

Abstract
The evolutionarily-conserved DNA-binding protein RBP-J directly interacts with the RAM domain and the ankyrin (ANK) repeats of the Notch intracellular region (RAMIC), and activates transcription of downstream target genes that regulate cell differentiation. In vitro binding assays demonstrate that the truncated N- and C-terminal regions of RBP-J bind to the ANK repeats but not to the RAM domain. Using an OT11 mouse cell line, in which the RBP-J locus is disrupted, we showed that RBP-J constructs mutated in the N- and C-terminal regions were defective in their transcriptional activation induced by either RAMIC or IC (the Notch intracellular region without the RAM domain) although they had normal levels of binding activity to DNA and the RAM domain. The studies using chimeric molecules between RBP-J and its homolog RBP-L showed that the N- and C-terminal regions of RBP-J conferred the IC- as well as RAMIC-induced transactivation potential on RBP-L, which binds to the same DNA sequence as RBP-J but fails to interact with RAMIC. Taken together, these results indicate that the interactions between the N- and C-terminal regions of RBP-J and the ANK repeats of RAMIC are important for transactivation of RBP-J by RAMIC.
INTRODUCTION
A ubiquitous DNA-binding protein RBP-J is evolutionarily conserved from nematode and fruit fly to human (1,2). Earlier studies in Drosophila revealed a pivotal role of Su(H), a Drosophila authlog of RBP-J, in neural development (3,4) and showed that it genetically interacts with the Notch receptor that is involved in cell fate determination (5). The mammalian Notch receptor also plays an important role in cell fate determination of many lineages including nerve, muscle, pancreas and lymphocytes (6–9). We have generated RBP-J knockout mice and demonstrated that the mice die before 10.5 days post-coitum with defects in somite and neural tube formations. The defects are quite similar to those observed in Notch1 knockout mice (10), indicating a conservation of the Notch signaling across the distantly-related species. It has recently been shown that the intracellular region of the Notch receptor (hereafter designated as RAMIC) is proteolytically cleaved by interaction with the ligand and is translocated into the nucleus (11–13). RBP-J directly interacts with RAMIC to activate transcription of downstream target genes such as mammalian homologs of Drosophila Hairy and Enhancer of split, HES-1 and HES-5 genes, which prevent cell differentiation (1). Thus, RBP-J is a critical effector molecule that functions in the Notch signaling pathway.
RBP-J also interacts with Epstein–Barr virus nuclear antigen 2 (EBNA2), an essential protein for virus-induced immortalization of human B lymphocytes, to activate transcription (14,15). Whereas RBP-J functions as a transcriptional activator in the presence of RAMIC or EBNA2 (16), it represses transcription in the absence of those interacting proteins (17,18). On the promoter of the adenoviral gene encoding the capsid protein polypeptide IX (pIX), RBP-J directly interacts with transcriptional co-activators of TFIID and TFIIA to perturb an optimal interaction between them, resulting in repression of the pIX promoter (19). The other type of repression by RBP-J has been attributed to co-repressors binding to the repression domain of RBP-J (18,20). Several molecules such as SMRT, N-CoR, CIR and KyoT2 have been reported as candidates for co-repressors of RBP-J to date, and some of them recruit the histone deacetylase to RBP-J to repress transcription (21–23).
RBP-J binds to two distinct domains of RAMIC, the RAM domain and the ankyrin (ANK) repeats. The RAM domain was originally isolated by yeast two-hybrid screening and was shown to strongly interact with the middle region (amino acids 196–372) of RBP-J (2,16). RAMIC competes with the co-repressors for binding to this middle region of RBP-J through the RAM domain (21,24). On the other hand, the ANK repeats of RAMIC interact with RBP-J weakly but significantly (25,26). IC activates transcription less strongly than RAMIC (25). Constitutively-active phenotypes by enforced expression of mouse Notch1 IC were abolished by missense mutations (M1 and M2) in the ANK repeats, which disrupted the weak interaction of the ANK repeats with RBP-J (25–27). We also showed that the transactivation activity of RAMIC was abrogated by the M1 mutation even though it still interacted with RBP-J via the RAM domain (28). This suggests that the interaction between RBP-J and the ANK repeats of RAMIC is important for transactivation, but it has not been determined which region of RBP-J is involved in this interaction.
A mutational analysis of RBP-J is useful to understand the mode of interaction between RBP-J and RAMIC in transactivation. However, it is difficult to assess transcriptional activities of exogenously-expressed RBP-J because RBP-J is constitutively expressed in all the cells and tissues that have been examined so far (29,30). To investigate the interaction between RBP-J and RAMIC in transactivation, we adopted an OT11 fibroblastic cell line that carries the homozygous (–/–) mutation of RBP-J and the transgene of H-2Kb-tsA58 (25). Using this RBP-J-deficient cell line, we examined the transcriptional activity of RBP-J mutants. Their ability to bind to DNA, to the two RBP-J-binding domains (ANK and RAM) of Notch, and to several co-repressor molecules were also analyzed. Finally, we examined the transactivation activity of chimeric proteins between RBP-J and its homolog RBP-L, which can bind to the same recognition sequence as RBP-J, but cannot interact with either the RAM domain or the ANK repeats (31,32). These results indicate that the interactions between the N- and C-terminal regions of RBP-J and the ANK repeats of RAMIC are important for transactivation of RBP-J by RAMIC, which is consistent with previous reports (25,28).
MATERIALS AND METHODS
Construction of plasmids
Most of the RBP-J mutants used, including deletion mutants, were in the pCDM8 expression vectors as generated by Chung et al. (33). Double mutants were combined from the mutants in pCDM8 and subcloned in pCMX (34). Deletion mutants were generated from pCDM8 RBP-J by the polymerase chain reaction (PCR) and subcloned in pCMX. For the chimera constructs of RBP-J and RBP-L, the fragments of Jn (amino acids 1–201 of RBP-J), Jm (202–372 of RBP-J), Jc (373–526 of RBP-J), Ln (1–200 of RBP-L), Lm (201–373 of RBP-L) and Ln (374–515 of RBP-L) were synthesized by PCR, combined to obtain the chimera constructs and subcloned in pCMX. For the GAL4-chimeras, the yeast GAL4 DNA binding region was derived from pGBT9 (Clontech), fused with the chimeras, and subcloned in pCMX. All RBP-J mutants, including the chimeras, were confirmed, by immunoblotting and immunofluorescent detection, as being expressed in the nuclei of OT11 cells. For GST-CID, the fragment of SMRT (amino acids 649–811) was generated by PCR and subcloned in pGEX4T-1 (Pharmacia). The other GST fusion proteins were prepared as reported previously (23,31,32). For the VP16 activator constructs, VP16 was derived from pCMX-VP16 (28), fused with RAM or ANK repeat fragments synthesized from pEFBOSneo RAMIC myc by PCR, and subcloned in pEFBOSneo.
Cell culture and transient transfections
OT11 cells were maintained in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum, 100 U/ml recombinant mouse interferon γ (Genzyme) and 100 U/ml penicillin (25). Cells confluently plated in 3.5 cm dishes were maintained without interferon γ for 24 h and co-transfected with plasmid DNA using LipofectAMINE (Gibco BRL). Cells were harvested ∼40 h after transfection and luciferase activities in the cell extracts were measured in a Berthold luminometer, LumatLB9501 according to the manufacturer’s instructions (Toyo Ink Corp.). Normalized luciferase activities (luciferase/β-galactosidase ratio) from all the samples carried out in triplicate were compared. To evaluate the maximal transcriptional activities of RBP-J mutants, 1 µg of pEFBOSneo RAMIC myc, 0.5 µg of pGa981-6, 0.25 µg of pCMXlacZ, and increasing amounts (0–128 ng) of pCDM8 RBP-J were introduced in a well of OT11 cells. Different amounts of expression plasmids were supplemented with vector plasmids to make the total amount of DNA in each sample equivalent. Reporter plasmids of pGa981-6 and TK-MH100×4-LUC, and a plasmid for normalization of pCMX lacZ were described previously (28).
In vitro binding assay with GST fusion proteins
Expression of glutathione S-transferase (GST) fusion proteins and in vitro interaction assays were performed as described previously (16,31). Relative radioactivities of in vitro-translated [35S]methionine-labeled proteins were pre-estimated by SDS–PAGE and the same molar quantities of labeled proteins were then reacted with the GST fusion proteins. To evaluate relative affinities of deleted mutants for GST fusion proteins, the results were adjusted to take into account the number of methionines within each protein. The reactions were confirmed to be within a linear range. All kinds of in vitro-translated [35S]methionine-labeled proteins in this study were confirmed to have no interaction with GST proteins.
Electrophoretic mobility shift assay (EMSA)
EMSA was performed as described previously (16,28). In vitro-translated [35S]methionine-labeled proteins of wild-type RBP-J and mutants were quantitated by SDS–PAGE and the same molar quantity of each protein was reacted with the 32P-labeled O54 probes containing the RBP-J binding sites (33). The binding reaction was performed for 30 min at 20°C and then electrophoresed in a 4% polyacrylamide gel at 130 V for 2 h at 4°C. The gel was dried and analyzed using an Imaging Analyzer BAS1500 (Fuji Film).
Immunoblotting
Transfected OT11 cells were scraped into PBS(–), resuspended in 1× sample buffer (6.25 mM Tris–HCl pH 6.8, 3% SDS, 5% 2-mercaptoethanol, 0.05% bromophenol blue and 10% glycerol), boiled for 5 min, subjected to 10% SDS–PAGE and electrophoretically-transferred onto a nitrocellulose membrane. Western blotting was performed as described previously (28) with anti-RBP-J rat monoclonal antibody (T6719) (33).
RESULTS
Characterization of RBP-J mutants in RBP-J-deficient OT11 cells
The endogenous expression of RBP-J has always hampered its mutational analysis in cultured cells. To overcome this problem, we used the fibroblastic cell line, OT11 carrying the homozygous (–/–) mutation of RBP-J and the transgene of H-2Kb-tsA58 (25). First, various RBP-J mutants described in our previous study (33) were transfected into OT11 cells, together with the expression construct of mouse Notch1 RAMIC (Fig. 1) and the pGa981-6 luciferase reporter plasmid that harbors the hexamized RBP-J binding sites (25). Increasing amounts of exogenous RBP-J augmented the transcriptional activity, reached a peak and then, in excess, reduced the activity. This is probably due to the competition of binding to RAMIC between DNA-bound and free RBP-J (7 and data not shown). To compare the transcriptional activity of mutant RBP-J constructs with that of wild-type RBP-J, their maximal activities, achieved with constant amounts of RAMIC and pGa981-6, were evaluated. Thirty-six RBP-J mutants exhibited peak activities ranging from 2 to 170% of that of wild-type RBP-J (Fig. 2A). We arbitrarily categorized the mutants into three groups according to their level of activity relative to wild-type: (i) high, activity >160% (one mutant); (ii) normal, activity 60–160% (22 mutants); (iii) low, activity <60% (13 mutants).
Figure 1.
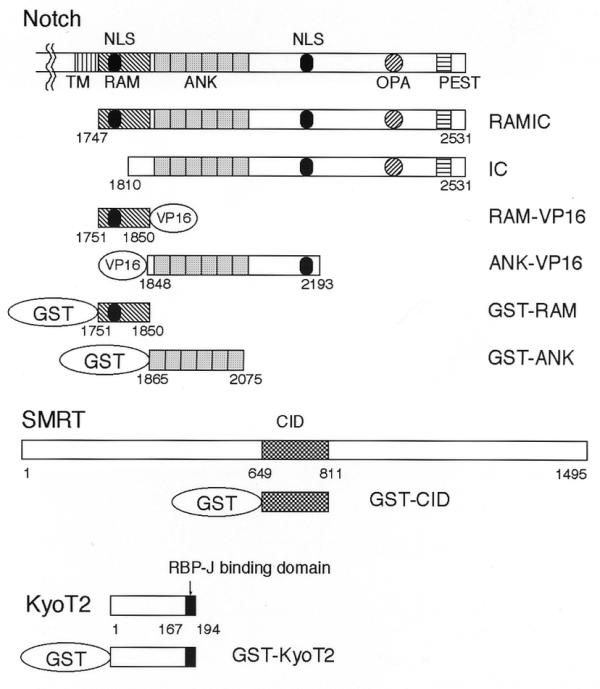
A schematic representation of Notch, SMRT and KyoT2. The GST fusion proteins and the activators used in this study are displayed. Vertically hatched, diagonally hatched, dotted, horizontally hatched, and checkered boxes indicate the transmembrane (TM), RAM, ANK (CDC10/ankyrin repeats) and PEST regions of Notch, and the CID region (CBF1/RBP-J interaction domain) of SMRT, respectively. The diagonally hatched circle and black ellipses indicate the OPA and nuclear localization signals (NLS), respectively. The numbers represent the amino acid positions of their ends.
Figure 2.

(Opposite) Relative transcriptional activities of wild-type RBP-J and mutants by RAMIC and their relative binding activities for interacting molecules. RBP-J mutants are represented by numbers, as follow: 1, InsSpl; 2, InsDra; 3, InsPvu; 4, RK162GS-T153A; 5a, KR185GS; 5b, KR185RS; 6, S194A; 7a, KK196GS; 7b, KK196RS; 8a, KV212GS; 8b, KV212RS; 9, R218H; 10, InsKpn; 11a, RY227GS; 11b, RY227RS; 12, H230G; 13, GA244GS; 14a, EE259GS; 14b, EE259RS; 15, EEF259AAA; 16, InsHind; 17, RL287GS; 18a, RK291GS; 18b, RK291RS; 19, KQ295GS; 20, DD303GS; 21, FY314GS; 22, InsRsa971; 23, ER329GS; 24a, KE341GS; 24b, KE341RS; 25, InsApa; 26, InsRsa1265. The designations 5a+6, 5a+7a and 6+7a indicate the double mutants of KR185GS+S194A, KR185GS+KK196GS and S194A+KK196GS, respectively. (A) The peak transcriptional activities of RBP-J mutants relative to wild-type RBP-J. RBP-J mutants with high (>160%), normal (60–160%) and low (<60%) transcriptional activities are shown as open, shaded and closed bars, respectively. (B) EMSA of mutants with low transcriptional activity in addition to KV212GS (mutant 8a) and FY314GS (mutant 21) are shown. The same molar quantities of proteins were reacted with the 32P-labeled O54 probes containing the RBP-J binding sites. Designations of type 2 mutants are explained in the text. Since 8a and 21 are not type 1 mutants, they are parenthesized. The asterisk represents non-specific bands. (C) The GST pull-down assay of EE259RS (14b), a mutant with high transcriptional activity, for GST, GST–RAM, GST–CID and GST–KyoT2 are displayed in comparison with wild-type RBP-J, EEF259AAA (15), and InsHind (mutant 16, a negative control for the RAM-binding activity). (D) The GST pull-down assay of the mutants as in (C) for GST–RAM, GST–ANK, GST–CID and GST–KyoT2 are shown, including 10% input proteins. The remaining mutants are not shown in the GST pull-down assays or EMSAs because they revealed activity very similar to wild-type RBP-J. (E) Schematic summation of the results in (A), (C) and (D). (a) The positions of mutations aligned along RBP-J with reduced transcriptional activity mutants as shown in (A). Closed ovals represent mutants with decreased (<60%) transcriptional activities. Types 1 and 2 are explained in the text. (b) DNA-binding activities of RBP-J mutants analyzed in (C) are shown. Closed, shaded and open ovals show significantly decreased (<20% of wild-type RBP-J), slightly decreased (20–50%) and normal affinities, respectively. (F) Expression levels of mutants with low transcriptional activity in OT11 cells are shown. An aliquot of 1 µg of pCDM8 RBP-J and mutants was transfected in a 3.5 cm dish of OT11 cells and one-tenth of lysate was applied, blotted and detected by anti-RBP-J antibody (T6719). Detected results were adjusted by background amounts and shown as relative amounts to wild-type RBP-J (with wild-type set at 100). Although anti-RBP-J antibody displays lower affinities for mutants 10 and 18a (33 and unpublished data), significant levels of all the type 2 mutants are expressed. All experiments were confirmed in triplicate.
To understand the molecular basis for the fluctuation in transcriptional activities of the mutants, we evaluated their binding to DNA and transcriptional cofactors. The DNA-binding activity of the mutant RBP-J in COS7 cells had previously been analyzed by EMSA (16) and an essentially similar result was obtained by using in vitro translation products (Fig. 2B). We also wanted to determine whether the binding activities to interacting molecules of RBP-J were affected by the mutations using in vitro-translated RBP-J mutants and the GST fusion proteins GST–RAM (the RAM domain of mouse Notch1), GST–CID (the CBF1/RBP-J interaction domain of SMRT), GST–KyoT2 (the RBP-J binding domain of KyoT2), and GST–ANK (the ANK repeats of mouse Notch1) (Figs 1 and 2C and D). This was because altered affinity for the activator (RAMIC) and/or co-repressors (SMRT or KyoT2) could be the cause of the differing transcriptional activity of RBP-J mutants.
EE259RS (mutant 14b), with an increased transcriptional activity (170%) displayed normal binding activity with DNA (16), GST–RAM and GST–KyoT2 but decreased binding activity with GST–CID (Fig. 2C). This suggests that SMRT could be implicated in repression of RBP-J in OT11 cells and the increased transcriptional activity of EE259RS may result from its decreased binding to SMRT. However, EEF259AAA (mutant 15), at the same site as EE259RS, displayed normal transcriptional activity, despite its decreased binding activity with GST–CID (Fig. 2C). This is probably because of the combined effects of its reduced binding to GST–RAM.
Two types of RBP-J mutants with reduced transactivation activity
We classified RBP-J mutants with decreased transcriptional activity into two subgroups: (i) mutants with decreased DNA-binding activity (type 1), or (ii) with normal DNA-binding activity (type 2). While all the type 1 mutations are found in the middle region of RBP-J (Jm, amino acids 202–370), all the type 2 mutations are located either in the N-terminal region (Jn, amino acids 1–201), or in the C-terminal region (Jc, amino acids 371–526) (Fig. 2E). Transcriptional activities of the type 2 mutants in the N-terminal region (5a, KR185GS; 6, S194A; and 7a, KK196GS) are lower than that of wild-type, but higher than those of type 1 mutants (Fig. 2A). To confirm their decreased transcriptional activity, we generated three constructs bearing double mutations. All double mutants (5a+6, KR185GS+S194A; 5a+7a, KR185GS+KK196GS; 6+7a, S194A+KK196GS) displayed a lower transcriptional activity (<40%), comparable with that of the other type 1 mutants, suggesting that the N-terminal region of RBP-J is important for transactivation by Notch1 RAMIC.
In most cases, decreased DNA-binding activity of the type 1 mutants correlates well with decreased binding activities to GST–RAM, GST–CID and GST–KyoT2, except that R218H (mutant 9) and RY227GS (mutant 11a) showed normal binding activity to the RAM domain (Fig. 2C and D). It is reasonable that the transcriptional activities of the type 1 mutants are low because of their decreased DNA-binding activity. In contrast, the type 2 mutants showed normal binding activities to DNA and the interacting molecules (Fig. 2 and Table 1), which leaves their reduced transcriptional activities unexplained.
Table 1. The interaction of type 2 RBP-J mutants with the ANK repeats of IC.
| DNA | GST pull-down assays | Transcriptional activity | |||||||
| |
|
binding |
ANK |
RAM |
CID |
KyoT2 |
RAMIC |
IC |
ANK–VP16 |
| wild-type RBP-J | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | |
| 5a | KR185GS | 104 | 109 | 114 | 92 | 108 | 71 | 56 | 83 |
| 6 | S194A | 92 | 92 | 103 | 90 | 102 | 41 | 39 | 47 |
| 7a | KK196GS | 102 | 135 | 101 | 90 | 99 | 49 | 53 | 67 |
| 5a+6 | KR185GS+S194A | 92 | 146 | 110 | 95 | 107 | 18 | 27 | 35 |
| 5a+7a | KR185GS+KK196GS | 106 | 116 | 108 | 81 | 98 | 19 | 38 | 73 |
| 6+7a | S194A+KK196GS | 104 | 95 | 107 | 80 | 97 | 23 | 34 | 55 |
| 26 | InsRsa1265 | 96 | 131 | 95 | 70 | 88 | 11 | 0 | 0 |
Relative transcriptional activities induced by Notch IC and ANK–VP16 (and RAMIC) of RBP-J mutants compared to wild-type RBP-J were evaluated by the same approach as described in Figure 1A (1 µg of pEFBOSneo IC or 0.25 µg of pEFBOSneo ANK–VP16 was reacted as an activator). Relative affinities of RBP-J mutants for GST fusion proteins were also evaluated in the GST pull-down assays as described in Figure 2D. To detect relative DNA-binding properties, the same amounts of in vitro-translated type 2 RBP-J mutants were reacted with the 32P-labeled O54 probes and evaluated by EMSA in a linear range (16). All activities are relative to wild-type RBP-J.
N- and C-terminal regions of RBP-J interact with the ANK repeats of RAMIC
When the binding activities of RBP-J deletion constructs to GST–ANK, GST–CID and GST–KyoT2 were examined with the pull-down assay (Fig. 3A), the strong binding region to the ANK repeats was mapped to amino acids 141–227 of RBP-J, which we designated the SAB (strong ANK-binding) domain, where most of the type 2 mutations were located (Fig. 3B). This SAB domain shows no sequence similarity to known proteins in the database and its function, other than the ANK-binding, has not yet been elucidated. Compared with the full-length form of RBP-J, mutants containing the SAB domain but not the middle region (ΔSph-Pvu, Dra-Kpn and Stop-Kpn) have much higher (>15-fold) affinities for the ANK repeats. These results suggest that the middle region of RBP-J may be inhibitory to the interaction of its N-terminal region with the ANK repeats. Deletion mutant DelRsa971, mainly composed of the C-terminal region, showed a comparable affinity to that of full-length RBP-J, and the RAM binding domain (amino acids 196–372), consisting of the middle region, showed a marginal affinity for the ANK repeats. These results demonstrate that the N- and C-terminal regions of RBP-J independently bind to the ANK repeats with different affinities. In agreement with replacement mutation analyses (Fig. 2D), KyoT2 and CID interacted with the Jm region.
Figure 3.

The affinities of deletion mutants of RBP-J for the ANK repeats of Notch, KyoT2 and SMRT. (A) The GST pull-down assays of deletion mutants of RBP-J for GST–ANK, GST–KyoT2 and GST–CID are demonstrated. Ten percent of input lysate of in vitro-translated [35S]methionine-labeled proteins of wild-type RBP-J and deletion mutants with almost the same molar quantities as each other were also applied. Notice that the shorter mutants, containing fewer methionines, revealed weaker band signals. Contaminated internal-methionine-translated proteins are marked with asterisks. (B) The relative affinities of various regions of wild-type RBP-J for GST–ANK, GST–KyoT2 and GST–CID (SMRT) in the GST pull-down assays are summarized. Full-length RBP-J (1–526), DelDra (67–526), DelKpn (229–526), DelRsa971 (321–526), RAM binding region (196–372), ΔSph-Pvu (1–40 and 141–227), Dra-Kpn (54–227), StopPvu (1–149), StopKpn (1–227), StopRsa971 (1–322) and StopRsa1265 (1–420) were examined. ND, not detected. All experiments were confirmed in triplicate.
To evaluate the in vivo interaction of the type 2 mutants with the ANK repeats of RAMIC, we measured their transcriptional activity induced by either IC or ANK-VP16 (containing ANK repeats and the transactivation domain of VP16) (Table 1). All the type 2 mutants, including doubles, displayed <60% wild-type transcriptional activity in association with IC and similarly-reduced levels in association with ANK-VP16. These in vivo results indicate that the decreased transcriptional activity of the type 2 mutants is due to reduced interaction with the ANK domain. The obvious contradiction with the GST pull-down assay (Fig. 2D) could be explained by the possibility that the in vivo interaction activity of RBP-J with IC or the ANK repeats may depend on interaction with other unidentified proteins and therefore differ from that in vitro.
Interaction between RBP-J and the ANK repeats is sufficient for transactivation
To address the functional significance of the interaction between RBP-J and the ANK repeats, several chimeric constructs of RBP-J and RBP-L were generated and their RAMIC-induced transcriptional activities were examined (Fig. 4). RBP-L is a lung-specific homolog of RBP-J, which binds to the same DNA recognition sequence as RBP-J, but cannot interact with either the RAM domain or the ANK repeats (31,32). To avoid the influence of the DNA-binding ability of each construct on transcriptional activity we also examined the GAL4 fusion construct of each chimera between RBP-J and RBP-L. As shown in Figure 4B, RBP-L shows no transcriptional activity, as reported previously (31). In contrast, the LJL construct, replacing the middle region of RBP-L with the same region of RBP-J, showed a transcriptional activity comparable with that of RBP-J, consistent with the previous report that the interaction between RBP-J and the RAM domain is sufficient for transactivation (24). Intriguingly, the JLJ construct, replacing both the N- and C-terminal regions of RBP-L with the same regions of RBP-J, showed an even stronger transcriptional activity, suggesting that the interaction between RBP-J and the ANK repeats would be sufficient and important for transactivation as well. The JLL construct, containing only the N-terminal region of RBP-J, did not bind to DNA and the LLJ construct, containing only the C-terminal region of RBP-J, failed to activate transcription despite its DNA-binding activity which was comparable to that of JLJ. Thus, the N-terminal region of RBP-J appears to play a crucial role in transactivation and the failure to activate transcription by the LLJ construct may be due to the absence of the SAB region that strongly binds to the ANK repeats (Fig. 3B).
Figure 4.
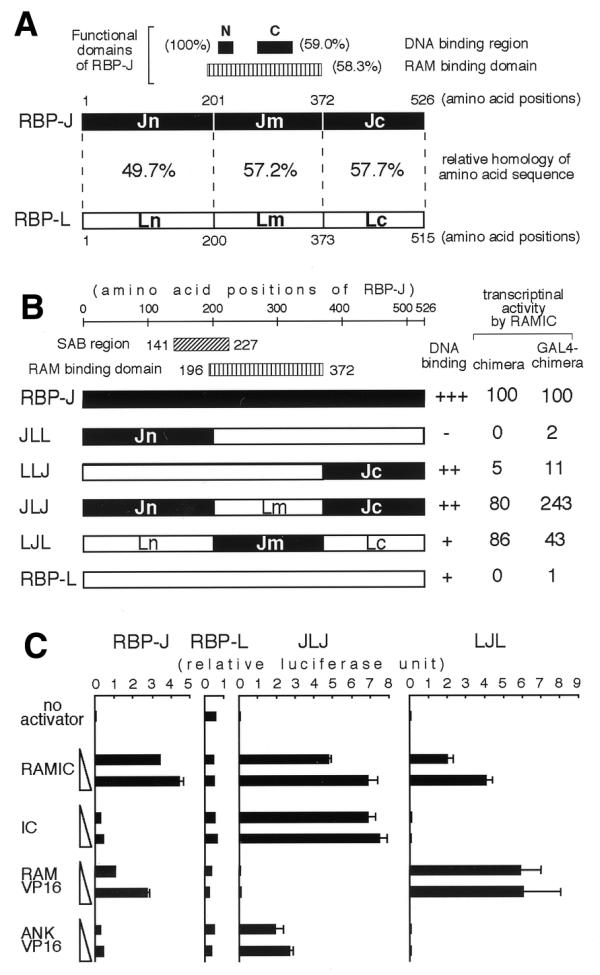
(A) A schematic comparison of amino acid sequences of RBP-J and RBP-L. Closed boxes represent DNA binding regions of RBP-J with the designations N (amino acids 212–227) and C (amino acids 275–323) (16,31). A vertically hatched box represents the RAM binding domain (amino acids 196–372) of RBP-J (16). The regions Jn, Jm and Jc are explained in the text: Ln, Lm and Lc are the corresponding regions in RBP-L. The percentage of each region represents the relative amino acid sequence homology between RBP-J and RBP-L. (B) Transcriptional activities of chimeras of RBP-J and RBP-L. Components from RBP-J and RBP-L are displayed as black and white bars, respectively. The numbers of amino acids correspond to their positions on RBP-J. RAMIC-dependent transcriptional activities of chimeras and GAL4-chimera fusions were evaluated by the same approach as shown in Figure 1 and are represented as relative to RBP-J and GAL4–RBP-J, respectively. (C) The transcriptional activities of chimeras by increasing amounts of activators are demonstrated. Aliquots of 10 ng of pCMX RBP-J, RBP-L or chimeras were constantly introduced into a well of OT11 cells with 0.5 µg of pGa981-6, 0.25 µg of pCMXlacZ and 0.25 or 1.0 µg of activators in pEFBOSneo. The scales of relative luciferase units in RBP-J, RBP-L, JLJ and LJL are equal to each other.
To reinforce the contribution of interaction between RBP-J and the ANK repeats in transactivation, we also examined the transactivation activity of the chimeric proteins co-transfected with IC, RAM–VP16 (a fusion protein of the RAM domain with VP16), or ANK–VP16 (Fig. 1). RBP-J, but not RBP-L, could transactivate the promoter by all the activators (Fig. 4C). LJL was activated by RAMIC and RAM–VP16, but not by IC or ANK–VP16, indicating that it does not interact with the ANK repeats but that its interaction with the RAM domain is essential for transactivation. Conversely, JLJ was activated by constructs containing the ANK repeats (RAMIC, IC and ANK–VP16), but not by RAM–VP16. Furthermore, the degrees of transactivation activity induced by RAMIC and IC are similar, although they are different in the case of RBP-J. These results strongly suggest that the interaction between the N- and C-terminal regions of RBP-J and the ANK repeats of RAMIC play an important role in transactivation.
DISCUSSION
It was reported that the Notch ANK repeats would play an important role in transactivation of RBP-J by RAMIC although their interaction with RBP-J is relatively weak (25,28). In this study, we showed that RBP-J interacts with the ANK repeats of Notch1 RAMIC via its N- and C-terminal regions. In addition, we identified the novel SAB domain in the N-terminal region of RBP-J. Mutational analyses of exogenously-expressed RBP-J mutants indicated that the N- and C-terminal regions of RBP-J were important for transactivation by RAMIC in RBP-J-deficient OT11 cells. Finally, we showed that the ANK-binding region of RBP-J conferred transactivation potential on RBP-L, which is intrinsically inactive, in transactivation by Notch1 RAMIC. Taken together, these results indicate that the interaction between RBP-J and the Notch ANK repeats is important for transactivation.
A role for the physical interaction between RBP-J and the ANK–repeats of RAMIC in transactivation was previously analyzed by employing the M1 mutation that disrupts the interaction with RBP-J (25). RAMIC with the M1 mutation fails to activate transcription although it does interact with RBP-J through the RAM domain (28). This suggests that the weak interaction between RBP-J and the ANK repeats of RAMIC would be important for transactivation and that the ANK repeats might be involved in displacing co-repressors from RBP-J (28). The fact that type 2 mutations of RBP-J show similarly-reduced transcriptional activity induced by IC or ANK-VP16 as well as by RAMIC (Fig. 1 and Table 1) seems to support the importance of the interaction between RBP-J and the ANK repeats in transactivation. The interaction of the type 2 mutants with the ANK repeats in RAMIC appears to be impaired in vivo although their in vitro binding activity to the ANK repeat alone is not altered. Obviously, we cannot exclude the possibility that their direct or indirect interaction with the unidentified co-activator molecule(s) might be affected. Recently, we reported the functional interactions between mouse Notch1 RAMIC and co-activator proteins with histone acetyltransferase activity, PCAF and GCN5 (35). The ANK repeats and the C-terminal transactivation domain of RAMIC are both required for interactions with PCAF and GCN5, and these interactions were also decreased by the M1 mutation of the ANK repeats. It is possible that the type 2 mutations affect the formation of a ternary complex of RBP-J, RAMIC and PCAF or GCN5. Further investigations are required to clarify this possibility.
Full-length RBP-J interacts with the ANK repeats much less strongly than its N-terminal region containing the SAB domain (Fig. 3). The fact that some mutations (8a, 18a, 21 and 22) in the middle region (amino acids 196–372) of RBP-J increased the binding activity to the ANK repeats (Fig. 2D) suggests that this region may have an inhibitory function on the interaction between the N-terminal region of RBP-J and the Notch ANK repeats. Two mutants in the Jm region, 8a (KV212GS) and 21 (FY314GS) retained normal transcriptional activity despite their decreased binding activities to DNA and GST–RAM. It is conceivable that their apparently normal transcriptional activities result from increased binding activity to the ANK repeats (Fig. 2). In fact, the JLJ construct, without the RAM-binding region (Jm), displayed transactivation activity with IC at the same level as with RAMIC (Fig. 3B), indicating that the absence of Jm augments interaction of the N- and C-terminal regions of RBP-J with the ANK repeats in IC. Although the molecular mechanism by which the interaction between the N-terminal region and the ANK repeats is weakened is currently unknown, the inhibitory regulation could be important for RAMIC-induced transactivation. Since the JLL construct, containing only the N-terminal region of RBP-J, lost DNA-binding activity, we constructed a fusion protein between JLL and the yeast GAL4 DNA binding domain (GAL4–JLL) and examined the transcriptional activity induced by RAMIC. However, it showed no transcriptional activity of the GAL4-dependent luciferase construct (Fig. 4B), suggesting that the N-terminal region alone is not sufficient for transactivation activity. The C-terminal region of RBP-J also interacts with the ANK repeats, but its affinity for the ANK repeats is lower than that of the SAB domain and comparable to that of full-length RBP-J. The C-terminal region alone did not confer RAMIC-induced transactivation potential on RBP-L, despite the fact that the chimeric protein (LLJ) could bind to DNA. These results indicate that both the N- and C-terminal regions of RBP-J are required for transactivation by RAMIC and suggest that the two regions may interact with the ANK repeats in a cooperative manner in vivo.
Acknowledgments
ACKNOWLEDGEMENTS
We gratefully acknowledge Ronald M.Evans for providing pCMX-SMRT. We thank R.Yamasaki and K.Saito for preparation of the manuscript. This work was supported by grants from the Ministry of Education, Science, Sports and Culture of Japan.
References
- 1.Honjo T. (1996) The shortest path from the surface to the nucleus: RBP–Jk/Su(H) transcription. Genes Cells, 1, 1–9. [DOI] [PubMed] [Google Scholar]
- 2.Tamura K., Taniguchi,Y., Minoguchi,S., Sakai,T., Tun,T., Furukawa,T. and Honjo,T. (1995) Physical interaction between a novel domain of the receptor Notch and the transcription factor RBP-Jκ/Su(H). Curr. Biol., 5, 1416–1423. [DOI] [PubMed] [Google Scholar]
- 3.Furukawa T., Maruyama,S., Kawaichi,M. and Honjo,T. (1992) The Drosophila homolog of the immunoglobulin recombination signal-binding protein regulates peripheral nervous system development. Cell, 69, 1191–1197. [DOI] [PubMed] [Google Scholar]
- 4.Schweisguth F. and Posakony,J.W. (1992) Suppressor of Hairless, the Drosophila homolog of the mouse recombination signal-binding protein gene, controls sensory organ cell fates. Cell, 69, 1199–1212. [DOI] [PubMed] [Google Scholar]
- 5.Fortini M.E. and Artavanis-Tsakonas,S. (1994) The suppressor of hairless protein participates in notch receptor signaling. Cell, 79, 273–282. [DOI] [PubMed] [Google Scholar]
- 6.Nye J.S., Kopan,R. and Axel,R. (1994) An activated Notch suppresses neurogenesis and myogenesis but not gliogenesis in mammalian cells. Development, 120, 2421–2430. [DOI] [PubMed] [Google Scholar]
- 7.Kuroda K., Tani,S., Tamura,K., Minoguchi,S., Kurooka,H. and Honjo,T. (1999) δ-induced Notch signaling mediated by RBP-J inhibits MyoD expression and myogenesis. J. Biol. Chem., 274, 7238–7244. [DOI] [PubMed] [Google Scholar]
- 8.Apelqvist A., Li,H., Sommer,L., Beatus,P., Anderson,D.J., Honjo,T., Hrabe de Angelis,M., Lendahl,U. and Edlund,H. (1999) Notch signalling controls pancreatic cell differentiation. Nature, 400, 877–881. [DOI] [PubMed] [Google Scholar]
- 9.Robey E., Chang,D., Itano,A., Cado,D., Alexander,H., Lans,D., Weinmaster,G. and Salmon,P. (1996) An activated form of Notch influences the choice between CD4 and CD8 T cell lineages. Cell, 87, 483–492. [DOI] [PubMed] [Google Scholar]
- 10.Oka C., Nakano,T., Wakeham,A., de la Pompa,J.L., Mori,C., Sakai,T., Okazaki,S., Kawaichi,M., Shiota,K., Mak,T.W. and Honjo,T. (1995) Disruption of the mouse RBP-Jκ gene results in early embryonic death. Development, 121, 3291–3301. [DOI] [PubMed] [Google Scholar]
- 11.Jarriault S., Brou,C., Logeat,F., Schroeter,E.H., Kopan,R. and Israel,A. (1995) Signalling downstream of activated mammalian Notch. Nature, 377, 355–358. [DOI] [PubMed] [Google Scholar]
- 12.Struhl G. and Adachi,A. (1998) Nuclear access and action of notch in vivo. Cell, 93, 649–660. [DOI] [PubMed] [Google Scholar]
- 13.Schroeter E.H., Kisslinger,J.A. and Kopan,R. (1998) Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature, 393, 382–386. [DOI] [PubMed] [Google Scholar]
- 14.Zimber-Strobl U., Strobl,L.J., Meitinger,C., Hinrichs,R., Sakai,T., Furukawa,T., Honjo,T. and Bornkamm,G.W. (1994) Epstein–Barr virus nuclear antigen 2 exerts its transactivating function through interaction with recombination signal binding protein RBP-Jκ, the homologue of Drosophila Suppressor of Hairless. EMBO J., 13, 4973–4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henkel T., Ling,P.D., Hayward,S.D. and Peterson,M.G. (1994) Mediation of Epstein–Barr virus EBNA2 transactivation by recombination signal-binding protein Jκ. Science, 265, 92–95. [DOI] [PubMed] [Google Scholar]
- 16.Sakai T., Taniguchi,Y., Tamura,K., Minoguchi,S., Fukuhara,T., Strobl,L.J., Zimber-Strobl,U., Bornkamm,G.W. and Honjo,T. (1998) Functional replacement of the intracellular region of the Notch1 receptor by Epstein–Barr virus nuclear antigen 2. J. Virol., 72, 6034–6039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dou S., Zeng,X., Cortes,P., Erdjument-Bromage,H., Tempst,P., Honjo,T. and Vales,L.D. (1994) The recombination signal sequence-binding protein RBP-2N functions as a transcriptional repressor. Mol. Cell. Biol., 14, 3310–3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsieh J.J. and Hayward,S.D. (1995) Masking of the CBF1/RBPJκ transcriptional repression domain by Epstein–Barr virus EBNA2. Science, 268, 560–563. [DOI] [PubMed] [Google Scholar]
- 19.Olave I., Reinberg,D. and Vales,L.D. (1998) The mammalian transcriptional repressor RBP (CBF1) targets TFIID and TFIIA to prevent activated transcription. Genes Dev., 12, 1621–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Waltzer L., Bourillot,P.Y., Sergeant,A. and Manet,E. (1995) RBP-Jκ repression activity is mediated by a co-repressor and antagonized by the Epstein–Barr virus transcription factor EBNA2. Nucleic Acids Res., 23, 4939–4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kao H.Y., Ordentlich,P., Koyano-Nakagawa,N., Tang,Z., Downes,M., Kintner,C.R., Evans,R.M. and Kadesch,T. (1998) A histone deacetylase corepressor complex regulates the Notch signal transduction pathway. Genes Dev., 12, 2269–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsieh J.J., Zhou,S., Chen,L., Young,D.B. and Hayward,S.D. (1999) CIR, a corepressor linking the DNA binding factor CBF1 to the histone deacetylase complex. Proc. Natl Acad. Sci. USA, 96, 23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taniguchi Y., Furukawa,T., Tun,T., Han,H. and Honjo,T. (1998) LIM protein KyoT2 negatively regulates transcription by association with the RBP-J DNA-binding protein. Mol. Cell. Biol., 18, 644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsieh J.J., Henkel,T., Salmon,P., Robey,E., Peterson,M.G. and Hayward,S.D. (1996) Truncated mammalian Notch1 activates CBF1/RBPJk-repressed genes by a mechanism resembling that of Epstein–Barr virus EBNA2. Mol. Cell. Biol., 16, 952–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kato H., Taniguchi,Y., Kurooka,H., Minoguchi,S., Sakai,T., Nomura-Okazaki,S., Tamura,K. and Honjo,T. (1997) Involvement of RBP-J in biological functions of mouse Notch1 and its derivative. Development, 124, 4133–4141. [DOI] [PubMed] [Google Scholar]
- 26.Aster J.C., Robertson,E.S., Hasserjian,R.P., Turner,J.R., Kieff,E. and Sklar,J. (1997) Oncogenic forms of NOTCH1 lacking either the primary binding site for RBP-Jκ or nuclear localization sequences retain the ability to associate with RBP-Jκ and activate transcription. J. Biol. Chem., 272, 11336–11343. [DOI] [PubMed] [Google Scholar]
- 27.Kopan R., Nye,J.S. and Weintraub,H. (1994) The intracellular domain of mouse Notch: a constitutively activated repressor of myogenesis directed at the basic helix–loop–helix region of MyoD. Development, 120, 2385–2396. [DOI] [PubMed] [Google Scholar]
- 28.Kurooka H., Kuroda,K. and Honjo,T. (1998) Roles of the ankyrin repeats and C-terminal region of the mouse notch1 intracellular region. Nucleic Acids Res., 26, 5448–5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hamaguchi Y., Yamamoto,Y., Iwanari,H., Maruyama,S., Furukawa,T., Matsunami,N. and Honjo,T. (1992) Biochemical and immunological characterization of the DNA binding protein (RBP-Jκ) to mouse Jκ recombination signal sequence. J. Biochem., 112, 314–320. [DOI] [PubMed] [Google Scholar]
- 30.Ling P.D., Hsieh,J.J., Ruf,I.K., Rawlins,D.R. and Hayward,S.D. (1994) EBNA-2 upregulation of Epstein–Barr virus latency promoters and the cellular CD23 promoter utilizes a common targeting intermediate, CBF1. J. Virol., 68, 5375–5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Minoguchi S., Taniguchi,Y., Kato,H., Okazaki,T., Strobl,L.J., Zimber-Strobl,U., Bornkamm,G.W. and Honjo,T. (1997) RBP-L, a transcription factor related to RBP-Jκ. Mol. Cell. Biol., 17, 2679–2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Minoguchi S., Ikeda,T., Itohara,S., Kaneko,T., Okaichi,H. and Honjo,T. (1999) Studies on the cell-type specific expression of RBP-L, a RBP-J family member, by replacement insertion of β-galactosidase. J. Biochem., 126, 738–747. [DOI] [PubMed] [Google Scholar]
- 33.Chung C.N., Hamaguchi,Y., Honjo,T. and Kawaichi,M. (1994) Site-directed mutagenesis study on DNA binding regions of the mouse homologue of Suppressor of Hairless, RBP-Jκ. Nucleic Acids Res., 22, 2938–2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Umesono K., Murakami,K.K., Thompson,C.C. and Evans,R.M. (1991) Direct repeats as selective response elements for the thyroid hormone, retinoic acid, and vitamin D3 receptors. Cell, 65, 1255–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kurooka H. and Honjo,T. (2000) Functional interaction between the mouse Notch1 intracellular region and histone acetyltransferases PCAF and GCN5. J. Biol. Chem., 275, 17211–17220. [DOI] [PubMed] [Google Scholar]