-ligand complex proceeds rapidly in both a matched and mismatched manner to provide two possible zwitterionic π–allyl Pd complexes 18 and ent-18, respectively. These two intermediates can interconvert through a rapid π–σ–π equilibration. Nucleophilic attack of the pyrrole nucleophile 2 on the π–allyl palladium species 18, at the more substituted terminus, provides the desired product 3.
Scheme 3.

Mechanistic rationale
In order to obtain high levels of enantioselectivity, a Curtin-Hammet situation must be established such that π–σ–π interconversion is rapid and much faster than the subsequent nucleophilic attack. By using a larger ligand, L2, a more sterically encumbered environment is established, which slows down the rate of nucleophilic attack and increases the time allowed for π–σ–π interconversion. An unfavorable steric interaction between one of the two diastereomeric π–allyl Pd intermediates determines which enantiomer of the product is formed, such that k1 is much larger than k2. The absolute configuration of the product from this transformation, as well as related reactions with vinyl epoxides, support this model.4
Pd-catalyzed allylic alkylation of monosubstituted π–allyl-Pd intermediates typically favors the achiral, linear product. To rationalize the exclusive regioselectivity for the branched product, we propose that nucleophilic attack proceeds through a five-membered transition state (Scheme 4). In this transition state, the amide anion of the vinyl aziridine directs the pyrrole nucleophile through a hydrogen bond with the pyrrole N-H, to the proximal end of the π–allyl-Pd moiety. Thus nucleophilic attack proceeds through the favored five-membered transition state 19, leading to the observed branched product. This would preclude the reaction proceeding through a strongly disfavored seven-membered transition state 20, which contains a trans-olefin and leads to the achiral linear product.
Scheme 4.

Rationale for observed regioselectivity
Conclusion
In conclusion, a highly chemo- regio- and enantioselective palladium-catalyzed asymmetric allylic alkylation between nitrogen heterocycles and vinyl aziridines has been developed. Both substituted pyrroles and indoles can be utilized as nucleophiles in the transformation to afford the N-alkylated branched products exclusively in high yield and % ee. The presence of an electron-withdrawing group on the nitrogen heterocycle was necessary for this reaction to proceed, but many functional groups on the pyrrole and indole nuclei were tolerated. No electron-withdrawing groups were needed on the vinyl aziridine to facilitate its opening, adding to the high generality of this process.
By substituting the heterocycles at the C2 position with an ester derivative, piperazinones were accessed readily through a formal asymmetric [3+3] cycloaddition. The indole-fused piperazinones were used as synthetic building blocks to access a series of drug leads that have received extensive attention from the medicinal chemistry community. Likewise, pyrrole-fused piperazinones were used as synthetic building blocks to rapidly assemble the bromopyrrole alkaloids longamide B, longamide B methyl ester, hanishin, agesamides A and B, and cyclooroidin.
Experimental Section
Typical Procedure for the Pd-Catalyzed Asymmetric Allylic Alkylation of Pyrroles with Vinyl Aziridines
To a flame-dried microwave vial equipped with a magnetic stir bar was added 1H-pyrrole-2-carbonitrile 2a (18.4 mg, 0.2 mmol) and vinyl aziridine 1a (48.2 mg, 0.22 mmol). The system was evacuated and filled with argon (3x) and dry degassed DCE (0.4 mL) was then added. In a separate flame-dried microwave vial equipped with a magnetic stir bar was added Pd2(dba)3•CHCl3 (4.1 mg, 4.0×10−3 mmol) and (R,R)-L2 (9.5 mg, 0.012 mmol). The system was evacuated and filled with argon (3x), and DCE (0.4 mL) was added. This vial was stirred at rt for 15–20 min until a bright orange color persisted and its contents were then cannulated to the first vial. The reaction mixture was stirred for 48 h at rt, diluted with CH2Cl2 (10 mL) and then poured onto water (10 mL). The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (3×10 mL). The combined organic fractions were dried over anhydrous sodium sulfate, filtered and concentrated en vacuo. The crude reaction mixture was purified by flash column chromatography on silica gel (30% ethyl acetate in petroleum ether) to provide 56 mg of a yellow oil (3a) (99% yield, 89% ee by HPLC, OJ-H column, 9:1 heptane/i-propanol, 1.0mL/min, 254 nm, rt = 27.4, 30.2 min). Rf = 0.30 (30% ethyl acetate/petroleum ether). 1H-NMR (CDCl3, 400 MHz): δ 7.20-7.17 (m, 2H), 6.92 (dd, J = 2.6, 1.7 Hz, 1H), 6.86-6.84 (m, 2H), 6.82 (dd, J = 3.9, 1.6 Hz, 1H), 6.22 (dd, J = 3.9, 2.8 Hz, 1H), 5.97 (ddd, J = 17.1, 10.5, 5.9 Hz, 1H), 5.28 (ddd, J = 10.5, 1.3, 0.7 Hz, 1H), 5.10 (ddd, J = 17.2, 1.5, 0.7 Hz, 1H), 4.97-4.92 (m, 1H), 3.79 (s, 3H), 3.72 (q, J = 12.2 Hz, 2H), 3.13-3.03 (m, 2H), 1.35 (s, 1H). 13C-NMR (CDCl3, 101 MHz): d 159.2, 135.3, 132.2, 129.7, 124.7, 120.5, 119.0, 114.4, 114.3, 110.3, 104.3, 61.5, 55.7, 53.1, 52.5. IR(film): 3335, 3123, 2917, 2836, 2214, 1610, 1511, 1457 cm−1. [α]D: −31.7 (CH2Cl2, c 1.00). HRMS (C17H19N3ONa): calculated (M + Na): 304.1426, found (M + Na): 304.1428.
Typical Procedure for the Pd-Catalyzed Asymmetric Allylic Alkylation of Indoles with Vinyl Aziridines
To a flame-dried microwave vial equipped with a magnetic stir bar was added methyl 1H-indole-3-carboxylate (35.0 mg, 0.2 mmol) and vinyl aziridine 1b (48.2 mg, 0.22 mmol). The system was evacuated and filled with argon (3x) and dry degassed DCE (0.4 mL) was then added. In a separate flame-dried microwave vial equipped with a magnetic stir bar was added Pd2(dba)3•CHCl3 (4.1 mg, 4.0×10−3 mmol) and (R,R)-L2 (9.5 mg, 0.012 mmol). The system was evacuated and filled with argon (3x), and DCE (0.4 mL) was added. This vial was stirred at rt for 15–20 min until a bright orange color persisted and its contents were then cannulated to the first vial. The reaction mixture was stirred for 48 h at rt, diluted with CH2Cl2 (10 mL) and then poured onto water (10 mL). The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (3×10 mL). The combined organic fractions were dried over anhydrous sodium sulfate, filtered and concentrated en vacuo. The crude reaction mixture was purified by flash column chromatography on silica gel (35% ethyl acetate in petroleum ether) to provide 66.0 mg of a clear oil (4i) (91% yield, 93% ee by HPLC, IA column, 95:5 heptane/i-propanol, 0.8 mL/min, 254 nm, rt = 28.6, 32.8 min.). Rf = 0.3 (45% ethyl acetate/petroleum ether). 1H-NMR (CDCl3, 400 MHz): δ 8.21-8.18 (m, 1H), 7.93 (s, 1H), 7.39-7.34 (m, 1H), 7.30-7.23 (m, 2H), 7.13-7.10 (m, 2H), 6.84-6.80 (m, 2H), 6.00 (ddd, J = 17.3, 10.6, 5.7 Hz, 1H), 5.27-5.24 (m, 1H), 5.10-5.06 (m, 2H), 3.91 (s, 3H), 3.77 (s, 3H), 3.73-3.66 (m, 2H), 3.16 (qd, J = 14.2, 6.8 Hz, 2H), 1.40 (s, 1H). 13C-NMR (CDCl3, 101 MHz): δ 165.9, 159.2, 137.1, 135.2, 132.5, 132.1, 129.6, 127.0, 123.2, 122.5, 122.2, 118.7, 114.3, 110.8, 108.1, 59.2, 55.7, 53.7, 51.8, 51.5. IR (film) 2995, 2948, 2835, 1697, 1612, 1532, 1512, 1460 cm−1. [α]D: −19.4 (CH2Cl2, c 1.03). HRMS (C22H24N2O3Na): calculated (M + Na): 387.1676, found (M + Na): 387.1685.
Supplementary Material
Acknowledgments
We thank the National Science Foundation and the National Institutes of Health (GM33049) for their generous support of our programs. M.O. thanks the John Stauffer Memorial Fellowship for financial support. G.D. thanks the Stanford Graduate Fellowship for financial support. Palladium salts were generously supplied by Johnson-Matthey.
Footnotes
Supporting Information Available: Experimental procedures and characterization data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Trost BM, Van Vranken DL. Chem Rev. 1996;96:395–422. doi: 10.1021/cr9409804. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Crawley ML. Chem Rev. 2003;103:2921–2944. doi: 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]; (c) Trost BM. J Org Chem. 2004;69:5813–5837. doi: 10.1021/jo0491004. [DOI] [PubMed] [Google Scholar]; (d) Trost BM, Machacek MR, Aponick A. Acc Chem Res. 2006;39:747–760. doi: 10.1021/ar040063c. [DOI] [PubMed] [Google Scholar]; (e) Trost BM, Fandrick DR. Aldrichimica Acta. 2007;40:59–72. [Google Scholar]
- 2.(a) Trost BM, Schroeder GM. J Am Chem Soc. 1999;121:6759–6760. [Google Scholar]; (b) Trost BM, Zhang T. Org Lett. 2006;8:6007–6010. doi: 10.1021/ol0624878. [DOI] [PubMed] [Google Scholar]; (c) Trost BM, Thaisrivongs DA. J Am Chem Soc. 2008;130:14092–14093. doi: 10.1021/ja806781u. [DOI] [PubMed] [Google Scholar]; (d) Trost BM, Thaisrivongs DA. J Am Chem Soc. 2009;131:12056–12057. doi: 10.1021/ja904441a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Trost BM, Kriche MJ, Grenzer EM. Org Lett. 2002;4:2005–2008. doi: 10.1021/ol020046s. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Dong G. Org Lett. 2007;9:2357–2359. doi: 10.1021/ol070742y. [DOI] [PubMed] [Google Scholar]; (c) Trost BM, Dong G. J Am Chem Soc. 2006;128:6054–6055. doi: 10.1021/ja061105q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trost BM, Bunt RC, Lemoine RC, Calkins TL. J Am Chem Soc. 2000;122:5968–5976. [Google Scholar]
- 5.(a) Somei M, Yamada F. Nat Prod Rep. 2005;22:73–103. doi: 10.1039/b316241a. [DOI] [PubMed] [Google Scholar]; (b) Thirumalairajan S, Pearce BM, Thompson A. Chem Commun. 2010;46:1797–1812. doi: 10.1039/b926045e. [DOI] [PubMed] [Google Scholar]
- 6.For a review on their total syntheses see: Weinreb SM. Nat Prod Rep. 2007;24:931–948. doi: 10.1039/b700206h.Christophersen C. In: The Alkaloids. Brossi A, editor. Vol. 24. Academic Press; Orlando: 1985. pp. 25–98.Hao E, Fromont J, Jardine D, Karuso P. Molecules. 2001;6:130–141.
- 7.(a) D’Ambrosio M, Guerriero A, Debitus C, Ribes O, Pusset J, Leroy S, Pietra F. J Chem Soc, Chem Commun. 1993:1305–1306. [Google Scholar]; (b) D’Ambrosio M, Guerriero A, Chiasera G, Pietra F. Helv Chim Acta. 1994;77:1895–1902. [Google Scholar]; (c) D’Ambrosio M, Guerriero A, Ripamonti M, Debitus C, Waikedre J, Pietra F. Helv Chim Acta. 1996;79:727–735. [Google Scholar]; (d) Maijer L, Thunnissen AM, White AW, Garnier M, Nikolic M, Tsai LH, Walter J, Cleverley KE, Salinas PC, Wu YZ, Biernat J, Mandelkov EM, Kim SH, Pettit GR. Chem Biol. 2000;2:51–63. doi: 10.1016/s1074-5521(00)00063-6. [DOI] [PubMed] [Google Scholar]
- 8.(a) Kinnel RB, Gehrken PH, Scheuer PJ. J Am Chem Soc. 1993;115:3376–3377. [Google Scholar]; (b) Seiple IB, Su S, Young IS, Lewis CA, Yamaguchi J, Baran PS. Angew Chem, Int Ed. 2010;49:1095–1098. doi: 10.1002/anie.200907112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Mancini I, Guella G, Amade P, Roussakis C, Pietra F. Tetrahedron Lett. 1997;38:6271–6274. [Google Scholar]; (b) Umeyama A, Ito S, Yuasa E, Arihara S, Yamada T. J Nat Prod. 1998;61:1433–1434. doi: 10.1021/np980207u. [DOI] [PubMed] [Google Scholar]; (c) Srinivasa Reddy N, Venkateswarlu Y. Biochem Syst Ecol. 2000;28:1035–1037. doi: 10.1016/s0305-1978(00)00016-8. [DOI] [PubMed] [Google Scholar]; (d) Fattorusso E, Taglialatela-Scafati O. Tetrahedron Lett. 2000;41:9917–9922. [Google Scholar]; (e) Tsuda M, Yasuda T, Fukushi E, Kawabata J, Sekiguchi M, Fromont J, Kobayashi J. Org Lett. 2006;8:4235–4238. doi: 10.1021/ol061464q. [DOI] [PubMed] [Google Scholar]
- 10.(a) Hebeisen P, Mattei P, Muller M, Richter H, Roever S, Taylor S. 02/072584. PCT Int Appl WO. 2002:A2.; (b) Jolidon S, Narquizian R, Norcross RD, Pinard E. 2006/0128712. PCT Int Appl WO US. 2006:A1.; (c) Nettekoven M, Plancher J-M, Richter H, Roche O, Taylor S. 2007/0135416. PCT Int Appl WO US. 2007:A1.
- 11.Dinsmore CJ, Beshore DC. Org Prep Proced. 2002;34:367–404. [Google Scholar]
- 12.For recent examples, see: Austin JF, Macmillan DWC. J Am Chem Soc. 2002;124:1172–1173. doi: 10.1021/ja017255c.Evans DA, Scheidt KA, Fandrick KR, Lam HW, Wu J. J Am Chem Soc. 2003;125:10780–10781. doi: 10.1021/ja036985c.Trost BM, Quancard J. J Am Chem Soc. 2006;128:6314–6315. doi: 10.1021/ja0608139.Trost BM, Müller C. J Am Chem Soc. 2008;130:2438–2439. doi: 10.1021/ja711080y.
- 13.Cui HL, Feng X, Peng J, Lei J, Jiang K, Chen YC. Angew Chem, Int Ed. 2009;48:5737–5740. doi: 10.1002/anie.200902093. [DOI] [PubMed] [Google Scholar]
- 14.Bandini M, Eichholzer A, Tragni M, Umani-Ronchi A. Angew Chem, Int Ed. 2008;47:3238–3241. doi: 10.1002/anie.200705685. [DOI] [PubMed] [Google Scholar]
- 15.Stanley LM, Hartwig JF. Angew Chem, Int Ed. 2009;48:5737–5740. [Google Scholar]
- 16.(a) Trost BM. Science. 1991;254:1471–1477. doi: 10.1126/science.1962206. [DOI] [PubMed] [Google Scholar]; (b) Trost BM. Angew Chem, Int Ed. 1995;34:259–281. [Google Scholar]
- 17.(a) Trost BM, Fandrick DR. J Am Chem Soc. 2003;125:11836–11837. doi: 10.1021/ja037450m. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Fandrick DR, Brodmann T, Stiles DT. Angew Chem, Int Ed. 2007;46:6123–6125. doi: 10.1002/anie.200700835. [DOI] [PubMed] [Google Scholar]
- 18.For Pd catalysis with vinyl aziridines, see: Butler ACD, Inman GA, Alper H. J Org Chem. 2000;65:5887–5890. doi: 10.1021/jo000608q.Dong C, Alper H. Tetrahedron: Asymmetry. 2004;15:1537–1540.Sebelius S, Olsson VJ, Szabó K. J Am Chem Soc. 2005;127:10478–10479. doi: 10.1021/ja052885q.Fontana F, Tron GC, Barbero N, Ferrini S, Thomas SP, Aggarwal VK. Chem Commun. 2010;46:267–269. doi: 10.1039/b920564k.
- 19.Kagawa N, Malerich JP, Rawal VH. Org Lett. 2008;10:2381–2384. doi: 10.1021/ol8006277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Banwell MG, Bray AM, Willis AC, Wong DJ. New J Chem. 1999;23:687–690. [Google Scholar]; (b) Patel J, Pelloux-Léon N, Minassian F, Vallée Y. J Org Chem. 2005;70:9081–9084. doi: 10.1021/jo051555l. [DOI] [PubMed] [Google Scholar]
- 21.(a) Papeo G, Gómez-Zurita Frau MA, Borghi D, Varasi M. Tetrahedron Lett. 2005;46:8635–8638. [Google Scholar]; (b) Patel J, Pelloux-Léon N, Minassian F, Vallée Y. Tetrahedron Lett. 2006;47:5561–5563. [Google Scholar]
- 22.Trost BM, Toste FD. J Am Chem Soc. 1999;121:4545–4554. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.