

The study of Mercer et al.1, published in this edition of Circulation Research, reports new evidence linking oxidative DNA damage, atherosclerosis and the metabolic syndrome. Although these relationships have been long proposed,2-4 many have criticized previous reports asking the rhetorical question of which came first “the chicken or the egg?” Or more specific to the topic of Mercer et al.'s study, does oxidative DNA damage actively promote atherosclerosis (and/or metabolic syndrome) or is DNA damage a result of these abnormalities?
Conceptually, the theory is attractive. DNA damage occurs often. Every time you walk outside from your office or laboratory to another building your skin is bombarded by UV irradiation. Were it not for the presence of robust and often redundant DNA repair systems, multiple layers of cells in your skin would be damaged. In some cases the cells apoptose. The causation between induced DNA damage and cellular apoptosis has been established for many different types of cells.5 In other cases, genomic DNA might be altered in such a way as to promote malignant transformation. Or, mitochondrial DNA could be damaged such that the cellular burden of reactive oxygen species results in further oxidative DNA damage to nuclear DNA.6,7
Why doesn't the same paradigm fit for oxidative DNA damage as a cause of atherosclerosis? First and foremost, the vasculature is not the skin and the connection between reactive oxygen species (ROS) and oxidative DNA damage (much less the connection to atherosclerosis and metabolic syndrome) is less straightforward to study than the effect of UV irradiation on keratinocytes and melanocytes. Directly measuring the impact of reactive oxygen species in the vasculature, or on the function of organs responsible for the cluster of metabolic abnormalities commonly referred to as the metabolic syndrome (liver, pancreas and adipose tissue), is not possible in the same way in which it is for the skin.
The notion that oxidative DNA damage contributes to atherosclerosis and its complications is far from new. Wallace was one of the first to suggest that mitochondrial DNA (mtDNA) mutations and/or damage correlate with human disease in 1992.8 Since that time, one consistent theme from the many laboratories studying oxidative DNA damage and atherosclerosis has been a focus on mtDNA damage. MtDNA lacks histone protection and mechanisms for repair of mtDNA damage are far less comprehensive than those that exist for nuclear DNA damage.9 A teleologic argument for this difference is that most cells have multiple mitochondria and damage to a small percentage of mitochondria is unlikely to adversely affect the cell in any major way. This is probably true, as major phenotypes emerge only when mitochondrial function is dramatically altered. Though a link between mtDNA damage and atherosclerosis has been established since the 1990s,3,8,10 the causality was not proven.
Enter Mercer et al.1 who chose to use the Ataxia Telangiectasia Mutated (ATM) protein defect as a model for studying whether oxidative mtDNA damage causes atherosclerosis and the metabolic syndrome. The rationale for these experiments was based on two different types of findings. First, some patients with Ataxia Telangiectasia have insulin resistance and presumably metabolic syndrome11 and various studies implicate ATM function in atherosclerosis.12 Secondly, ATM is a serine/threonine kinase that plays a role in DNA repair, mtDNA content, mitochondrial biogenesis and glucose homeostasis.13,14
For the present study, Mercer et al.1 used mice that were ApoE null and either ATM haplodeficient or normal in ATM function. In the ApoE-/- background, ATM haplodeficiency was associated with hyperlipidemia, hypertension, weight gain, increased numbers of adipocytes and inflammatory changes in the liver, as well as other features consistent with the metabolic syndrome. These mice also displayed mtDNA damage and mitochondrial dysfunction in multiple organs.
An initial impression is that this study hardly overcomes the burden of proof of causality between oxidative DNA damage and metabolic syndrome and atherosclerosis. Indeed, a significant limitation of the present study is that the authors did not address mtDNA damage and mitochondrial dysfunction in the aortas from ApoE-/-/ATM+/- mice. These studies could have provided additional insight on the relative roles of nuclear DNA and mtDNA damage in vascular dysfunction.
However, in the breadth of their studies the authors do, in our opinion, provide a level of evidence consistent with mtDNA damage causing metabolic syndrome and atherosclerosis - well surpassing that of prior studies. They were able to define a complex phenotype consisting of histologic change in the aorta, advanced atherosclerosis, metabolic changes with abnormal function of liver and pancreas, and mtDNA damage. Further, they show that accelerated atherosclerosis in ApoE-/-/ATM+/- mice compared with ApoE-/- mice was partially reversed in bone marrow transplant experiments indicating that ATM deficiency enhances atherosclerosis by stimulating stress-activated signaling pathways in macrophages12 (Figure). Incidentally, activation of these signaling pathways and impairment of phosphoinositide 3-kinase/Akt pathway have been implicated in the development of insulin resistance in ApoE-/-/ATM+/- mice.12
Figure.
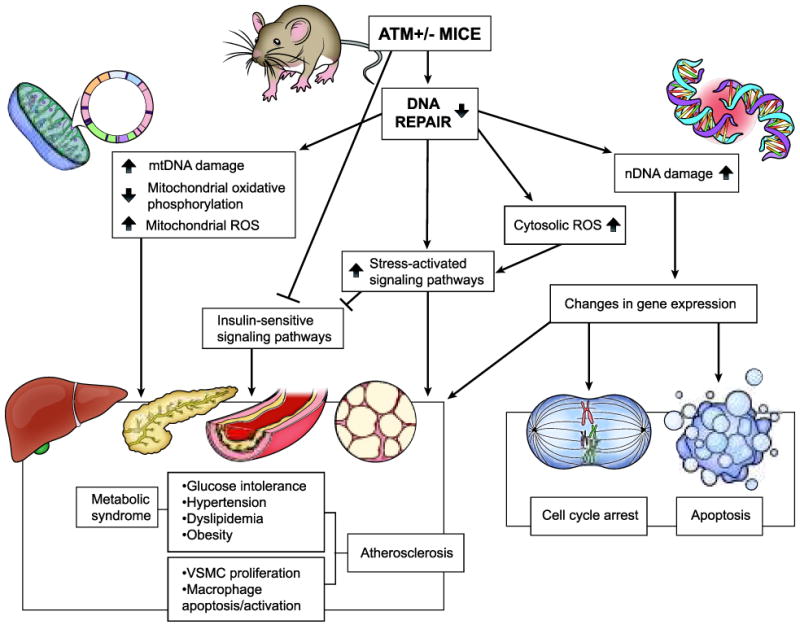
Schematic diagram depicting molecular pathways that regulate atherosclerosis and metabolic syndrome in ATM haplodeficient ApoE-/- mice. Thick up and down arrows indicate increase and decrease, respectively, whereas ┤ indicates inhibition.
Plaque macrophages in ApoE-/-/ATM+/- mice demonstrated increased apoptosis, consistent with the findings of mtDNA damage in these cells. Macrophage apoptosis has been demonstrated to lead to necrotic core formation in the plaque and ultimately plaque instability. It should be noted, however, that while the ApoE-/-/ATM+/- genotype had accelerated atherosclerosis there was also an increase in proliferating cells and reduction in apoptotic cells in the plaque area. The impact of the BMT experiments on this phenotype was unclear.
The authors also extensively characterized metabolic and metabolomic changes related to ATM haplodeficiency in these mice. Their studies on tissues and cells isolated from these mice included demonstration of the impact of ATM haplodeficiency on relevant signaling pathways involved in DNA repair. Additionally, the increased mtDNA damage observed in the insulin-sensitive tissues such as liver, skeletal muscle and pancreas of ApoE-/-/ATM+/- mice relative to ApoE-/-/ATM+/+ mice may have contributed to metabolic syndrome and atherosclerosis (Figure). Impaired glucose tolerance in ApoE-/-/ATM+/- mice, relative to ApoE-/-/ATM+/+ mice (with no difference in serum insulin levels and insulin-stimulated glucose clearance) could indicate impaired liver and/or pancreatic function due to mitochondrial deletions or attenuation of signaling pathways involved in membrane translocation of glucose transporter 4 involved in glucose uptake15 (Figure). Furthermore, decreased mitochondrial complex I activity in the liver of ApoE-/-/ATM+/- mice suggests mitochondrial dysfunction and a feed-forward increase in mitochondrial ROS. The authors propose with reasonable evidence that these events lead to impaired lipid metabolism (β-hydroxybutyrate and lipid accumulation) and reduced glycolysis and eventually to development of insulin intolerance and signs of metabolic syndrome.
Despite these very convincing experiments, it is important to consider a number of important questions.
Are the phenotypic changes described in the ATM+/-/ApoE-/- mice attributable solely to mtDNA damage (and hence mitochondrial dysfunction) or are these changes due at least in part to genomic DNA damage causing yet to be delineated molecular changes?
There is also increasing evidence that ATM plays signaling role in signaling pathways other than those involving direct DNA damage. Are there hormones or cytokines present in the oxidative milieu that may activate or inhibit ATM?
Because both genomic and mtDNA damage are present in ATM+/- mice, is genomic DNA damage inducing mtDNA damage and/or dysfunction and is this important in the metabolic abnormalities described by Mercer et al.1?
Finally, although H2O2 production is important in this model, as evidenced by the measurement of 2′,7′-dichlorohydrofluorescein diacetate (H2DCFDA), what role does superoxide play in cellular dysfunction.
For all these reasons, one must accept that even with the strength of the findings of Mercer et al.1 many questions remain regarding the causative role of mtDNA damage in atherosclerosis and metabolic syndrome. This is, however, the hallmark of a well done study. It generates a host of additional questions that can only be answered by further experimentation. The study of Mercer et al.1 fits our criteria for an important, innovative and well done study and we look forward to more information on this important topic in the future.
Acknowledgments
Sources of Funding: Supported in part by National Institutes of Health grants HL-57352 and AG 024282.
Non-standard Abbreviations and Acronyms
- ApoE
apolipoprotein E
- ATM
Ataxia telangiectasia mutated
- MtDNA
mitochondrial DNA
- nDNA
nuclear DNA
- ROS
reactive oxygen species
- VSMCs
vascular smooth muscle cells
Footnotes
Disclosures: None.
References
- 1.Mercer JR, Cheng KK, Figg N, Gorenne I, Mahmoudi M, Griffin J, Vidal-Puig A, Logan A, Murphy MP, Bennett M. DNA Damage Links Mitochondrial Dysfunction to Atherosclerosis and the Metabolic Syndrome. Circ Res. 2010;107:XXX–XXX. doi: 10.1161/CIRCRESAHA.110.218966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Madamanchi NR, Runge MS. Mitochondrial dysfunction in atherosclerosis. Circ Res. 2007;100:460–473. doi: 10.1161/01.RES.0000258450.44413.96. [DOI] [PubMed] [Google Scholar]
- 3.Ballinger SW, Patterson C, Knight-Lozano CA, Burow DL, Conklin CA, Hu Z, Reuf J, Horaist C, Lebovitz R, Hunter GC, McIntyre K, Runge MS. Mitochondrial integrity and function in atherogenesis. Circulation. 2002;106:544–549. doi: 10.1161/01.cir.0000023921.93743.89. [DOI] [PubMed] [Google Scholar]
- 4.Satoh M, Ishikawa Y, Takahashi Y, Itoh T, Minami Y, Nakamura M. Association between oxidative DNA damage and telomere shortening in circulating endothelial progenitor cells obtained from metabolic syndrome patients with coronary artery disease. Atherosclerosis. 2008;198:347–353. doi: 10.1016/j.atherosclerosis.2007.09.040. [DOI] [PubMed] [Google Scholar]
- 5.Roos WP, Kaina B. DNA damage-induced cell death by apoptosis. Trends Mol Med. 2006;12:440–450. doi: 10.1016/j.molmed.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 6.Rasmussen AK. Identification of factors interacting with hMSH2 and HMLH1 in the fetal liver and investigations of how mitochondrial dysfunction creates a mutator phenotype. http://hdl.handle.net/1800/458.
- 7.Rasmussen AK, Chatterjee A, Rasmussen LJ, Singh KK. Mitochondria-mediated nuclear mutator phenotype in Saccharomyces cerevisiae. Nucleic Acids Res. 2003;31:3909–3917. doi: 10.1093/nar/gkg446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corral-Debrinski M, Shoffner JM, Lott MT, Wallace DC. Association of mitochondrial DNA damage with aging and coronary atherosclerotic heart disease. Mutat Res. 1992;275:169–180. doi: 10.1016/0921-8734(92)90021-g. [DOI] [PubMed] [Google Scholar]
- 9.Ballinger S, Patterson C, Yan C, Doan R, Burow D, Young C, Yakes F, van Houten B, Ballinger C, Freeman B, Runge M. Hydrogen peroxide and peroxynitrite inducedmitochondrial DNA damage and dysfunction in vascular endothelial and smooth muscle cells. Circ Res. 2000;86:960–966. doi: 10.1161/01.res.86.9.960. [DOI] [PubMed] [Google Scholar]
- 10.Chuang GC, Yang Z, Westbrook DG, Pompilius M, Ballinger CA, White CR, Krzywanski DM, Postlethwait EM, Ballinger SW. Pulmonary ozone exposure induces vascular dysfunction, mitochondrial damage, and atherogenesis. Am J Physiol Lung Cell Mol Physiol. 2009;297:L209–L216. doi: 10.1152/ajplung.00102.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schalch DS, McFarlin DE, Barlow MH. An unusual form of diabetes mellitus in ataxia telangiectasia. N Engl J Med. 1970;282:1396–1402. doi: 10.1056/NEJM197006182822503. [DOI] [PubMed] [Google Scholar]
- 12.Schneider JG, Finck BN, Ren J, Standley KN, Takagi M, Maclean KH, Bernal-Mizrachi C, Muslin AJ, Kastan MB, Semenkovich CF. ATM-dependent suppression of stress signaling reduces vascular disease in metabolic syndrome. Cell Metab. 2006;4:377–389. doi: 10.1016/j.cmet.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 13.Eaton JS, Lin ZP, Sartorelli AC, Bonawitz ND, Shadel GS. Ataxia-telangiectasia mutated kinase regulates ribonucleotide reductase and mitochondrial homeostasis. J Clin Invest. 2007;117:2723–2734. doi: 10.1172/JCI31604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miles PD, Treuner K, Latronica M, Olefsky JM, Barlow C. Impaired insulin secretion in a mouse model of ataxia telangiectasia. Am J Physiol Endocrinol Metab. 2007;293:E70–E74. doi: 10.1152/ajpendo.00259.2006. [DOI] [PubMed] [Google Scholar]
- 15.Halaby MJ, Hibma JC, He J, Yang DQ. Cell Signal. ATM protein kinase mediates full activation of Akt and regulates glucose transporter 4 translocation by insulin in muscle cells. Cell Signal. 2008;20:1555–1563. doi: 10.1016/j.cellsig.2008.04.011. [DOI] [PubMed] [Google Scholar]