

Abstract
The Ca2+ dependant interaction between troponin I (cTnI) and troponin C (cTnC) triggers contraction in heart muscle. Heart failure is characterized by a decrease in cardiac output, and compounds that increase the sensitivity of cardiac muscle to Ca2+ have therapeutic potential. The Ca2+-sensitizer, levosimendan, targets cTnC; however, detailed understanding of its mechanism has been obscured by its instability. In order to understand how this class of positive inotropes function, we investigated the mode of action of two fluorine containing novel analogues of levosimendan; 2’ ,4’-difluoro(1,1’-biphenyl)-4-yloxy acetic acid (dfbp-o) and 2’ ,4’-difluoro(1,1’-biphenyl)-4-yl acetic acid (dfbp). The affinities of dfbp and dfbp-o for the regulatory domain of cTnC were measured in the absence and presence of cTnI by NMR spectroscopy, and dfbp-o was found to bind more strongly than dfbp. Dfbp-o also increased the affinity of cTnI for cTnC. Dfbp-o increased the Ca2+-sensitivity of demembranated cardiac trabeculae in a manner similar to levosimendan. The high resolution NMR solution structure of the cTnC-cTnI-dfbp-o ternary complex showed that dfbp-o bound at the hydrophobic interface formed by cTnC and cTnI making critical interactions with residues such as Arg147 of cTnI. In the absence of cTnI, docking localized dfbp-o to the same position in the hydrophobic groove of cTnC. The structural and functional data reveal that the levosimendan class of Ca2+-sensitizers work by binding to the regulatory domain of cTnC and stabilizing the pivotal cTnC-cTnI regulatory unit via a network of hydrophobic and electrostatic interactions, in contrast to the destabilizing effects of antagonists such as W7 at the same interface.
Keywords: Levosimendan, troponin C, troponin I, Ca2+-sensitizer, NMR spectroscopy
Introduction
Cardiac muscle contraction is regulated by a Ca2+-triggered cascade of thin and thick filament protein-protein interactions. The thin filament is comprised of three molecular units: troponin, tropomyosin, and f-actin. Troponin is a heterotrimeric protein complex consisting of troponin C (cTnC), a Ca2+-binding protein with two EF-hand motifs in each terminal domain, troponin I (cTnI), the subunit which inhibits contraction via its interaction with f-actin, and troponin T (cTnT), a scaffolding protein that tethers troponin to the thin filament through association with cTnI and tropomyosin. The C-terminal, or “structural”, domain of cTnC (cCTnC) is bound to two divalent cations (Mg2+ or Ca2+) throughout the contraction-relaxation cycle of muscle contraction. Its primary function is to keep cTnC mounted on the thin filament via its tight association with the “anchoring” region of cTnI (cTnI34–71). Like cCTnC, the regulatory domain of cTnC (cNTnC) contains two EF-hand motifs; however, Ca2+-binding site I is defunct, leaving only site II available for Ca2+ association. During systole, Ca2+ enters the cytosol and binds to the N-domain of cTnC (cNTnC•Ca2+), which prompts a slight opening of cNTnC and the resultant interaction with the “switch” region of cTnI (cTnI147–163). The association of cTnI147–163 with cNTnC•Ca2+ is coupled with the separation of the inhibitory (cTnI128–147) and C-terminal (cTnI163–181) regions of cTnI from f-actin. Once cTnI dissociates from f-actin, tropomyosin changes its orientation to reveal the myosin binding surface on f-actin, the actin-myosin cross-bridge can subsequently form, and the ATPase dependant contraction occurs (for reviews on the regulation of contraction by troponin see [1–3]).
Heart failure is a condition that is characterized by compromised blood supply through the body. There are numerous drug therapies; however, many tend to have undesirable side effects. For example, therapeutic strategies which involve the increase of Ca2+ levels within the cardiomyocyte may result in arrhythmia, tachycardia, and mortality. In response to the negative long-term performance of these drugs, another treatment scheme being explored is to increase the Ca2+-sensitivity of the thin filament, and thus improve contractility without modulating Ca2+ concentration. This class of pharmaceuticals is referred to as Ca2+-sensitizers [4–6]. Levosimendan is a positive inotrope that binds cTnC to elicit its Ca2+-sensitizing function [7–9]. One current theory is that levosimendan functions by stabilizing the open conformation of cNTnC•Ca2+, so that it is in a favorable conformation to bind cTnI147–163 [10]. Thus, levosimendan would increase the affinity of cTnI147–163 for cNTnC•Ca2+ and as a result, the Ca2+-sensitivity of the thin filament. Although the structure of levosimendan bound to cTnC has been modeled [11], there have not been any high resolution structures of levosimendan bound to cTnC or cTnC-cTnI solved. The unstable nature of levosimendan [12] is the major barrier preventing the determination of these structures and consequently is thwarting a complete understanding of its molecular mechanism.
In order to investigate the Ca2+-sensitizing mechanism of levosimendan, we have elected to study the fluorine containing analogues, 2’ ,4’-difluoro(1,1’-biphenyl)-4-yl acetic acid (dfbp) and 2’ ,4’-difluorobiphenyl-4-yloxy acetic acid (dfbp-o). We conducted contractility measurements with demembranated trabeculae from rat ventricles and saw an increase in the Ca2+-sensitivity of the cardiac muscle in the presence of dfbp-o. Thus, the chemical structure of dfbp-o constitutes a potential scaffold for the development of novel Ca2+-sensitizers. Titration experiments done in vitro suggest that this enhanced contractility is caused by an increase in the affinity of cTnI147–163 for cNTnC•Ca2+ by dfbp-o. In addition to determining the functional effects of dfbp-o, we investigated the structure of dfbp-o bound to cNTnC•Ca2+ and cNTnC•Ca2+•cTnI144–163. The structures highlight important pharmacophores of Ca2+-sensitizing molecules and provide insight into the molecular mechanism of Ca2+-sensitizers that target troponin.
Material and Methods
Sample preparation
Recombinant human cNTnC (residues 1–89) was used in this study. The engineering of the expression vector and the expression of 15N- and 13C,15N-labeled proteins in E. coli were as described previously [13]. The two cTnI peptides, cTnI147–163, acetyl-RISADAMMQALLGARAK-amide, and cTnI144–163, acetyl-RRVRISADAMMQALLGARAK-amide, were synthesized by GL Biochem Ltd. (Shanghai, China). Peptide quality was verified by HPLC and ESI-Mass Spectrometry. Dfbp-o was purchased from Sigma-Aldrich and dfbp was purchased from Amatek Co. and purities and chemical structures were verified by NMR spectroscopy, ESI-Mass Spectrometry, MS/MS, and Infrared Spectroscopy. Stock solutions of the compounds in DMSO-d6 (Cambridge Isotopes Inc.) were prepared and the vials containing the solutions were wrapped in aluminum foil to protect the molecules from light catalyzed degradation. All NMR samples prepared in 5 mm NMR tubes had a volume of 500 μL. The protein samples were solubilized in 100 mM KCl, 10 mM imidazole, and 0.2–0.25 mM 2,2-dimethyl-2-silapentane-5-sulfonate sodium salt (DSS) (Chenomix) in 95% H2O/5% D2O, 5–10 mM CaCl2 (Fluka) and the pH was maintained at ~7.0. For the paramagnetic studies, 3 mm NMR tubes were used with a final volume of 200 μL, and the NMR buffer conditions were 100 mM KCl, 10 mM imidazole, and 0.2–0.25 mM DSS, 5–10 mM CaCl2, 0.5 mM trifluoroacetic acid (TFA) in 99.9% D2O and the pD was maintained at ~7.3.
Ab initio calculation details
The minimum energy conformations and electronic properties were calculated with Gaussian03[14]. The parameters implemented for the calculations on dfbp, dfbp-o, and levosimendan were identical. Geometry optimization was performed using Becke’s three-parameter Lee, Yang, Parr (B3LYP) hybrid functional with a split-valence basis set and polarization d-orbitals added (B3LYP/6-31G(d). The optimized geometries were subsequently used to calculate the total density and electrostatic potential (ESP), using the B3LYP/6-311+G(d, p) basis set in aqueous solvent. Final contour surfaces were represented using GaussView 3.0.
Computational Docking
Autodock 4.0[15] was used to localize the binding site of dfbp-o on cNTnC. Autodock uses a genetic search algorithm as a global optimizer and energy minimization as a local search method. The target protein was kept rigid, and dfbp-o was given torsional flexibility around the ether bond. The spacing of 0.375 Å was used to generate the affinity grid maps. The Lamarckian genetic algorithm (LGA) and pseudo-Solis and Wets method were used for minimization. Default parameters were used for docking, unless otherwise specified. For the binding-site-centered docking, a grid map of 78 × 66 × 54 centered at 57.192, 2.138, and 0.939 was generated with each LGA job that consisted of 200 runs with 270000 generations in each run and maximum number of energy evaluations of 5.0 × 106. The resulting docked orientations with an rmsd of 2.0 Å were clustered together. AutoDockTools (http://www.scripps.edu/~sanner) was used to generate all of the necessary files and to analyze the results. The structure of dfbp-o used for the docking was generated by Gaussian03. Partial atomic charges were assigned automatically in AutoDockTools using the Gasteiger-Marsili method[16]. The protein coordinates used in the calculation were taken from the X-ray structure of cTnC•3bepridil (PDB 1DTL)[17]. The coordinates for the three bepridil molecules, the C-domain of cTnC (92–160), and the two Ca2+ bound to the C-domain were removed prior to docking. Kollman united-atom partial charges and solvation parameters were added to the protein file using AutoDockTools.
NMR spectroscopy
Titration Analysis
All NMR experiments were run on either a Varian Inova 500 MHz spectrometer or a Unity 600 MHz spectrometer. All data were collected at 30 °C. Both spectrometers are equipped with a triple resonance 1H13C15N probe and z-pulsed field gradients. Protein concentration was determined by amino acid analysis and by 1D 1H,15N-HSQC NMR spectroscopy. Stock solutions of dfbp, dfbp-o, cTnI147–163, and cTnI144–163 were prepared in DMSO-d6 and concentrations were calibrated by comparing their 1D 1H NMR spectra with that of a DSS standard. In contrast to levosimendan[12], there was no noticeable degradation of dfbp-o and dfbp in the presence of DTT. The binding of the various ligands to cNTnC and to cNTnC in complex with a ligand was monitored by 2D-1H,15N-HSQC NMR spectroscopy. At each point in a titration, a 2D-1H, 15N-HSQC spectrum was acquired, and chemical shift changes were used to calculate the dissociation constant.
We titrated 0.17 mM 15N-cNTnC with 0.11, 0.23, 0.45, 0.67, 1.22, 1.75, 2.28, 2.79, and 3.30 mM of dfbp. We titrated 0.08 mM 15N-cNTnC in ~2× excess cTnI144–163 with 0.23, 0.45, 0.67, 0.89, 1.43, 1.96, 2.49, 3.00, and 3.50 mM of dfbp. We titrated 0.24 mM 15N-cNTnC with 0.10, 0.30, 0.69, 1.27, and 1.56 mM of dfbp-o. We titrated 0.34 mM 15N-cNTnC in ~2× excess cTnI144–163 with 0.05, 0.14, 0.28, 0.46, 0.64, 1.01, and 1.55 mM of dfbp-o. We titrated 0.61 mM 15N-cNTnC in ~4× excess cTnI147–163 with 0.09, 0.19, 0.28, 0.37, 0.46, 0.55, 0.64, and 1.09 mM of dfbp-o. We used a larger excess of cTnI147–163 than cTnI144–163 because cTnI147–163 has a lower affinity for cNTnC, and we wanted to obtain saturation of cNTnC. We titrated 0.29 mM 15N-cNTnC with 0.02, 0.03, 0.06, 0.12, 0.27, 0.44, 0.63, and 0.74 mM of cTnI144–163. We titrated 0.3 mM 15N-cNTnC in ~4× excess dfbp-o with 0.03, 0.06, 0.18, 0.28, 0.37, 0.53, and 0.60 mM of cTnI144–163. We titrated 0.11 mM 15N-cNTnC in ~4× excess dfbp-o with 0.01, 0.02, 0.03, 0.05, 0.07, 0.09, 0.11, 0.14, 0.17, and 0.23 mM of cTnI147–163. We titrated 0.10 mM 15N-cNTnC with 0.01, 0.03, 0.05, 0.08, 0.11, 0.14, 0.17, 0.20, and 0.25 mM of cTnI147–163. The titrations involving cTnI147–163 were done at lower concentrations because it is difficult to get a concentrated stock solution of cTnI147–163. During the titrations with the ligands the pH was maintained by adding small aliquots of NaOH to the samples.
The binding of the dfbp, dfbp-o, cTnI147–163, and cTnI144–163 to the target molecules or complexes were fit with a 1:1 stoichiometry. We corrected the concentrations of 15N-cNTnC, cTnI147–163, cTnI144–163, dfbp, and dfbp-o for sample dilution that occurred during the various titrations. The dissociation constants were calculated by averaging the normalized individual chemical shifts as a function of the ligand to protein ratios and fitting was done using xcrvfit (www.bionmr.ualberta.ca/bds/software/xcrvfit). The amide resonances that were perturbed larger than the mean plus one standard deviation were chosen for the KD calculation. The KD was determined by fitting the data to the equation:
NMR experiments for Assignment and Structure Calculation
The backbone atoms were assigned by the 2D-1H,15N-HSQC, 3D-CBCACONNH and 3D-HNCACB experiments and the side chain resonances were assigned by making use of the 2D-1H,13C-HSQC and 3D-CCONH and 3D-HCCONH experiments. Aromatic resonances were assigned by running a 2D-NOESY of the complex in D2O. The HNHA experiment was run to unambiguously assign the α-protons of most residues. Intramolecular distance restraints for cNTnC were obtained by the 3D-NOESYCHSQC and 3D-NOESYNHSQC experiments. We chose a mixing time of 100 ms for the 3D-NOESYCHSQC experiment and 150 ms for the 3D-NOESYNHSQC experiment. Structural distance restraints for cTnI144–163 and dfbp-o were obtained with the 2D-13C, 15N filtered NOESY (mix = 200 ms) and the 2D-13C,15N filtered TOCSY (mix = 60 ms) experiments [18–20]. Assignments of cTnI144–163 were guided by the previously made assignments for cTnI147–163 in the cNTnC•Ca2+•cTnI147–163•bepridil complex [21]. The two fluorine nuclei of dfbp-o were assigned by running 1D 19F and 2D 1H,19F HOESY – NMR spectroscopy [22, 23]. In order to obtain distance restraints between the unlabeled ligands and labeled protein we employed the 13C-edited, filtered HMQCNOESY experiment [24, 25] with a mixing time of 250 ms. Contacts between methionine methyls were crucial for the structure determination of this complex. They were assigned by the 13C-edited NOESYHSQC and by comparing these assignments with those determined by Krudy et al. for the cTnC-cTnI complex[26] by the employment of site-specific mutagenesis of Met to Leu[27].
We used paramagnetic relaxation enhancement (PRE) to provide restraints between the metal ion bound to cNTnC and dfbp-o. The basic principle behind PRE is that the relaxation rates of nuclei surrounding the paramagnetic metal, such as gadolinium (Gd3+), will be enhanced by the large contribution from the unpaired 4f electrons of the metal. The difference in relaxation rates between the diamagnetic and paramagnetic bound complexes can provide us with distance restraints to dfbp-o. In order to extract distances from the relaxation enhancement by Gd3+ we followed the procedures outlined by Gariépy et al. [28]. Since Gd3+ has seven unpaired 4f electrons, the metal is a very efficient broadening agent and we did not need very much to elicit a strong response to dfbp-o. We prepared a stock solution of 0.3 mM cNTnC•Gd3+•cTnI144–163 and titrated it into an NMR tube containing 0.8 mM dfbp-o acquiring 1D 19F or 1D 1H spectra at each titration point. We repeated the titration with the diamagnetic lanthanide (Ln3+), lanthanum (La3+), in complex with cNTnC and cTnI144–163 to test for any diamagnetic contribution to the relaxation enhancement of dfbp-o. We found that diamagnetic relaxation enhancement was negligible at the low cNTnC•La3+•cTnI144–163 concentrations we were working with. We also added free Gd3+ into dfbp-o to see if excess Gd3+ (unbound to cNTnC•cTnI144–163) would contribute to broadening via specific coordination with the carboxylate moiety of dfbp-o. Dfbp-o signals were broadened in the presence Gd3+; however, only at concentrations greatly exceeding the cNTnC•Ln3+•cTnI144–163 concentration range we were working in. Since free Gd3+ only enhanced relaxation of dfbp-o nuclei at high concentrations and that ~1:1 cNTnC:Gd3+ complexes were prepared in order to limit the amount of free Gd3+, we assumed that all paramagnetic effects were from bound dfbp-o. In order to determine the amount of dfbp-o bound, we took advantage of the situation where the total concentration of dfbp-o ([L]T) greatly exceeds the concentration of the paramagnetic protein ([P]T). The fraction of dfbp-o bound is then determined by the simple equation:
This relationship requires that the total ligand concentration is approximately equal to free ligand. We determined the KD of dfbp-o for cNTnC•Ca2+•cTnI144–163, and assumed that the affinity of dfbp-o for cNTnC•Gd3+•cTnI144–163 was unchanged.
Structure determination by NMR spectroscopy
Generation of the structure file of dfbp-o
The PRODRG web server[29] was used to create a PDB file for dfbp-o and XPLO-2D was used to create a topology file, a parameter file, and an XPLOR-NIH script which was used to generate a structure file. The structure file of dfbp-o was manually altered to include the atomic charges predicted by Gaussian3.0 for water refinement.
Data processing and structure calculation
VNMRJ (Varian Inc.) was used for the analysis of 1D NMR spectra, necessary for the assignment of dfbp-o and for the PRE measurements. All 2D and 3D NMR data were processed with NMRPipe[30]. The assignment of chemical shifts was done with NMRView [31], and sequential assignment was done with the program SmartNoteBook [32]. The ψ and ϕ dihedral angle restraints predicted by TALOS [33] were used for both cNTnC and cTnI144–163. Six distance restrains derived from x-ray crystallographic data of Ca2+-chelating oxygen atoms to Ca2+ were included, as well as, two distance restraints between Asp65 and Gly70 included to hold the Ca2+-binding loop together. In order to help assign the 3D-NOESYCHSQC and 3D-NOESYNHSQC experiments, structures of cNTnC•Ca2+ were generated using the program CYANA[34]. Distance restraints were calibrated in CYANA with an upper limit of 6 Å. Assignments were made manually and kept during the first four CYANA calculation cycles, after which they were open for automatic assignment with the “noeassign” command of CYANA. CYANA was used to calculate 100 structures, of which the 20 conformers with the lowest target function were used to further refine the structure. Following the CYANA refinement, peaklists were read back into NMRView, reviewed manually, and converted into XPLOR-NIH [35] format. The median method was used to calibrate interproton distances in the 3D-NOESYCHSQC and 3D-NOESYNHSQC experiments. The simulated annealing protocol of XPLOR-NIH was used, with 10,000 high temperature steps and 6000 cooling steps. The structure of cNTnC•Ca2+ was initially optimized, followed by the cNTnC•Ca2+•cTnI144–163 structure, and finally the cNTnC•Ca2+•cTnI144–163•dfbp-o complex. Intermolecular proton-proton distance restraints were calculated using loose restraints (1.8–6.0 Å). There were 8 intermolecular NOE restraints between dfbp-o and cNTnC, 3 intramolecular NOEs of dfbp-o, 6 PRE distance restraints between dfbp-o and Gd3+, and 4 intermolecular NOEs between dfbp-o and cTnI144–163. We measured 30 intermolecular NOEs between cTnI144–163 and cNTnC as well as 24 intramolecular NOEs. 200 structures were calculated and the 50 lowest energy structures were refined in water with a water box edge length of 18.8 Å[36]. The final ensemble is represented by the 20 structures after the water refinement with the fewest dihedral violations, ordered by lowest energy prior to water refinement (see Supplementary Table II for statistics).The ensemble was validated by Procheck[37] available with the online Protein Structure Validation Software (PSVS) suite (http://psvs-1_4-dev.nesg.org/). The ensemble has been deposited in the protein data bank (www.rcsb.org) with the accession code of 2L1R.pdb.
Force measurement in ventricular trabeculae
The methods for working with the ventricular trabeculae are the same as in Sun et al. [38]. Wistar rats (200–250 g) were stunned and killed by cervical dislocation (Schedule 1 procedure in accordance with UK Animal (Scientific Procedures) Act 1986). The hearts were removed and rinsed free of blood in Krebs solution containing (mM): NaCl, 118; NaHCO3, 24.8; Na2HPO4, 1.18; MgSO4, 1.18; KCl, 4.75; CaCl2, 2.54; glucose, 10; bubbled with 95% O2–5% CO2; pH 7.4 at 20 °C. Suitable trabeculae (free running, unbranched, diameter < 250 μm) were dissected from the right ventricle in Krebs solution containing 25 mM 2,3-butanedione-monoxime, permeabilised in relaxing solution (see below) containing 1% Triton X-100 for 30 min, and stored in relaxing solution containing 50% (vol/vol) glycerol at −20 °C for experiments, normally within 2 days of dissection. Demembranated trabeculae were mounted, via aluminium T-clips between a force transducer (AE 801) and a fixed hook in a 60 μl glass trough containing relaxing solution. The sarcomere length of the relaxing muscle was set to 2.1–2.2 μm by laser diffraction. The experimental temperature was 20–22 °C. Experimental solutions contained 25 mM imidazole, 5 mM MgATP, 1 mM free Mg2+, 10 mM EGTA (except pre-activating solution), 0–10 mM total calcium, 1 mM dithiothreitol and 0.1% (vol/vol) protease inhibitor cocktail (P8340, Sigma). All solutions also included an ATP backup system consisting of 15 mM phosphocreatine (P7936, Sigma) and 1 mg/ml creatine phosphokinase (C3755, Sigma). Ionic strength was adjusted to 200 mM with potassium propionate; pH was 7.1 at 20 °C. The concentration of free Ca2+ was calculated using the program WinMAXC V2.5 (http://www.stanford.edu/~cpatton/maxc.html). When required, 0.5 mM dfbp-o was added from a 500 mM stock solution in DMSO and the pH was re-adjusted. Each trabecular activation was preceded by a 1-min incubation in pre-activating solution. Isometric force and fluorescence intensities were measured after steady-state force had been established in each activation. The dependence of force on [Ca2+] was fit to data from individual trabeculae using nonlinear least-squares regression to the Hill equation:
where EC50 is the [Ca2+] corresponding to half-maximal change in Y, and nH is the Hill coefficient. All values are shown in Supplementary table I and given as mean ± standard error except where noted, with n representing the number of trabeculae.
Results
Structural Description of the Levosimendan Analogues
The short lifespan of levosimendan in aqueous solution has made it difficult to gain a thorough understanding of its mode of action. We chose to study two compounds (dfbp and dfbp-o) that were originally developed as analogues of the anti-inflammatory drug, flobufen [39]. Although dfbp and dfbp-o are ostensibly structurally unique from levosimendan, we propose that these differences are mostly superficial, and the features that are exclusive to dfbp and dfbp-o may in fact contribute to their attractiveness as new lead compounds. Levosimendan, dfbp, and dfbp-o all contain a central phenyl constituent attached to a substituted ring structure and an electronegative moiety (figure 1). We propose that the difluorophenyl group of dfbp and dfbp-o replaces of the pyridazinone ring of levosimendan and the carboxyl moiety, the malonodinitrile hydrazone group. Since fluorine has a van der Waals radius of 1.44 Å [40] it is isosteric with oxygen [41]. Aromatic fluorine has a large dipole moment, similar to the carbonyl of levosimendan; however, unlike the carbonyl, it is not significantly polarizable. Therefore, although aromatic fluorine can take part in electrostatic interactions it does not take part in hydrogen bonding and consequently increases the overall lipophilicity of a molecule [41, 42]. We propose that the second fluorine at the 2’ position replaces the methyl on the pyridazinone ring of levosimendan. Given that levosimendan is predicted to bind to the hydrophobic pocket of cNTnC [11, 43] and that the substitution of a methyl with a fluorine can increase the metabolic stability of a compound [44], the replacement of the carbonyl and methyl groups with fluorines suggests these compounds may represent a novel chemical platform for the development of more suitable troponin targeting Ca2+-sensitizers. The malonodinitrile group of levosimendan may be replaced by the carboxylate of dfbp and dfbp-o, and the aromatic ether of dfbp-o may serve as a hydrogen bond acceptor in a similar manner as the hydrazone group of levosimendan. In order to investigate the therapeutic prospect of these compounds, we monitored their interaction with the regulatory domain of cTnC in both the absence and presence of cTnI. Following these titrations, we chose the most potent of the ligands to pursue with a structural and functional analysis.
Figure 1.

The chemical structures of (A) dfbp (B) dfbp-o and (C) levosimendan. Distinct chemical units of dfbp-o and levosimendan are labeled.
The Comparison of Dfbp and Dfbp-o
We monitored the binding of dfbp and dfbp-o to 15N-labeled cNTnC ± cTnI144–163 by acquiring 1H,15N-heteronuclear single quantum coherence (HSQC) NMR spectra at each point in the titrations. These data were used to determine the dissociation constants and stoichiometry of the ligands for the cNTnC•Ca2+ and the cNTnC•Ca2+•cTnI144–163 complexes (unless otherwise stated, all cTnC states are Ca2+-saturated and thus the Ca2+ will be omitted). The 1H,15N-HSQC spectra also contribute information about changes in the chemical environment at each 15N-labeled amide nucleus. When a ligand binds to a protein, the amide resonances of residues near the bound ligand will experience a change in chemical shift. A chemical shift change may also be induced by a conformational change in the protein rather than by direct contact with the ligand. In either scenario, the change in chemical shift may be used to characterize protein-ligand interactions. In this study, we calculated dissociation constants using all amide chemical shifts that were perturbed greater than the mean shift change plus one standard deviation. The data from these shifts were than normalized and averaged to calculate a final dissociation constant. We found that dfbp bound to cNTnC with a dissociation constant of 7150 ± 1160 μM, whereas dfbp-o bound to cNTnC with a KD of 820 ± 190μM. Dfbp bound to the•cNTnC cTnI144–163 complex with a KD = 2820 ± 480 μM, and dfbp-o bound to the same complex with a KD = 270 ± 30μM (figure S1). Since dfbp-o binds to cNTnC and cTnC•cTnI144–163 ~10-fold tighter than dfbp we focused the rest of the study on characterizing its function.
The Inotropic Mechanism of Dfbp-o
In vitro analysis by NMR spectroscopy
The regulation of muscle contraction is linked to the opening and closing of cNTnC. The Ca2+-triggered opening of cNTnC leads to a slight increase in the solvent exposure of a core hydrophobic patch. In the Ca2+-saturated state, cNTnC does not fully open as is the case with sTnC [45], but rather its conformational equilibrium is shifted from the closed state towards the open state in the presence of Ca2+ [46, 47]. Following Ca2+-binding, the switch region of cTnI binds to cNTnC and stabilizes the open form of cNTnC [48]. The development of ligands that likewise stabilize the open conformation of cNTnC might strengthen the interaction of cTnI with cNTnC, and thus augment contractility [10]. In order to test whether dfbp-o increased the affinity of cTnI144–163 for cNTnC, we titrated cTnI144–163 into cNTnC in the absence and presence of dfbp-o. The affinity of cTnI144–163 for cNTnC was measured to be 26 ± 4 μM and in the presence of dfbp-o was measured to be 19 ± 10 μM. Since it is difficult to measure dissociation constants of low micromolar ranges by NMR spectroscopy, we chose to investigate an alternative cTnI segment with a lower affinity for cNTnC. The affinity of cTnI147–163 for cNTnC was calculated to be 130 ± 10 μM and 60 ± 10 μM for cNTnC•dfbp-o (figure 2A, B). The dissociation constant of dfbp-o was also enhanced by the presence of cTnI147–163 from 820 ± 190 μM to 380 ± 80 μM (figure 2C, D). The results indicate that dfbp-o and cTnI147–163 bind reciprocally to cNTnC. We fit the data to a ternary complex model (figure 2E), which is defined by a contribution of four equilibrium constants, two for dfbp-o (reactions C and D) and two for cTnI147–163 (reactions A and B). The data meet the thermodynamic constraints of this model (C/D = 2.16 ± 0.95; A/B = 2.17 ± 0.53) and the cNTnC-cTnI interaction appears to be stabilized by dfbp-o.
Figure 2.

(A) Titration of cTnI147–163 into cNTnC. (B) Titration of cTnI147–163 into cNTnC•dfbp-o (4 fold excess of dfbp-o). (C) Titration of dfbp-o into cNTnC. (D) Titration of dfbp-o into cNTnC•cTnI147–163 (2–3 fold excess of cTnI147–163). The same expanded region of the two dimensional 1H,15N- HSQC NMR spectra of cNTnC for each ligand is shown. Spectra taken at the various titration points are superimposed. Arrows indicate the direction of chemical shift perturbation induced by the ligand. V28 is present in the spectrum (D) but not narrow enough to be seen in the other spectra. (E) The thermodynamic cycle of dfbp-o and cTnI147–163 binding to cNTnC.
In situ analysis by cardiac trabeculae contractility
The Ca2+-sensitizing properties of dfbp-o were monitored using demembranated cardiac trabeculae from rat ventricles. Similar to previous results found for levosimendan [9, 49, 50], dfbp-o did not increase the maximal force of the trabeculae; however, at sub-maximal Ca2+ concentrations (1.0–1.5 μM) the force of contraction was significantly increased (figure 3). The dependence of force on free [Ca2+] was fitted by the Hill equation. The [Ca2+] giving half-maximum force (EC50) was decreased from 1.42 ± 0.03 μM to 1.23 ± 0.06 μM (SEM, n=4, P < 0.05, paired t-test) from the control to in the presence of 500 μM dfbp-o. The corresponding pCa (pCa50; pCa=−log10[Ca2+]) was 5.85 ± 0.01 and 5.91± 0.02 in the absence and presence of dfbp-o (table SI), this change is similar to that induced by 10 μM levosimendan, where the pCa increased from 5.88 to 5.98[49]. As the relationship between force and [Ca2+] is steep in the cardiac trabeculae, with Hill coefficients (nH) of 5.7 and 4.6 in the absence and presence of dfbp-o, a decrease of 0.06 pCa units for pCa50 would result in an increase of force up to 20% of the maximum value during sub-maximal activation (dashed line in figure 3). Similar results were also found with levosimendan [50].
Figure 3.

Effect on Ca2+-sensitivity of skinned cardiac trabeculae by dfbp-o. The relationship between [Ca2+] and relative force at various [Ca2+] was determined in the absence (O) and presence of 500 μM dfbp-o (Δ). In the presence of dfbp-o, the relative force at Ca2+ concentrations of 1.0 μM and 1.5 μM were significantly higher than the control. Mean results were fit to the Hill equation and shown as dashed (control) or solid (dfbp-o) lines. The fine dashed line represents the change in force in the presence of dfbp-o.
The observation that dfbp-o has no influence on the force during relaxation and maximal Ca2+ activation suggests that dfbp-o is not altering the myosin-actin cross-bridge formation, and that dfbp-o would not impair relaxation. We chose to study trabeculae contraction in the presence of 500 μM dfbp-o, which is higher than the 10 μM of levosimendan used in previous studies [9, 49]. This was done because the affinity of dfbp-o (820 ± 190 μM) for cNTnC is lower than what has been estimated for levosimendan (~ 10 μM [10, 51]).
Structural Details of the Dfbp-o - Troponin Interaction
Dfbp-o binds to the hydrophobic surface of cNTnC
There are a number of structures of small molecules bound to cNTnC that have been solved by NMR spectroscopy and X-ray crystallography. Despite the markedly different chemical structures of the ligands, they all bind to the hydrophobic core of cNTnC and stabilize the open form of cNTnC. In this study, we titrated dfbp-o into cNTnC and followed the chemical shifts (1H, 15N) of each backbone amide resonance. The chemical shifts of all residues were averaged, and any resonance that moved greater than the mean change was mapped on the surface of cNTnC (figure 4A). The results imply that dfbp-o binds in the hydrophobic groove of cNTnC, as does bepridil (1dtl.pdb) [17, 52], trifluoperazine (1wrk.pdb and 1wr1.pdb) [52, 53], and W7 (2kfx.pdb) [54].
Figure 4.

(A) Mapping of the chemical shifts induced by the binding of dfbp-o to cNTnC onto the cartoon (left) and surface (right) representations of cNTnC taken from the cTnC•3Bep structure (1dtl.pdb). All residues perturbed more than the average chemical shift change are colored in slate, with the unperturbed residues colored in green; the Ca2+ is shown as a black sphere. (B) Localization of the binding site of dfbp-o on cNTnC by AutoDock 4.0. The lowest energy binding mode of dfbp-o (stick representation) is shown docked onto the cartoon (left) and surface (right) representations of cNTnC. (C) Bar diagram plotting the relative chemical shift perturbations of cNTnC induced by cTnI147–163 (red bars) or dfbp-o (slate bars). Residues marked with a star were exchange broadened during the dfbp-o titration, and therefore final perturbed distances were not measured.
In conjunction with the chemical shift mapping, we used AutoDock 4.0 [15] to predict the binding site and pose of dfbp-o on cNTnC. We docked dfbp-o onto the structure of the N-domain of the bepridil bound form of cTnC. We used a binding-site-centered docking approach for dfbp-o on the hydrophobic patch of cNTnC since the results from the chemical shift mapping identified this location as the binding site. To predict the binding mode of dfbp-o, the docking calculations generated clusters of binding modes and subsequently ranked them based on a lowest energy solution determined by the minimum docking energy. The lowest energy cluster predicted that the difluorophenyl ring binds in the hydrophobic core of cNTnC with the carboxyl moiety projecting towards the C-terminus of the D-helix (figure 4B). AutoDock calculates the binding energy of each docked structure, and this energy can be converted to a binding constant. For the lowest energy cluster, the program predicted a KD of 604 ± 145 μM, which is close to the experimentally determined KD of 820 ± 190 μM, and thus gives additional credence to predicted binging mode.
To investigate if dfbp-o induced an opening of cNTnC as do other ligands, we compared the relative chemical shift changes of dfbp-o with the relative chemical shift changes induced by cTnI147–163 (figure 4C). Although the total chemical shift changes induced by dfbp-o are smaller than what is caused by cTnI147–163 (figure 2), the patterns are quite similar. We propose that dfbp-o stabilizes the open state of cNTnC in a similar manner as cTnI147–163. Therefore, the increase in the affinity of cNTnC•dfbp-o for cTnI147–163 may be explained, in part, by the increase solvent exposure of the hydrophobic residues in the core of cNTnC. One goal of the design of Ca2+-sensitizers is to select for ligands that increase the affinity of cTnI147–163 for cNTnC, it has been proposed that the target molecule should be the cNTnC-cTnI complex [55]. Accordingly, we have determined the structure of the cNTnC•cTnI144–163•dfbp-o complex by NMR spectroscopy.
Solution Structure of cNTnC•cTnI144–163•dfbp-o
The structure of cNTnC•cTnI144–163•dfbp-o was determined by solution state NMR spectroscopy. See the online Methods for details on the NMR experiments we ran and references therein. For some of the less standard NMR experiments a brief discussion of the theory and expected results is included. Our discussion of the structure will start by considering each unit of the complex individually and then we will summarize the overall implications of the structure. The cNTnC•cTnI144–163•dfbp-o structure discussed in the text is the lowest energy structure from the deposited ensemble of 20 structures (see figure S2 and table SII for statistics).
Structural features of cNTnC
The structure of cNTnC in the cNTnC•cTnI144–163•dfbp-o complex is made up of five helical elements (N, A, B, C, D) and a short anti-parallel β-sheet (figure 5A,B). This overall fold is conserved throughout all known structures of cNTnC and indicates that dfbp-o is not inducing any large structural perturbation of cNTnC. The backbone rmsd for cNTnC (residues 5–85) in this complex with the X-ray structure cNTnC•cTnI144–163 (1j1d.pdb)[56] is 1.154 A and with the NMR structure cNTnC•cTnI147–163 (1mxl.pdb)[48] is 1.556 Å (figure 5C). The only difference between cNTnC•cTnI144–163•dfbp-o and the other structures is in the orientation of the C-helix, which has shifted slightly away from the D-helix to accommodate dfbp-o.
Figure 5.

Structure of dfbp-o bound to cNTnC•cTnI144–163. For all representations of cNTnC•cTnI144–163•dfbp-o the backbone atoms for cNTnC are colored in grey and in orange for cTnI144–163; dfbp-o is shown in stick representation with its carbon atoms colored in magenta, oxygen in red, and fluorine in aqua; and Ca2+ is shown as a black sphere. (A) Cartoon representation of the lowest energy structure of cNTnC•cTnI144–163•dfbp-o. (B) The structure of cNTnC•cTnI144–163•dfbp-o with cNTnC shown as a surface and cTnI144–163 shown as a mesh. (C) Overlay of cNTnC•cTnI144–163•dfbp-o with the X-ray crystal structure cNTnC•cTnI144–163 (blue; 1j1d.pdb) and the NMR structure cNTnC•cTnI147–163 (green; 1mxl.pdb). (D) Overlay of cNTnC•cTnI144–163•dfbp-o with cNTnC•dfbp-o (forest green; predicted by AutoDock). The docked version of dfbp-o is colored in slate, and the dfbp-o from the NMR structure is shown in magenta.
Structural features of cTnI144–163
The structure of cTnI144–163 is bound in a similar conformation as in the crystal structure of cNTnC•cTnI144–163 (1j1d.pdb) and the NMR structure of cNTnC•cTnI147–163 (1mxl.pdb) with backbone rmsds for residues 150–157 of 0.504 Å and 1.278 Å, respectively. The assignment of the 1H resonances of cTnI144–163 when bound to cNTnC were obtained by the 13C, 15N-filtered nuclear overhauser effect spectroscopy (NOESY) and the 13C, 15N-filtered total correlation spectroscopy (TOCSY)[18–20]. These experiments filter signals from the labeled-protein, while keeping signals from unlabeled ligands. We assigned 24 intramolecular NOEs belonging to cTnI144–163 in the 13C, 15N-filtered NOESY. We determined the location of cTnI relative to cNTnC via the 13C-edited, filtered HQMCNOESY NMR experiment [24, 25]. This NMR experiment measures NOEs between a 13C-labeled protein and an unlabeled ligand. We measured 30 intermolecular NOEs between cTnI144–163 and 13C, 15N-labeled cNTnC (for examples see figure S3), and the position of cTnI144–163 relative to cNTnC is similar to that of cNTnC•cTnI144–163. Since neither the structure of cTnI144–163, nor the position of cTnI144–163 is significantly perturbed by the presence of dfbp-o, it appears that dfbp-o is not competing for the same binding site on cNTnC. This is in contrast to W7[57] and bepridil[21], which sterically clash with cTnI and subsequently perturb the position of cTnI on cNTnC.
Structural features of dfbp-o
The binding site of dfbp-o is located in a well defined pocket formed by hydrophobic residues from cNTnC and cTnI144–163. We used the filtered NOESY and TOCSY experiments to assign the 1H chemical shifts of dfbp-o and we assigned the 19F chemical shifts by one-dimensional 19F and two-dimensional 1H-19F heteronuclear overhauser spectroscopy (HOESY) experiments[23]. In addition to assigning the 1H and 19F chemical shifts of dfbp-o, the NOESY and HOESY experiments provided structural restraints for dfbp-o. In total we used three intramolecular distance restraints in the structure calculation, including one between F2’ and H6/H2 and two between the phenyl protons (H2/H6 and H3/H5) and the CH2 group. An important characteristic of dfbp-o is that its phenyl ring is symmetric, and so NOEs to H2 and H6 or H3 and H5 are indistinguishable and therefore were ambiguously assigned. The filtered NOESY also revealed NOEs between dfbp-o and cTnI144–163. The terminal methyls of Met153 and Ile148 make NOE contacts with the aromatic H2/H6 and H3/H5 of dfbp-o (figure S4). These NOEs added structural restraints between dfbp-o and cTnI144–163, and gave us information into the relative orientations of the two molecules. Intermolecular NOEs between residues from cNTnC and dfbp-o helped position dfbp-o. We used the 13C-edited, filtered HMQCNOESY NMR experiment to observe contacts between the dfbp-o H2/H6 and H3/H5 and the terminal methyls of Met60 and Met45 of 13C,15N-cNTnC. We also measured intermolecular contacts between Met80 and H6’ and H3’ as well as between Leu41 or Val72 and H3’ and H5’ (figure S5).
The ambiguity of the NOE restraints and the lack of distance restraints between the fluorine atoms of dfbp-o and cNTnC left the orientation of dfbp-o poorly resolved. In an effort to improve the resolution of dfbp-o, we used paramagnetic relaxation enhancement (PRE). Lanthanide paramagnetism has recently been employed by the Otting group to determine the structure of thymidine bound to the ε exonuclease subunit of DNA polymerase III [58]. Briefly, we used the principle that unpaired electrons from paramagnetic molecules will increase the relaxation rate of nuclei in a distance dependant manner. We titrated free dfbp-o with cNTnC•Gd3+•cTnI144–163 and cNTnC•La3+•cTnI144–163 and compared the relaxation rates of the two 19F signals as well as the 1H signals from H2/H6, H6’ , and CH2 in the paramagnetic (Gd3+) and diamagnetic (La3+) systems. The broadening of the 1D 19F and 1H spectra of dfbp-o are shown in figure S6. The differences in the relaxation rates were then used to calculate distances between the protons and the fluorines of dfbp-o and the Gd3+.
The structure indicates that dfbp-o is bound in the hydrophobic pocket formed between cTnI144–163 and cNTnC. The F4’ of dfbp-o points towards the anti-parallel β-sheet of cNTnC and F2’ points away from cTnI144–163. The central phenyl ring of dfbp-o is near the N-terminus of cTnI144–163 with the negatively charged carboxylate of dfbp-o positioned near the positively charged side-chains of Arg147 and Arg83. The overall orientation of dfbp-o is similar to the binding mode of dfbp-o in complex with cNTnC as predicted by AutoDock (figure 5D). In summation, the structure reveals that dfbp-o binds deep enough within the hydrophobic pocket as to avoid sterically clashing with cTnI144–163 and the negatively charged moiety of dfbp-o is in the vicinity of the positively charged N-terminus of cTnI144–163.
Comparison of dfbp-o binding and other NTnC ligands
There have been several structures determined of small molecules bound to the cTnC-cTnI complex. We have overlaid the secondary structures of these complexes with cNTnC•Ca2+•cTnI144–163•dfbp-o (figure 6) and found that the most similar was the sNTnC•sTnI112–131•anapoe (1ytz.pdb) X-ray crystal structure [59], with a backbone rmsd of 1.253 A. The overlay of cNTnC•cTnI144–163•dfbp-o with cNTnC•cTnI147–163•bepridil (1lxf.pdb) [21] and cNTnC•cTnI147–163•W7 (2krd.pdb) [57] yielded much larger rmsds (2.459 Å and 3.061 Å, respectively). These ligands all bind at the interface formed between cNTnC and cTnI, but only the Ca2+-sensitizing agents, dfbp-o and anapoe do not perturb the quaternary structure of the troponin complex.
Figure 6.

Overlay of the secondary structure backbone nuclei of cNTnC•cTnI144–163•dfbp-o with (A) sNTn•sTnI112–131•anapoe (1ytz.pdb), (B) cNTnC•cTnI147–163•W7 (2krd.pdb), and (C) cNTnC•cTnI147–163•bepridil (1lxf.pdb). Backbone atoms for cNTnC are colored in grey (cNTnC•cTnI144–163•dfbp-o), brown (sNTnC•sTnI112–131•anapoe), yellow (cNTnC•cTnI147–163•W7), cyan (cNTnC•cTnI147–163•bepridil). Only in cNTnC•cTnI144–163•dfbp-o was cTnI colored differently than cNTnC (orange). Dfbp-o is shown in stick representation, with carbon atoms in magenta, oxygen in red, and fluorine in aqua. All other ligands are also shown in stick representation with carbon atoms colored in blue, oxygen in red, and nitrogen atoms in dark blue. Ca2+ ions are shown as black spheres.
Discussion
The efficacy of levosimendan in the treatment of heart failure has garnered much attention over the past few decades. Despite intense efforts to understand the molecular mechanism of levosimendan, its instability has circumvented knowledge of its precise mode of action in vivo. In this report, we described the molecular mechanism of the Ca2+-sensitizing agent, dfbp-o, a stable analogue of levosimendan. The chemical structures of dfbp-o and levosimendan highlight several pharmacophores required for targeting the cTnC-cTnI complex. Firstly, a substituted ring system; in levosimendan it is a pyridazinone ring which contains a methyl and carbonyl oxygen, and in dfbp-o it is a phenyl ring with two fluorine atoms. The second functional moiety is a phenyl ring that serves as a hydrophobic scaffold to connect the substituted ring to a hydrophilic extension. The hydrophilic group contains a central H-bond acceptor (the ether oxygen in dfbp-o and the hydrazone nitrogen in levosimendan) and an electronegative moiety (the carboxylate in dfbp-o and malonodinitrile in levosimendan) coplanar with the central ring (figure 7A–C). It was reported by Levijoki and coworkers that the hydrazone constituent of levosimendan is crucial for its full cardiotonic function [43]. The authors removed the hydrogen accepting nitrogen from the hydrazone so that instead of NH-N it was NH-C and saw a drastic decrease in the Ca2+-sensitizing and cTnC binding functionalities. Consistent with these findings, dfbp, which lacks the H-bond acceptor at this location has a ten-fold lower affinity for cTnC and the cTnC-cTnI complex than dfbp-o. In a recent study, Amin et al. identified a few more pharmacophores necessary to elicit cardiotonic activity [60]. They found that carbonyl groups attached to the hydrazone greatly improved the cardiotonic activity of the compounds. The location of the carbonyl groups is approximately in the same position as the carboxyl oxygen atoms of dfbp-o. All these data suggest that dfbp-o has a molecular framework similar to other well studied Ca2+-sensitizers and presents a novel scaffold for the design of new Ca2+-sensitizing agents.
Figure 7.

The electron density isosurfaces of (A) dfbp, (B) dfbp-o, and (C) levosimendan calculated using the quantum chemistry program Gaussian03. The compounds are positioned so that the fluorine atoms of dfbp and dfbp-o are in a similar orientation as the methyl and carbonyl oxygen groups of levosimendan as in figure 1. The mapped electrostatic potential is colored on the isosurfaces (negative – red, positive – blue, neutral – green). Electrostatic surface representation of the structure of cNTnC•cTnI144–163•dfbp-o. (D) cTnI144–163 has been omitted from the structure to indicate the location of dfbp-o. (E) Full ternary complex. (F) Close-up view of the electrostatic environment surrounding the carboxyl group of dfbp-o with Arg83 and Arg147 labeled. The electrostatic potential is shown on the surface of cNTnC and cTnI144–163 as a color gradient from negatively charged (red) to positively charged (blue). Dfbp-o is shown in stick representation, with carbon atoms in magenta, oxygen in red, and fluorine in aqua.
Heart muscle experiments showed that dfbp-o has a Ca2+-sensitizing function and NMR spectroscopy was employed to uncover the mechanism by which dfbp-o acts to enhance contractility. The muscle studies indicated that dfbp-o increased the Ca2+-sensitivity of the muscle fiber without increasing the maximum force of contraction. These results are consistent with previous findings for levosimendan [9, 49, 50], and the lack of an increase in the maximal force suggests that dfbp-o targets troponin instead of the myosin-actin system. NMR spectroscopy showed that the affinity of cTnI147–163 for cNTnC was enhanced by the presence of dfbp-o, and cTnI147–163 bound to cNTnC also improved the binding of dfbp-o. These data indicate that dfbp-o may increase contractility by strengthening the cNTnC-cTnI interaction.
The docked structure of cNTnC•dfbp-o and the NMR structure of cNTnC•cTnI144–163•dfbp-o have identified several of the important features of a Ca2+-sensitizing agent that targets cNTnC. In the docked structure, dfbp-o is bound with its difluorophenyl ring in the hydrophobic core of cNTnC. The difluorophenyl ring comes in close contact with Leu41, Phe27, Phe77, Ile36, Val72, Met60 and Met80. The central phenyl component of dfbp-o is near Met60 and Met80, while the carboxylate forms an electrostatic interaction with Arg83 of cNTnC. The structure of levosimendan bound to cNTnC, has not been solved; however, there has been a data-driven model of levosimendan in complex with cNTnC [11]. Although both levosimendan and dfbp-o models indicate the importance of the D-helix in forming the binding cleft, the binding site of dfbp-o is predicted to be located between helices B, C, and D; whereas, the binding location of levosimendan was predicted to be nearer to the NAD helix bundle. The primary binding sites of W7 [54], bepridil [17], and trifluoperazine [53] are consistent to that predicted for dfbp-o, and it seems likely that the binding site of levosimendan is also in a similar location.
In the NMR ensemble of cNTnC•cTnI144–163•dfbp-o, the fluorine-substituted phenyl ring of dfbp-o is buried deep in the hydrophobic pocket of cNTnC. The orientations of dfbp-o in the docked and the NMR structures are consistent, which emboldens the AutoDock model of cNTnC•dfbp-o. The biphenyl component of dfbp-o makes hydrophobic contacts to the same residues of cNTnC as in the docked model as well as the side chains of Met153 and Ile148 from cTnI144–163. The carboxylate moiety is directed outward, away from the hydrophobic pocket towards the N-terminus of cTnI144–163. The carboxylate is encapsulated by the positively charged side-chains of Arg83 from cNTnC and Arg147 from cTnI144–163 (figure 7). The distances between the carboxylate carbon of dfbp-o and the gaunidinyl group for Arg83 and Arg147 (CZ) over the ensemble are 8.3 ± 2.8 Å and 8.3 ± 2.5 Å, respectively. Given electrostatic potential can be exerted by a charged particle over a long range (≥ 20 A) these electrostatic interactions may be critical for the Ca2+-sensitizing mechanism of dfbp-o. In addition to the importance of Arg83 and Arg147, Arg145 and Arg144 are also near dfbp-o: 8.0 ± 4.1 Å and 11.6 ± 3.9 Å away from its carboxylate carbon. The malonodinitrile of levosimendan is also slightly electronegative (figure 7C) and therefore the network of charge-charge interactions we observed between dfbp-o and cTnI might be the pervasive mode of action of all cNTnC-targeting Ca2+-sensitizers.
The structural data we have presented here point towards a two-pronged mechanism of dfbp-o. Firstly, dfbp-o stabilizes a more open state of cNTnC and thus cNTnC is primed to bind cTnI147–163. However, several structural studies have indicated that simply opening cNTnC does not necessarily promote binding of cTnI147–163. W7 and bepridil were found to bind to the hydrophobic surface of cNTnC and promote an opening similar to that induced by cTnI147–163 [17, 54], yet they decreased the affinity of cTnI147–163 [21, 57]. Both W7 and bepridil have positively charged moieties that repel the positively charged N-terminal Arg147 of cTnI147–163. In the same theme, the negatively charged carboxylate of dfbp-o may attract Arg147, therefore explaining how dfbp-o increases the affinity of cTnI147–163 for cNTnC and enhances cardiac trabeculae Ca2+-sensitivity.
The conclusion that levosimendan functions as a Ca2+-sensitizer by the same mechanism as dfbp-o hinges on our assumption that dfbp-o is a good analogue of levosimendan; however, the affinity of dfbp-o for cNTnC was weaker than previously measured for levosimendan. The differences in affinities between dfbp-o and levosimendan may have several explanations. We initially proposed that the fluorine at the F2’ position of dfbp-o may substitute for the stereospecific methyl group of levosimendan; however, the van der Waals radius of fluorine is significantly smaller than a methyl group and therefore may not be an optimal substitution. The second noteworthy and perhaps more critical difference between the two molecules is the hydrophilic region. The carboxyl group of dfbp-o is much more electronegative than the malonodinitrile moiety of levosimendan. While the negatively charged carboxyl moiety of dfbp-o may contribute to the increase in cTnI147–163 affinity, it most likely decreases the affinity of dfbp-o for cNTnC, since cNTnC has a predominantly negative electrostatic surface (figure 7A). Levosimendan is slightly longer than dfbp-o, and this may also contribute to its increase in affinity for cNTnC by promoting a stronger interaction with cTnI, or along the D-helix of cNTnC. For example, the side-chain of Cys84 has been implicated in being essential for levosimendan binding to cNTnC [12].
Conclusion
We have described the molecular mechanism of a novel Ca2+-sensitizer, dfbp-o. We used in silico methods to compare aspects of dfbp-o with the well-known Ca2+-sensitizing agent, levosimendan. Functional studies with cardiac trabeculae indicated that dfbp-o increased the Ca2+-sensitivity in a similar manner as levosimendan. We also used NMR spectroscopy to show that dfbp-o enhanced the affinity of the switch region of cTnI for the Ca2+-saturated N domain of cTnC. Chemical shift mapping and automated docking identified the hydrophobic core of cNTnC as the likely binding site of dfbp-o. The solution structure of cNTnC•cTnI144–163•dfbp-o implicates an electrostatic attraction between dfbp-o and cTnI as a possible explanation for the Ca2+-sensitizing effect of dfbp-o. The structure also gives insight into the possible orientation levosimendan might adopt when bound to troponin. Overall, these results indicate that Ca2+-sensitizing agents may function by binding the hydrophobic surface of cNTnC, stabilizing its open conformation, and promoting the cTnI - cNTnC interaction. The data presented here contribute knowledge into the molecular mechanism of cTnC targeting inotropes, such as levosimendan, and highlight several of the key pharmacophores that direct their function.
Supplementary Material
(A) Titration of dfbp into cNTnC. (B) Titration of dfbp into cNTnC•cTnI144–163. (C) Titration of dfbp-o into cNTnC. (D) Titration of dfbp-o into cNTnC•cTnI144–163.
Cross-eye stereo view of the ensemble of 20 structures of cNTnC•cTnI144–163•dfbp-o. cNTnC (grey) and cTnI144–163 (orange) are shown in ribbon representation, dfbp-o (magenta) shown in stick representation, and Ca2+ is represented as a black sphere.
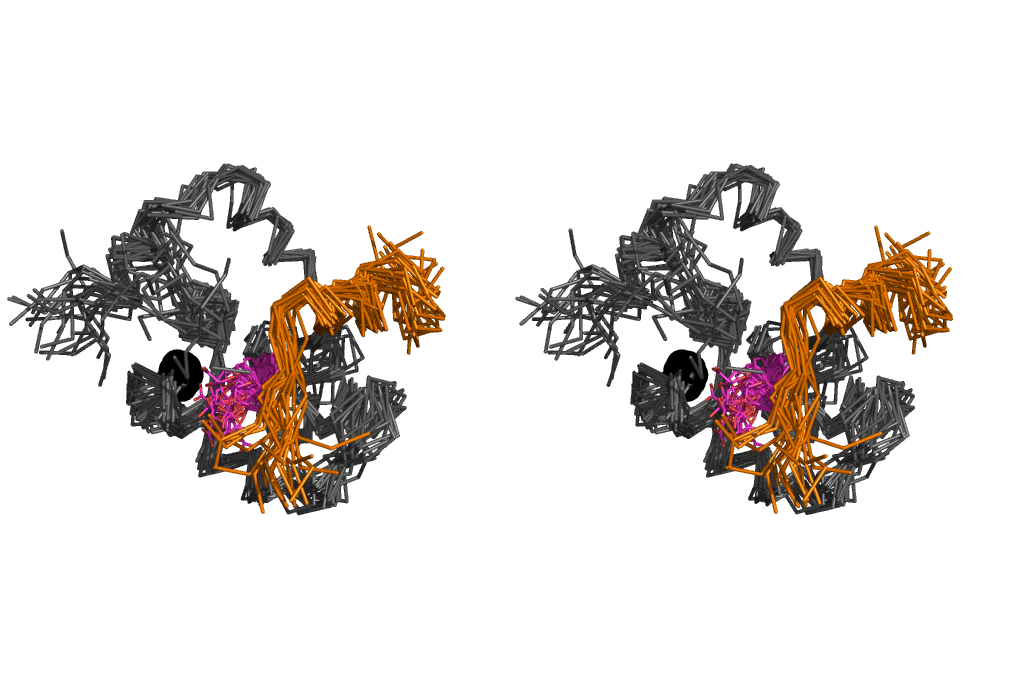{kind=link}
13C, 15N-labeled cNTnC. Intermolecular NOEs between cTnI144–163 and The right side of the spectra correspond to proton resonances from 13C, 15N-labeled cNTnC, and the vertical axis corresponds to proton resonances from cTnI144–163.
Intermolecular NOEs between cTnI144–163 and dfbp-o. The right side of the spectra correspond to proton resonances from cTnI144–163, and the vertical axis corresponds to proton resonances from dfbp-o.
13C, 15N-labeled cNTnC. The Intermolecular NOEs between dfbp-o and right side of the spectra correspond to proton resonances from 13C, 15N-labeled cNTnC, and the vertical axis corresponds to proton resonances from dfbp-o.
Paramagnetic relaxation enhancement of signals of dfbp-o by Gd3+ bound to cNTnC-cTnI144–163. The stacked 1D 19F spectrum of dfbp-o is shown in the upper left, and the stacked 1D 1H spectrum of dfbp-o is shown at the bottom. Both stacked spectra are shown as a function of percentage dfbp-o bound to cNTnC•Gd3+•cTnI144–163. The numbering of dfbp-o is shown on its chemical structure as a reference.
Acknowledgments
Special thanks to Dr. Liang Li for his help in characterizing dfbp-o and dfbp. We would like to thank Reify Co. for preliminary thoughts on drug analogs; Dr. Malcolm Irving and Dr. Marta Oleszczuk for insightful discussions; Dr. Leo Spyracopoulos for help with Gaussian03; Dr. Pascal Mercier and Olivier Julien for help with NMRView and structure calculation. The authors would also like to thank David Corson and Melissa Crane for protein expression and purification; Robert Boyko and Nick Shaw for spectrometer maintenance; and Robert Boyko for in-house software development.
This work is supported by grants from the Canadian Institutes of Health Research (FRN 37760), the National Institutes of Health (R01 HL-085234), and the Heart and Stroke Foundation of Canada to B.D.S., from the Alberta Heritage Foundation for Medical Research to I.M.R., and from the British Heart Foundation to Y.-B.S.
Abbreviations
- dfbp
2’ ,4’-difluoro(1,1’-biphenyl)-4-yl acetic acid
- dfbp-o
2’ ,4’-difluoro(1,1’-biphenyl)-4-yloxy acetic acid
- cTnC
cardiac troponin C
- cNTnC
N-domain of cardiac troponin C
- cCTnC
C-domain of cardiac troponin C
- cTnI
cardiac troponin I
- short ‘switch’ peptide region of cTnI
cTnI147–163
- long ‘switch’ peptide region of cTnI
cTnI144–163
- sNTnC
N-domain of skeletal troponin C
- sTnI
skeletal troponin I
- DTT
dithiothreitol
- NMR
nuclear magnetic resonance
- NOE
nuclear overhauser effect
- HOESY
heteronuclear overhauser effect spectroscopy
- HSQC
heteronuclear single quantum correlation
- PRE
paramagnetic relaxation enhancement
- NOESY
nuclear overhauser effect spectroscopy
- TOCSY
total correlation spectroscopy
- DSS
2,2-dimethyl-2-silapentane-5-sulfonate sodium salt, TFA
- TFA
trifluoroacetic acid
Footnotes
Disclosures:
None declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Li MX, Wang X, Sykes BD. Structural based insights into the role of troponin in cardiac muscle pathophysiology. J Muscle Res Cell Motil. 2004;25:559–79. doi: 10.1007/s10974-004-5879-2. [DOI] [PubMed] [Google Scholar]
- 2.Parmacek MS, Solaro RJ. Biology of the troponin complex in cardiac myocytes. Prog Cardiovasc Dis. 2004;47:159–76. doi: 10.1016/j.pcad.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 3.Gomes AV, Potter JD, Szczesna-Cordary D. The role of troponins in muscle contraction. IUBMB Life. 2002;54:323–33. doi: 10.1080/15216540216037. [DOI] [PubMed] [Google Scholar]
- 4.Endoh M. Could Ca2+ sensitizers rescue patients from chronic congestive heart failure? Br J Pharmacol. 2007;150:826–8. doi: 10.1038/sj.bjp.0707163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kass DA, Solaro RJ. Mechanisms and use of calcium-sensitizing agents in the failing heart. Circulation. 2006;113:305–15. doi: 10.1161/CIRCULATIONAHA.105.542407. [DOI] [PubMed] [Google Scholar]
- 6.Li MX, Robertson IM, Sykes BD. Interaction of cardiac troponin with cardiotonic drugs: a structural perspective. Biochem Biophys Res Commun. 2008;369:88–99. doi: 10.1016/j.bbrc.2007.12.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haikala H, Nissinen E, Etemadzadeh E, Linden IB, Pohto P. Levosimendan increases calcium sensitivity without enhancing myosin ATPase activity and impairing relaxation. J Mol Cell Cardiol. 1992;24(Suppl 5):S97. [Google Scholar]
- 8.Haikala H, Kaivola J, Nissinen E, Wall P, Levijoki J, Linden IB. Cardiac troponin-C as a target protein for a novel calcium sensitizing drug, levosimendan. J Mol Cell Cardiol. 1995;27:1859–66. doi: 10.1016/0022-2828(95)90009-8. [DOI] [PubMed] [Google Scholar]
- 9.Haikala H, Nissinen E, Etemadzadeh E, Levijoki J, Linden IB. Troponin C-mediated calcium sensitization induced by levosimendan does not impair relaxation. J Cardiovasc Pharmacol. 1995;25:794–801. doi: 10.1097/00005344-199505000-00016. [DOI] [PubMed] [Google Scholar]
- 10.Sorsa T, Pollesello P, Solaro RJ. The contractile apparatus as a target for drugs against heart failure: Interaction of levosimendan, a calcium sensitiser, with cardiac troponin C. Mol Cell Biochem. 2004;266:87–107. doi: 10.1023/b:mcbi.0000049141.37823.19. [DOI] [PubMed] [Google Scholar]
- 11.Pollesello P, Ovaska M, Kaivola J, Tilgmann C, Lundstrom K, Kalkkinen N, et al. Binding of a new Ca2+ sensitizer, levosimendan, to recombinant human cardiac troponin-C - a molecular modeling, fluorescence probe, and proton nuclear-magnetic-resonance study. J Biol Chem. 1994;18(269):28584–90. [PubMed] [Google Scholar]
- 12.Sorsa T, Heikkinen S, Abbott MB, Abusamhadneh E, Laakso T, Tilgmann C, et al. Binding of levosimendan, a calcium sensitizer, to cardiac troponin C. J Biol Chem. 2001;276:9337–43. doi: 10.1074/jbc.M007484200. [DOI] [PubMed] [Google Scholar]
- 13.Li MX, Saude EJ, Wang X, Pearlstone JR, Smillie LB, Sykes BD. Kinetic studies of calcium and cardiac troponin I peptide binding to human cardiac troponin C using NMR spectroscopy. European Biophysics Journal with Biophysics Letters. 2002;31:245–56. doi: 10.1007/s00249-002-0227-1. [DOI] [PubMed] [Google Scholar]
- 14.Frisch MJGWT, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JAT, Jr, et al. Gaussian. 3. Wallingford CT: Gaussian, Inc; 2004. [Google Scholar]
- 15.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–91. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gasteiger J, Marsili M. Iterative partial equalization of orbital electronegativity - a rapid access to atomic charges. Tetrahedron. 1980;36:3219–28. [Google Scholar]
- 17.Li Y, Love ML, Putkey JA, Cohen C. Bepridil opens the regulatory N-terminal lobe of cardiac troponin C. Proc Natl Acad Sci U S A. 2000;97:5140–5. doi: 10.1073/pnas.090098997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gemmecker G, Olejniczak ET, Fesik SW. An improved method for selectively observing protons attached to 12C in the presence of 1H-13C spin pairs. J Magn Reson. 1992;96:199–204. [Google Scholar]
- 19.Ikura M, Bax A. Isotope-filtered 2d NMR of a protein peptide complex - study of a skeletal-muscle myosin light chain kinase fragment bound to calmodulin. J Am Chem Soc. 1992;114:2433–40. [Google Scholar]
- 20.Ogura K, Terasawa H, Inagaki F. An improved double-tuned and isotope-filtered pulse scheme based on a pulsed field gradient and a wide-band inversion shaped pulse. J Biomol NMR. 1996;8:492–8. doi: 10.1007/BF00228150. [DOI] [PubMed] [Google Scholar]
- 21.Wang X, Li MX, Sykes BD. Structure of the regulatory N-domain of human cardiac troponin C in complex with human cardiac troponin I147–163 and bepridil. J Biol Chem. 2002;277:31124–33. doi: 10.1074/jbc.M203896200. [DOI] [PubMed] [Google Scholar]
- 22.Yu C, Levy GC. Solvent and intramolecular proton dipolar relaxation of the 3 phosphates of ATP - a heteronuclear 2d NOE study. J Am Chem Soc. 1983;105:6994–6. [Google Scholar]
- 23.Rinaldi PL. Heteronuclear 2d-NOE spectroscopy. J Am Chem Soc. 1983;105:5167–8. [Google Scholar]
- 24.Lee W, Revington MJ, Arrowsmith C, Kay LE. A pulsed-field gradient isotope-filtered 3d 13C HMQC-NOESY experiment for extracting intermolecular NOE contacts in molecular-complexes. FEBS Lett. 1994;350:87–90. doi: 10.1016/0014-5793(94)00740-3. [DOI] [PubMed] [Google Scholar]
- 25.Robertson IM, Spyracopoulos L, Sykes BD. The evaluation of isotope editing and filtering for protein-ligand interaction elucidation by NMR. Biophysics and the Challenges of Emerging Threats. 2009:101–19. [Google Scholar]
- 26.Krudy GA, Kleerekoper Q, Guo XD, Howarth JW, Solaro RJ, Rosevear PR. NMR-studies delineating spatial relationships within the cardiac troponin-I troponin-C complex. J Biol Chem. 1994;269:23731–5. [PubMed] [Google Scholar]
- 27.Lin X, Krudy GA, Howarth J, Brito RMM, Rosevear PR, Putkey JA. Assignment and calcium-dependence of methionyl εC and εH Resonances in Cardiac Troponin-C. Biochemistry. 1994;33:14434–42. doi: 10.1021/bi00252a009. [DOI] [PubMed] [Google Scholar]
- 28.Gariepy J, Kay LE, Kuntz ID, Sykes BD, Hodges RS. Nuclear magnetic resonance determination of metal proton distances in a synthetic calcium-binding site of rabbit skeletal troponin-C. Biochemistry. 1985;24:544–50. doi: 10.1021/bi00323a045. [DOI] [PubMed] [Google Scholar]
- 29.Schuttelkopf AW, van Aalten DMF. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr Sect D Biol Crystallogr. 2004;60:1355–63. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 30.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRpipe - a multidimensional spectral processing system based on unix pipes. J Biomol NMR. 1995;6:277–93. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 31.Johnson BA, Blevins RA. NMRView - a computer-program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4:603–14. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 32.Slupsky CM, Boyko RF, Booth VK, Sykes BD. Smartnotebook: A semi-automated approach to protein sequential NMR resonance assignments. J Biomol NMR. 2003;27:313–21. doi: 10.1023/a:1025870122182. [DOI] [PubMed] [Google Scholar]
- 33.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 34.Guntert P. Automated NMR structure calculation with CYANA. Methods Mol Biol. 2004;278:353–78. doi: 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- 35.Schwieters CD, Kuszewski JJ, Clore GM. Using Xplor-NIH for NMR molecular structure determination. Prog Nucl Magn Reson Spectrosc. 2006;48:47–62. [Google Scholar]
- 36.Linge JP, Williams MA, Spronk CAEM, Bonvin AMJJ, Nilges M. Refinement of protein structures in explicit solvent. Proteins-Structure Function and Bioinformatics. 2003;50:496–506. doi: 10.1002/prot.10299. [DOI] [PubMed] [Google Scholar]
- 37.Laskowski RA, Rullmann JAC, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–86. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 38.Sun YB, Lou F, Irving M. Calcium- and myosin-dependent changes in troponin structure during activation of heart muscle. Journal of Physiology-London. 2009;587:155–63. doi: 10.1113/jphysiol.2008.164707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lapka R, Smolik S, Rejholec V. Pharmacokinetics of flobufen and its main active metabolite in the rat. Drug Metab Dispos. 1990;18:1060–4. [PubMed] [Google Scholar]
- 40.Williams DE, Houpt DJ. Fluorine nonbonded potential parameters derived from crystalline perfluorocarbons. Acta Crystallographica Section B-Structural Science. 1986;42:286–95. [Google Scholar]
- 41.Smart BE. Fluorine substituent effects (on bioactivity) J Fluorine Chem. 2001 Jun;109(1):3–11. [Google Scholar]
- 42.DiMagno SG, Sun HR. The strength of weak interactions: Aromatic fluorine in drug design. Curr Top Med Chem. 2006;6:1473–82. doi: 10.2174/156802606777951127. [DOI] [PubMed] [Google Scholar]
- 43.Levijoki J, Pollesello P, Kaivola J, Tilgmann C, Sorsa T, Annila A, et al. Further evidence for the cardiac troponin C mediated calcium sensitization by levosimendan: Structure-response and binding analysis with analogs of levosimendan. J Mol Cell Cardiol. 2000;32:479–91. doi: 10.1006/jmcc.1999.1093. [DOI] [PubMed] [Google Scholar]
- 44.Bohm HJ, Banner D, Bendels S, Kansy M, Kuhn B, Muller K, et al. Fluorine in medicinal chemistry. Chembiochem. 2004;5:637–43. doi: 10.1002/cbic.200301023. [DOI] [PubMed] [Google Scholar]
- 45.Spyracopoulos L, Li MX, Sia SK, Gagne SM, Chandra M, Solaro RJ, et al. Calcium-induced structural transition in the regulatory domain of human cardiac troponin C. Biochemistry. 1997;36:12138–46. doi: 10.1021/bi971223d. [DOI] [PubMed] [Google Scholar]
- 46.McKay RT, Saltibus LF, Li MX, Sykes BD. Energetics of the induced structural change in a Ca2+ regulatory protein: Ca2+ and troponin I peptide binding to the E41A mutant of the N-domain of skeletal troponin C. Biochemistry. 2000;39:12731–8. doi: 10.1021/bi001240u. [DOI] [PubMed] [Google Scholar]
- 47.Paakkonen K, Sorsa T, Drakenberg T, Pollesello P, Tilgmann C, Permi P, et al. Conformations of the regulatory domain of cardiac troponin C examined by residual dipolar couplings. Eur J Biochem. 2000;267:6665–72. doi: 10.1046/j.1432-1327.2000.01763.x. [DOI] [PubMed] [Google Scholar]
- 48.Li MX, Spyracopoulos L, Sykes BD. Binding of cardiac troponin-I147–163 induces a structural opening in human cardiac troponin-C. Biochemistry. 1999;38:8289–98. doi: 10.1021/bi9901679. [DOI] [PubMed] [Google Scholar]
- 49.Szilagyi S, Pollesello P, Levijoki J, Kaheinen P, Haikala H, Edes I, et al. The effects of levosimendan and OR-1896 on isolated hearts, myocyte-sized preparations and phosphodiesterase enzymes of the guinea pig. Eur J Pharmacol. 2004;486:67–74. doi: 10.1016/j.ejphar.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 50.Edes I, Kiss E, Kitada Y, Powers FM, Papp JG, Kranias EG, et al. Effects of levosimendan, a cardiotonic agent targeted to troponin-C, on cardiac-function and on phosphorylation and Ca2+ sensitivity of cardiac myofibrils and sarcoplasmic-reticulum in guinea-pig heart. Circ Res. 1995;77:107–13. doi: 10.1161/01.res.77.1.107. [DOI] [PubMed] [Google Scholar]
- 51.Sorsa T, Pollesello P, Rosevear PR, Drakenberg T, Kilpelainen L. Stereoselective binding of levosimendan to cardiac troponin C causes Ca2+-sensitization. Eur J Pharmacol. 2004;486:1–8. doi: 10.1016/j.ejphar.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 52.Kleerekoper Q, Liu W, Choi D, Putkey JA. Identification of binding sites for bepridil and trifluoperazine on cardiac troponin C. J Biol Chem. 1998;273:8153–60. doi: 10.1074/jbc.273.14.8153. [DOI] [PubMed] [Google Scholar]
- 53.Igarashi T, Takeda S, Mori H. Crystal structure of the N-terminal domain of human cardiac troponin C in complex with a calcium-sensitizer; trifluoperazine. J Mol Cell Cardiol. 2005;39:1016. [Google Scholar]
- 54.Hoffman RMB, Sykes BD. Structure of the inhibitor W7 bound to the regulatory domain of cardiac troponin C. Biochemistry. 2009;48:5541–52. doi: 10.1021/bi9001826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Robertson IM, Baryshnikova OK, Li MX, Sykes BD. Defining the binding site of levosimendan and its analogues in a regulatory cardiac troponin C-troponin I complex. Biochemistry. 2008;47:7485–95. doi: 10.1021/bi800438k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takeda S, Yamashita A, Maeda K, Maeda Y. Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- 57.Oleszczuk M, Robertson IM, Li MX, Sykes BD. Solution structure of the regulatory domain of human cardiac troponin C in complex with the switch region of cardiac troponin I and W7: The basis of W7 as an inhibitor of cardiac muscle contraction. J Mol Cell Cardiol. 2010;48:925–33. doi: 10.1016/j.yjmcc.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.John M, Pintacuda G, Park AY, Dixon NE, Otting G. Structure determination of protein-ligand complexes by transferred paramagnetic shifts. J Am Chem Soc. 2006;128:12910–6. doi: 10.1021/ja063584z. [DOI] [PubMed] [Google Scholar]
- 59.Vinogradova MV, Stone DB, Malanina GG, Karatzaferi C, Cooke R, Mendelson RA, et al. Ca(2+)-regulated structural changes in troponin. Proc Natl Acad Sci U S A. 2005;102:5038–43. doi: 10.1073/pnas.0408882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Amin EN, Abdel-Alim AAM, Abdel-Moty SG, El-Shorbagi ANA, Abdel-Rahman MS. Synthesis of new 4,5-3(2H)pyridazinone derivatives and their cardiotonic, hypotensive, and platelet aggregation inhibition activities. Arch Pharmacal Res. 2010;33:25–46. doi: 10.1007/s12272-010-2222-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Titration of dfbp into cNTnC. (B) Titration of dfbp into cNTnC•cTnI144–163. (C) Titration of dfbp-o into cNTnC. (D) Titration of dfbp-o into cNTnC•cTnI144–163.
Cross-eye stereo view of the ensemble of 20 structures of cNTnC•cTnI144–163•dfbp-o. cNTnC (grey) and cTnI144–163 (orange) are shown in ribbon representation, dfbp-o (magenta) shown in stick representation, and Ca2+ is represented as a black sphere.
13C, 15N-labeled cNTnC. Intermolecular NOEs between cTnI144–163 and The right side of the spectra correspond to proton resonances from 13C, 15N-labeled cNTnC, and the vertical axis corresponds to proton resonances from cTnI144–163.
Intermolecular NOEs between cTnI144–163 and dfbp-o. The right side of the spectra correspond to proton resonances from cTnI144–163, and the vertical axis corresponds to proton resonances from dfbp-o.
13C, 15N-labeled cNTnC. The Intermolecular NOEs between dfbp-o and right side of the spectra correspond to proton resonances from 13C, 15N-labeled cNTnC, and the vertical axis corresponds to proton resonances from dfbp-o.
Paramagnetic relaxation enhancement of signals of dfbp-o by Gd3+ bound to cNTnC-cTnI144–163. The stacked 1D 19F spectrum of dfbp-o is shown in the upper left, and the stacked 1D 1H spectrum of dfbp-o is shown at the bottom. Both stacked spectra are shown as a function of percentage dfbp-o bound to cNTnC•Gd3+•cTnI144–163. The numbering of dfbp-o is shown on its chemical structure as a reference.