


Abstract
Proliferating cell nuclear antigen (PCNA), a processivity factor for DNA polymerases δ and ɛ, is involved in DNA replication as well as in diverse DNA repair pathways. In quiescent cells, UV light-induced bulky DNA damage triggers the transition of PCNA from a soluble to an insoluble chromatin-bound form, which is intimately associated with the repair synthesis by polymerases δ and ɛ. In this study, we investigated the efficiency of PCNA complex formation in response to ionizing radiation-induced DNA strand breaks in normal and radiation-sensitive Ataxia telangiectasia (AT) cells by immunofluorescence and western blot techniques. Exposure of normal cells to γ-rays rapidly triggered the formation of PCNA foci in a dose-dependent manner in the nuclei and the PCNA foci (40–45%) co-localized with sites of repair synthesis detected by bromodeoxyuridine labeling. The chromatin-bound PCNA gradually declined with increasing post-irradiation times and almost reached the level of unirradiated cells by 6 h. The PCNA foci formed after γ-irradiation was resistant to high salt extraction and the chromatin association of PCNA was lost after DNase I digestion. Interestingly, two radiosensitive primary fibroblast cell lines, derived from AT patients harboring homozygous mutations in the ATM gene, displayed an efficient PCNA redistribution after γ-irradiation. We also analyzed the PCNA complex induced by a radiomimetic agent, Bleomycin (BLM), which produces predominantly single- and double-strand DNA breaks. The efficiency and the time course of PCNA complex induced by BLM were identical in both normal and AT cells. Our study demonstrates for the first time that the ATM gene product is not required for PCNA complex assembly in response to DNA strand breaks. Additionally, we observed an increased interaction of PCNA with the Ku70 and Ku80 heterodimer after DNA damage, suggestive of a role for PCNA in the non-homologous end-joining repair pathway of DNA strand breaks.
INTRODUCTION
Proliferating cell nuclear antigen (PCNA) is a processivity factor for DNA polymerases δ and ɛ (pol δ and ɛ) (1,2) and is an essential protein for replication of chromosomal DNA (3,4). PCNA forms a toroidal trimer in S phase with replication factor-C (RF-C) and DNA in an ATP-dependent manner and enables the loading of DNA pol δ and ɛ onto the complex (3). The close association of PCNA with kinase complexes involved in cell cycle machinery (4) indicates that PCNA has a regulatory role in cell cycle progression (5–8). PCNA also participates in the processing of branched intermediates that arise during the lagging strand DNA synthesis (9,10). The amount of PCNA in eukaryotic cells is ∼10-fold more than that of pol δ and RF-C, indicating that PCNA has additional roles other than being a processivity factor. In support, PCNA has been demonstrated to be an integral component of diverse DNA repair pathways such as nucleotide excision repair (NER; 11–13), base excision repair (BER; 14–18) and mismatch repair (19). Reconstitution of an NER reaction in vitro using purified factors indicates a critical role for PCNA in the repair resynthesis step (1).
Two forms of PCNA exist in cells: (i) a detergent-insoluble trimeric form stably associated with the replicating forks during S phase and (ii) a soluble form in quiescent cells in G1 and G2 phases (20). Treatment of quiescent cells with DNA damaging agents like UV-C irradiation (21–23), X-irradiation (24), alkylating agents (25) and hydrogen peroxide (25,26) triggers the redistribution of PCNA from a soluble to an insoluble chromatin-bound complex analogous to that found in S phase cells. The functional transition of PCNA in quiescent cells after DNA damage is an indicator for the repair activity mediated by DNA pol δ and ɛ. This approach has been successfully used to determine the efficiency of the NER pathway in cells derived from patients belonging to different genetic complementation groups (A–G) of the human photosensitive disorder, xeroderma pigmentosum (XP). NER deficient human XP-A, XP-F and XP-G cells fail to show any detectable PCNA complex after UV-irradiation (21,27). PCNA complex formation therefore correlates with NER activity in human cells. A good correlation also exists between PCNA complex formation and the BER activity in human cells following oxidative DNA damage induced by hydrogen peroxide (26). A direct link between PCNA and cellular DNA repair processes comes from the observation that PCNA mutations cause increased sensitivity to DNA damaging agents in budding and fission yeast (28–31).
In the present study, we examined the formation of PCNA complex in human cells in vivo after treatment with ionizing radiation (IR) and a radiomimetic drug, Bleomycin (BLM). Both these agents induce single-strand breaks (SSBs) and double-strand breaks (DSBs) in the genomic DNA. We compared the PCNA complex formation of normal cells with radiation sensitive cells derived from Ataxia telangiectasia (AT) patients. Our studies show that PCNA complex formation, indicative of DNA pol δ and ɛ activity, was efficiently induced by γ-radiation and BLM. The PCNA–chromatin association formed fairly rapidly after DNA damage and almost reached the level of untreated cells by 6 h, suggestive of the completion of repair events. AT cells showed an identical response to that seen in normal cells, indicating that the assembly of PCNA–chromatin complex in response to DNA strand breaks occurs independent of the ATM gene product. Efficient PCNA complex formation in AT cells despite their abnormal p53 induction further suggests that PCNA-dependent repair activity is independent of p53 associated pathway(s) in human cells.
MATERIALS AND METHODS
Cell lines and culture conditions
Normal human dermal fibroblasts (NHDF) were obtained from Clonetics. Human normal (WI38 and MRC5) and AT (GM5823C and GM2052C) primary fibroblast cells were procured from Coriell Cell Repository (Camden, NJ). All the cells were routinely maintained in 2× Eagle’s minimal essential medium supplemented with 15% fetal bovine serum, vitamins, essential amino acids, non-essential amino acids and antibiotics (Gibco BRL). The cultures were maintained at 37°C in a humidified 5% CO2 atmosphere.
Treatment of cells with IR and BLM
Normal and AT cells were synchronized at G0/G1 phase by growth in low serum (0.5%) containing medium for 2–3 days prior to treatment with DNA damaging agents. Alternatively, cells were synchronized at G0/G1 by growing them to confluence and maintaining them in this state for a week. Flow cytometric analysis revealed that >95% of the cells were in G1 phase of the cell cycle. γ-Ray irradiation was done using a 137Cs source delivering a dose rate of 0.98 Gy/min (Gamma cell 40, Atomic Energy of Canada). Cells were irradiated with different doses (5–30 Gy) and incubated for various post-incubation times. For BLM treatment, cells were washed twice with phosphate-buffered saline (PBS) containing 1 mM CaCl2 and permeabilized with 40 µg/ml of l-α-lysophosphatidylcholine (Sigma) in PBS–CaCl2 for 2 min on ice. The solution was carefully removed and the cells were treated with BLM (Calbiochem; 10 mU/ml) in serum free medium for 30 min at 37°C. After treatment, the medium was removed and the cells were washed once with PBS. The cells were incubated in complete medium for different recovery times.
Detection of PCNA complex formation by immunohistochemistry
Cells were grown on glass chamber slides for immunofluorescence (Labtek). The cells were exposed to either γ-rays or BLM and incubated for the indicated times. The slides were treated with a hypotonic lysis solution (buffer I) containing 10 mM Tris–HCl pH 7.4, 2.5 mM MgCl2, 1 mM PMSF and 0.5% Nonidet P-40 for 8 min on ice. In some cases, the slides were treated with buffer II (25 mM sodium phosphate buffer pH 7.4, 0.5 M NaCl, 1 mM EDTA, 0.5% Triton X-100, 10% glycerol, 5 mM MgCl2 and 1 mM PMSF) for 10 min on ice. They were then washed once in PBS and fixed in ice-cold acetone–methanol (1:1). The slides were washed in TBST (20 mM Tris–HCl pH 7.4, 137 mM NaCl and 0.2% Tween-20) and incubated for 30 min in TBST containing 5% non-fat dried milk (NFDM). The slides were washed in TBST and incubated with mouse monoclonal PCNA antibody (1:10 dilution in TBST–5% NFDM; Santa Crutz Biotechnology) for 1 h at 37°C. The slides were washed three times (5 min each) in TBST buffer, incubated for 1 h with goat anti-mouse IgG2a antibody conjugated with fluorescein isothiocyanate (FITC; Boehringer Mannheim) diluted 1:20 in TBST–5% NFDM. The slides were washed in TBST, counterstained for DNA with DAPI (0.1 µg/ml prepared in Vectashield mounting medium; Vector laboratories) and covered with a cover glass. The variously fluorochromed images were captured using a Cytovision imaging system equipped with a charged coupled device camera and the PCNA fluorescence intensity per cell was measured using Adobe Photoshop.
Co-localization of PCNA- and bromodeoxyuridine (BrdU)-labeled repair sites in interphase nuclei
MRC5 cells in G1 phase were irradiated with 20 Gy of γ-rays and incubated in the presence of 100 µM BrdU for 3 h at 37°C. The slides were immunostained for PCNA and fixed in 2% formaldehyde in PBS containing 50 mM MgCl2 for 10 min at room temperature followed by denaturation in 0.07 M NaOH in 70% ethanol for 2 min and dehydrated through cold ethanol series. The slides were immunostained using mouse primary BrdU antibody (1:50 dilution, 1 h at 37°C; Boehringer Mannheim) followed by biotinylated anti-mouse IgG (1:100 dilution, 1 h at 37°C; Vector Laboratories) and Texas red-conjugated Avidin (1:100 dilution, 1 h at 37°C; Vector Laboratories). The slides were washed thoroughly in TBST buffer, counterstained for DNA with DAPI (0.1 µg/ml prepared in Vectashield mounting medium; Vector laboratories) and covered with a cover glass. The images were captured using an Axioplan2 Imaging microscope (Zeiss) using the Isis software programme of MetaSystems.
Protein extraction, SDS–PAGE and western blot analysis
Soluble and insoluble proteins were isolated following the protocol of Prosperi et al. (32). At different post-incubation times, 6–8 × 106 treated and mock treated cells were lysed for 8 min on ice in 500 µl of buffer I. The cell lysates were centrifuged at 735 g (Eppendorf Centrifuge 5415C) for 5 min and the soluble proteins (supernatant) were transferred to a fresh tube. The pellet fraction containing the detergent insoluble proteins was lysed for 20 min with 200 µl of buffer II. The proteins were recovered by centrifugation at 11 750 g for 5 min. Protein concentration was determined by Pierce protein assay kit. Aliquots of 20 µg of soluble and insoluble proteins were fractionated by 4–20% polyacrylamide gradient gel electrophoresis and blotted onto PVDF membrane following the standard protocol (Novex). The membrane was incubated with TBST buffer containing 5% NFDM for 60 min at room temperature. It was then incubated with mouse monoclonal antibody to PCNA (PC10, 1:1000 dilution in TBST–5% NFDM; Santa Cruz Biotechnology) for 1 h and washed three times (5 min each) in TBST buffer. The membrane was incubated with horseradish peroxidase (HRP)-conjugated anti-mouse IgG antibody (1:2000 dilution in TBST–5% NFDM; Vector Laboratories) for 1 h followed by repeated washing in TBST buffer. The signal was detected using the enhanced chemiluminescence technique (ECL) following the manufacturer’s protocol (Amersham). Immunodetection of actin (Mouse IgM, 1:2000 dilution; Oncogene) and p53 (D0-1, Mouse IgG, 1:1000 dilution; Santa Cruz Biotechnology) was carried out as described above. The PCNA band intensity was measured using the Scion Image Programme (National Institutes of Health).
Preservation of PCNA trimeric structure by protein crosslinking
The trimeric structure of PCNA was preserved for SDS–PAGE by the addition of glutaraldehyde to a final concentration of 0.1% to the insoluble protein extracts. The samples were kept on ice for 20 min and the reaction was terminated by addition of lysine to a final concentration of 0.1 M (33). The samples were briefly heated at 80°C for 5 min prior to electrophoresis.
Immunoprecipitation and Ku70/80 heterodimer detection
Total cellular proteins were extracted using RIPA buffer (10 mM sodium phosphate buffer pH 7.2, 2 mM EDTA, 50 mM sodium fluoride, 150 mM NaCl, 0.2 mM sodium vanadate, 0.1% SDS, 1% sodium deoxycholate, 1% Nonidet P-40 and 10 U/ml Aprotinin) from control and γ-irradiated (20 Gy) WI38 cells. Proteins (500 µg) were pre-cleared with normal mouse serum and 25 µl of protein G agarose beads (Boehringer Mannheim) for 1 h at 4°C with constant mixing. The samples were spun at 8160 g for 20 s and the supernatant was carefully transferred to new Eppendorf tubes. PCNA antibody–agarose conjugate (25 µl; Santa Cruz Biotechnology) was added to the samples and mixed constantly for 4 h at 4°C. The immunoprecipitated complex was pelleted down by centrifugation and washed four times in RIPA buffer under stringent conditions. The pellet was resuspended in SDS–PAGE loading buffer (100 µl), boiled for 5 min, vortexed and centrifuged. Supernatant (25 µl) was loaded in a 4–20% gradient gel and transferred to PVDF membrane using the manufacturer’s specifications (Novex). The membranes were probed with either Ku70 or Ku80 antibody (1:1000 dilution; Neomarkers) followed by HRP-conjugated secondary antibody (1:2000 dilution; Vector Laboratories). The signal was detected using the ECL method (Amersham).
Alkaline single cell gel electrophoresis
The treatment of NHDF cells with BLM (10 mU/ml) was done essentially as described earlier and the cells were collected by trypsinization at 0 min and 3 h after treatment. Alkaline single gel electrophoresis was done following the method described by Angelis et al. (34).
RESULTS
γ-Irradiation triggers the PCNA complex formation efficiently in normal cells
In order to determine the involvement of PCNA with IR-induced DNA damage, normal human primary fibroblast cells synchronized at G1 phase were irradiated with γ-rays and the chromatin-bound PCNA complex was analyzed by both immunofluorescence and western blot techniques. Fixation of cells with methanol revealed both soluble and insoluble forms of PCNA. In the initial experiments, cells were fixed and stained for PCNA without hypotonic buffer I treatment. The PCNA intensity detected by immunofluorescence and western blot was comparable between control and γ-irradiated (5 Gy) cells (Table 1, Fig. 1A). To determine the chromatin-bound PCNA after DNA damage, cells were treated with buffer I containing 0.5% Nonidet P-40 to release the unbound PCNA. In the untreated cells, a major proportion of PCNA was in the soluble form and the chromatin-bound PCNA was barely detectable. In cells irradiated with 5 Gy of γ-rays, intense PCNA staining was observed in the nuclei of all the cells (Fig. 1B). The PCNA complex appeared to form rapidly during irradiation, as the cells fixed immediately after 5 Gy of γ-irradiation (irradiation time, 5 min; no recovery time) also showed intense chromatin-bound PCNA foci (data not shown). Quantitation of PCNA fluorescence intensity in control and irradiated cells revealed that the PCNA complex formed after IR was resistant to high salt extraction (buffer II), indicating a very tight binding of PCNA to DNA (Table 1). The chromatin association of PCNA was lost by DNase I treatment evidenced by greatly diminished staining of PCNA (data not shown). The time course kinetics of PCNA foci formation was next determined in normal WI38 cells exposed to 10 Gy of γ-rays (Fig. 2A–F). For this purpose, 50 randomly chosen cells were analyzed for each time point. Microscopic examination of cells under high magnification (1000×) revealed a nice speckled pattern of 567 ± 10.3 (mean ± SE) focal sites of PCNA per cell immediately after irradiation (0 min recovery). The number of foci then declined to 166 ± 1.6/cell and 53 ± 0.7/cell after 3 and 6 h, respectively. A representative nucleus from each time point showing the time course distribution of PCNA foci is shown in Figure 2 (Fig. 2A and B, 0 min recovery; Fig. 2C and D, 3 h recovery; Fig. 2E and F, 6 h recovery).
Table 1. Fluorescence intensity of PCNA in control and γ-ray irradiated NHDF cells.
| Treatment | Fluorescence intensity (arbitrary units) |
| Control (no extraction) | 80.24 ± 8.50 |
| 5 Gy (no extraction) | 77.17 ± 7.13 |
| Control (buffer I extracted) | 23.35 ± 1.60 |
| 5 Gy (buffer I extracted) | 83.50 ± 5.50 |
| Control (buffer II extracted) | 18.86 ± 1.00 |
| 5 Gy (buffer II extracted) | 93.30 ± 7.20 |
Images of 15–20 randomly chosen NHDF cells were captured using a Zeiss epifluorescence microscope equipped with a charged coupled device camera and the relative fluorescence intensity of PCNA in control and γ-ray treated cells was measured using Adobe Photoshop and expressed in arbitrary units as mean ± SE.
Figure 1.

Immunofluorescence analysis of PCNA complex triggered by γ-ray irradiation in NHDF cells. Cells synchronized at G1 stage were irradiated (5 Gy), untreated or treated with buffer I and fixed after 30 min in a mixture of acetone:methanol (1:1). The cells were indirectly immunostained with a primary mouse monoclonal antibody to PCNA and a FITC-conjugated secondary antibody to mouse IgG2a. In order to determine whether or not the total cellular PCNA level is elevated by IR, the cells were immediately fixed in acetone:methanol without the extraction of soluble proteins (A). Immunofluorescence of PCNA in control and γ-treated cells without buffer I extraction (a) and the western blot detection of PCNA in the total cellular proteins isolated by RIPA buffer (b) show similar levels of PCNA. A histogram shows the intensity of PCNA band in control and irradiated NHDF cells (c). NDHF cells were extracted with buffer to reveal the chromatin-bound PCNA formation (B). The cells stained with DAPI are given in gray scale.
Figure 2.

Time-dependent distribution of PCNA foci induced by γ- ray irradiation in the interphase nuclei of WI38 cells (A–F). WI38 cells in G1 phase were irradiated with 10 Gy and the cells were incubated for different post-incubation times. The cells were extracted with buffer I and immunostained for PCNA with FITC conjugated antibody. The pattern of PCNA foci (green, FITC) induced by γ-rays at 0 min (A), 3 h (C) and 6 h (E) after irradiation is shown in the left panel and the combination of DNA and PCNA staining of the same cell (blue, DAPI; green, FITC) is shown in the right panel (B, D and F). MRC5 cells in G1 phase were irradiated with 20 Gy of γ-rays and the cells were incubated in complete medium containing 100 µM BrdU for 3 h. BrdU sites were detected with Avidin-Texas red (red) and the PCNA foci (green) with FITC. Control cells did not show any BrdU labeling (G), while irradiated cells show a punctuated pattern of repair foci detected by BrdU antibody (H). The cells were counterstained with DAPI to show both DNA (blue) and BrdU labeling (red). A single representative nucleus of an irradiated cell under high magnification shows the spatial distribution of PCNA foci (I), repair foci (J) and a combination of repair and PCNA foci (K). The merging of all the three colors (DNA, blue; PCNA, green; BrdU, red) is shown in (L). The co-localization of PCNA and repair foci appears as yellow spots due to the merging of green and red colors (K and L).
Co-localization of PCNA- and BrdU-labeled repair sites in interphase nuclei
In order to determine whether the PCNA foci associate with repair sites, co-localization of PCNA- and BrdU-labeled repair sites was carried out by immunofluorescence microscopy. With the exception of S phase cells, BrdU repair incorporation was not detected in the unirradiated cells (Fig. 2G). Cells exposed to 20 Gy of γ-rays displayed repair foci detected with BrdU antibody after continuous labeling of cells with BrdU for 3 h after irradiation (Fig. 2H and J). As BrdU labeling shorter than 3 h resulted in less consistent labeling patterns, co-localization of PCNA and BrdU sites could not be done earlier than 3 h after irradiation. In order to co-localize PCNA- and BrdU-labeled repair sites, PCNA (Fig. 2I) and BrdU sites (Fig. 2J) were detected by FITC- and Texas red-conjugated antibodies, respectively. Analysis of 50 randomly chosen cells revealed that each interphase nucleus exhibited an average of 223 ± 2.6 (mean ± SE) PCNA foci and 183 ±1.5 BrdU-labeled repair sites. The merging of PCNA foci and BrdU sites showed the regions of overlap as yellow color (Fig. 2K and L). Microscopic analysis indicates that 40–45% of PCNA foci co-localized with repair sites labeled with BrdU.
The response of PCNA to IR- and BLM-induced DNA damage occurs independently of the ATM gene product
AT cells are extremely radiosensitive and exhibit defects in the regulation of different cell cycle phases after damage. The gene mutated in AT patients (ATM) is considered to play an important role in radiation-induced damage-signaling pathways in mammalian cells. We therefore wished to determine whether or not ATM plays a role in regulating the efficiency of PCNA complex formation in response to DNA damage. The PCNA complex formation after IR was studied in two AT cell lines that carry homozygous mutations (indexed in the Coriell Cell Repository Catalogue). Immunofluorescence analysis of PCNA complex in buffer I extracted control and irradiated (5 Gy) cells revealed identical staining patterns in the two normal (NHDF and WI38) and the two AT (GM5823C and GM2052C) cell lines (Fig. 3). In order to determine whether the PCNA complex formation exhibits a dose-dependent response, normal (NHDF) cells were irradiated with 5, 10, 20 and 30 Gy of γ-rays and the proteins isolated 30 min after irradiation were analyzed by western blot. The enrichment of PCNA in the insoluble pool of proteins showed a 3-, 7.8-, 10- and 11.4-fold increase over control for 5, 10, 20 and 30 Gy of γ-irradiation, respectively (Fig. 4A and B) illustrating a dose-dependent PCNA complex formation.
Figure 3.

Immunofluorescence analysis of IR-induced PCNA complexes in the interphase nuclei of normal and AT cells. The normal (NHDF and WI38) and AT (GM5823C and GM2052C) cells in G1 phase were exposed to 5 Gy of γ-rays and incubated for 30 min. The cells were extracted with hypotonic buffer I and fixed in acetone:methanol. The cells were indirectly immunolabeled for insoluble PCNA as described before.
Figure 4.
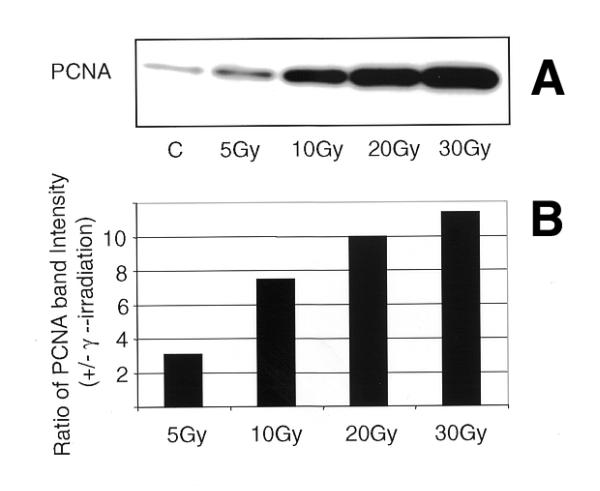
Western blot analysis of insoluble proteins isolated from NHDF cells exposed to increasing doses of γ-rays (A). NHDF cells in G1 phase were irradiated (0, 5,10, 20 and 30 Gy) and the insoluble proteins were extracted after 30 min. The proteins were size fractionated on 4–20% SDS–PAGE, transferred to PVDF membrane and probed with antibody to PCNA. The signal was detected by the ECL method. (B) Histogram showing the PCNA band intensity in the insoluble proteins.
The time course kinetics of PCNA complex formation were then analyzed by western blot in all four cell lines. The PCNA complex was readily detectable soon after irradiation (irradiation time, 10 min; no recovery time; Fig. 5A and B) in the insoluble proteins and gradually declined to the control level (≥1) by 6 h in WI38 and two AT cell lines (Fig. 5B). The NHDF cell line, however, showed a 2.7-fold increase in the induction of PCNA over control at 6 h, which declined to 1.9-fold at 24 h. Although there was variation among the cell lines, the kinetics of PCNA complex formation (peaking at ≥0 min recovery and declining at 6 h post-irradiation) was essentially similar in all the four cell lines. The time-dependent association of PCNA with chromatin appears to coincide with the fast kinetics of DSB repair reported during the first 30 min after IR (35,36). In order to verify the equal loading of proteins, the same membranes were stripped and immunoreacted with actin antibody (Fig. 5A).
Figure 5.
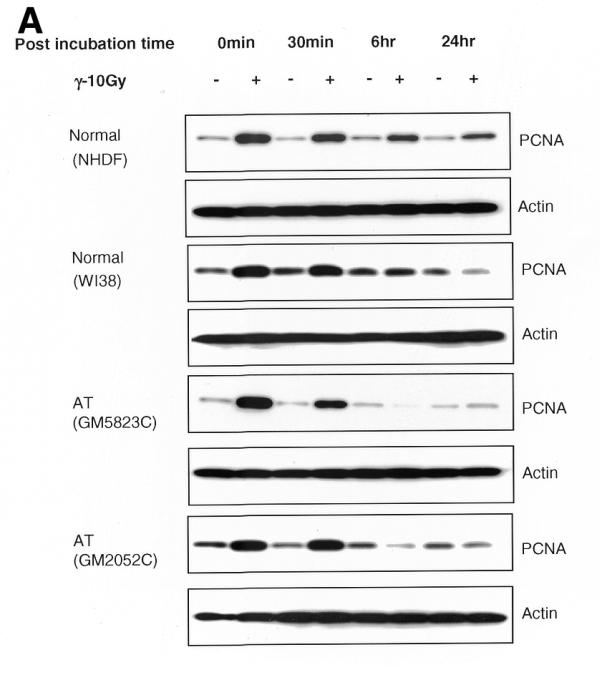

(A) Time course kinetics of PCNA complex formation induced by IR in normal and AT cell lines. The normal and AT cells in G1 phase were exposed to γ-rays (10 Gy) and incubated for different recovery times. The insoluble proteins were extracted, size fractionated and transferred to PVDF membrane. The membranes were immunoreacted with PCNA antibody and the signal was detected by the ECL method. The 0 time point represents an irradiation time of 10 min only. The PCNA complex analyzed at 15 min post-irradiation was essentially similar to 0 min while that analyzed at 3 h showed a decline in PCNA band intensity (data not shown) consistent with the immunofluorescence results (Fig. 2C and D). In order to verify equal loading of proteins, the same membranes were stripped off and reprobed with actin antibody. (B) Histogram showing the intensity of PCNA in normal and AT cell lines at different post-incubation times.
Irradiation with γ-rays induces a wide spectrum of lesions including SSBs, DSBs, oxidative base damage and DNA–protein crosslinks. To evaluate a direct involvement of PCNA in the repair of DNA strand breaks, PCNA complex was analyzed in both normal and AT cells following treatment with an anti-tumor drug, BLM, which induces DNA strand breaks. As the BLM uptake may be variable among cells, BLM treatment was carried out in lysolecithin permeabilized cells. NHDF cells were treated for 30 min with different concentrations of BLM (0, 5, 10, 20, 30 and 40 mU/ml) and the soluble and insoluble proteins were analyzed for PCNA. The extent of DSBs induced by the highest concentration of BLM (40 mU/ml) is expected to approximately equal that generated by 15 Gy of X-rays (37). BLM effectively triggered PCNA redistribution at all concentrations but, unlike IR, no dose-dependent increase in PCNA complex was observed (Fig. 6). It is likely that the PCNA complex formation reached a saturation level even at the lowest BLM concentration. As there was no dose response, a concentration of 10 mU/ml was chosen for subsequent experiments. Western blots of insoluble proteins extracted from normal and AT cells showed a 4–5-fold increase in PCNA induction as compared to control cells immediately after 30 min BLM treatment (0 min; no recovery) and this PCNA level persisted up to 1 h after treatment (Fig. 7A and B). The PCNA level was reduced to 1.7-fold by 3 h after treatment. In order to verify equal loading of proteins, the same membranes were stripped and probed with actin antibody. Alkaline single gel electrophoresis of BLM-treated (10 mU/ml) NHDF cells was carried out to determine the initial induction and repair of DNA strand breaks. The initial induction of strand breaks expressed as Olive tail movement was found to be 6.8 at 0 min after BLM treatment, which then declined to 1.8 after 3 h of repair, demonstrating the repair of 74% of the DNA strand breaks. Thus, there is a good correlation between the time course of PCNA complex and the repair kinetics of DNA strand breaks. The efficient PCNA complex formation induced by both IR and BLM in AT cells suggest that the ATM gene product is not required for PCNA complex assembly in response to DNA strand breaks.
Figure 6.
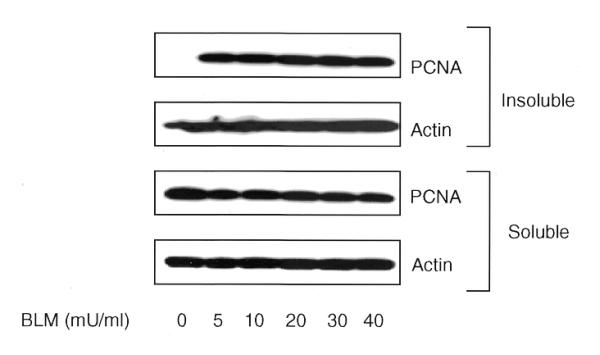
Immunoblot analysis of soluble and insoluble proteins isolated from NHDF cells after treatment with different concentrations of BLM for 30 min at 37°C. The proteins were size fractionated on 4–20% SDS–PAGE, transferred to PVDF membrane and probed with antibody to PCNA. The signal was detected by the ECL method. Equal loading of proteins was verified by actin antibody.
Figure 7.

(A) Time course kinetics of PCNA complex formation induced by BLM treatment in normal and AT cells. The cells were treated with 10 mU/ml of BLM for 30 min and incubated for various recovery times. The insoluble proteins isolated from control and treated cells were electrophoresed and transferred to PVDF membrane. The membrane was reacted with antibodies to PCNA, p53 and actin. (B) Histogram showing the time course kinetics of PCNA complex in normal and AT cells.
PCNA complex formation does not correlate with p53 induction
A recent study has indicated that p53 regulates the expression of PCNA in rat embryo fibroblast cells upon exposure to IR (38). In order to verify this possibility in the human cells, time course induction of p53 was compared with that of PCNA in normal and AT cells after γ-rays and BLM. In γ-irradiated normal cells, maximal induction of p53 was observed between 2 and 6 h and the p53 level declined at 8 and 24 h (Fig. 8). In addition to p53, a low molecular weight protein was detected with D0-1 antibody in NHDF cells. This band was observed in NHDF cells but not in MRC5 and WI38 cells. In contrast to p53 induction, the PCNA level peaks immediately after irradiation and drops to the level of unirradiated cells by 6 h in both normal and AT cells (Fig. 5A and B). In spite of a greatly attenuated p53 induction (Figs 7A and 8), AT cells showed the PCNA complex formation immediately after irradiation like that observed in normal cells. Likewise, p53 accumulation did not correlate with PCNA complex formation after BLM treatment (Fig. 7A), indicating that the p53-dependent pathway is not critical for DNA damage-dependent PCNA response.
Figure 8.
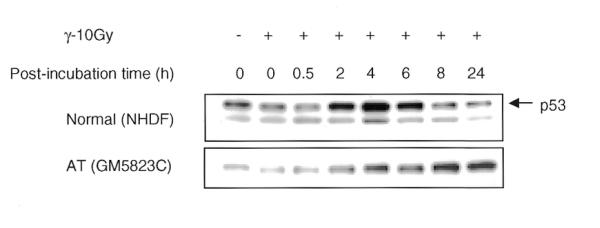
Time course kinetics of p53 protein induction by IR in normal and AT cells. The cells in G1 phase were exposed to 10 Gy of γ-rays and incubated for various recovery times. The insoluble proteins were isolated, size fractionated and transferred to PVDF membrane. The membranes were probed for p53 and the signal was detected by the ECL method.
Functional PCNA trimeric structures accumulate in the chromatin-bound proteins of damaged cells
The functional form of PCNA is often phosphorylated and exists as a trimeric structure, which is loaded onto DNA template in the presence of RF-C (20). In order to determine whether the chromatin-bound PCNA formed after DNA damage also occurs in a functional trimeric form, the insoluble proteins isolated from NHDF cells were crosslinked with glutaraldehyde and the PCNA trimeric structure was analyzed by western blot using PCNA antibody. The PCNA trimeric structures were rapidly triggered after treatment (0 min; no recovery) and the induction was 4- and 6-fold more than that of control cells in γ-rays and BLM-treated cells, respectively (Fig. 9). The PCNA trimeric structures constituted 22 and 37% of the total insoluble PCNA detected immediately after exposure to γ-rays and BLM, respectively, and the remaining fraction was in the monomeric form. The specific enrichment of PCNA trimer in a time- and damage-dependent manner in normal cells indicates the involvement of PCNA in strand break repair activity.
Figure 9.

Analysis of PCNA trimeric structures induced by IR and BLM in NHDF cells. The G1 cells were exposed to either 10 Gy of γ-rays or 10 mU/ml of BLM. Prior to electrophoresis, the PCNA trimeric structures in the pool of insoluble proteins were preserved by crosslinking with 0.1% glutaraldehyde for 10 min on ice. The reaction was terminated by addition of glycine to a final concentration of 0.1 M. The samples were heated at 80°C for 5 min, electrophoresed, transferred to PVDF membrane and immunoreacted with PCNA. The film was exposed longer to reveal the trimeric structures induced by γ-rays, which has resulted in the darkening of the PCNA band at the monomer position.
Interaction of PCNA with Ku70/80 heterodimer is increased after DNA damage
The damage-dependent redistribution of PCNA in normal and AT cells clearly reflects its role in the repair of DNA strand breaks. In order to determine whether the repair function of PCNA is mediated through its interaction with known DSB repair proteins such as Ku70 and Ku80, immunoprecipitation studies were carried out using PCNA antibody. Western analysis indicates that Ku70 protein was much more abundant than Ku80 and the levels of Ku70 and Ku80 remained grossly similar in both control and irradiated samples (Fig. 10A). Western blot analyses of immunoprecipitated complexes using antibodies to Ku70 and Ku80 indicated a tight physical interaction of PCNA with the Ku70/80 heterodimer and this interaction was increased at 3 h after irradiation (Fig. 10A). The immunoprecipitation was highly specific as omission of either primary or secondary antibody did not result in the precipitation of Ku70/80 proteins. Immunoprecipitation done using p53 antibody also did not precipitate the Ku proteins (data not shown). The observed interaction of PCNA with Ku70/80 heterodimer raises the possibility that PCNA may be involved in non-homologous end joining (NHEJ) pathway. Immunoprecipitation results were confirmed by immunological co-localization of PCNA and Ku70/80 heterodimer in WI38 cells 3 h after irradiation (Figure 10B).
Figure 10.
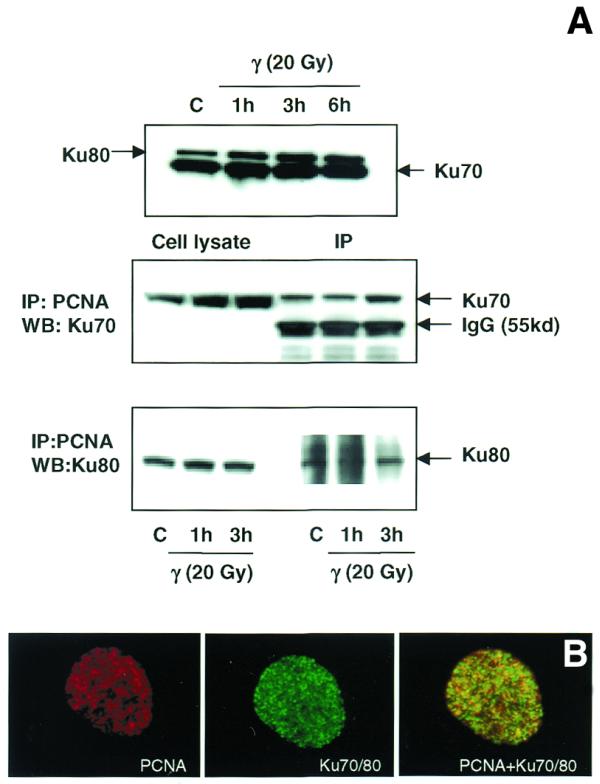
(A) Immunoblot analysis of the association of PCNA complex with Ku70/80 heterodimer by immunoprecipitation. Total cellular proteins were isolated from control and γ-irradiated (20 Gy) WI38 cells. Western blot analysis of Ku70 and Ku80 proteins in control and irradiated cells is shown in the top panel. Protein (500 µg) was immunoprecipitated using agarose-conjugated PCNA antibody (see Materials and Methods). The immunoprecipitated complex was size fractionated by 4–20% SDS–PAGE and transferred to PVDF membrane. The membranes were immunoreacted with mouse monoclonal antibodies to Ku70 (middle panel) and Ku80 (lower panel) and the signal was detected using the ECL kit. Ku70 antibody detected a cross-reacting immunoglobulin band of 55 kDa (middle panel). (B) Immunological co-localization of PCNA and Ku70/80 heterodimer in the interphase nucleus after γ-irradiation. WI38 cells in G1 phase were irradiated with 10 Gy of γ-rays, post-incubated for 3 h and fixed in acetone:methanol (1:1) after extraction with hypotonic buffer. The cells were sequentially immunostained for PCNA (detected by Texas red-conjugated secondary antibody to mouse IgG) and Ku70/80 heterodimer (detected by FITC-conjugated secondary antibody to mouse IgG). The immunostaining of PCNA (red), Ku70/80 heterodimer (green) and the merging of both PCNA and Ku70/80 heterodimer (yellow) are shown.
DISCUSSION
The chromatin association of PCNA was observed in earlier studies following oxidative damage (25,26), and bulky DNA adducts (21,25,39,40) induced by hydrogen peroxide, UV and alkylating agents. The functional transition of PCNA from a soluble to an insoluble form observed after DNA damage is considered to reflect the repair activities mediated by pol δ and ɛ (20). We have studied the efficiency and time course kinetics of PCNA complex assembly in response to DNA strand breaks induced by γ-rays and BLM. Both agents triggered rapid PCNA complex formation in a time-dependent manner in human primary fibroblast cells. The maximal level of chromatin-bound PCNA observed in cells sampled immediately after exposure indicates that the process occurs even during the treatment time. Similar rapid formation of PCNA complex in radiosensitive ATM deficient cells indicates that ATM gene product is not required for this phenomenon. PCNA relocation was observed in the nuclei of damaged cells but not in mock treated cells, suggesting that the process is highly specific in relation to DNA repair activities.
The spatial distribution of PCNA foci in γ-rays and BLM-treated cells was homogenous throughout the nucleoplasm and this pattern was very similar to that obtained after UV damage (22,39). In both NER and BER pathways, PCNA is implicated in the repair resynthesis step and the repair patches vary from 5–7 to 25–30 bp for BER and NER, respectively (41,42). Unlike UV, which induces bulky DNA adducts, γ-rays and BLM predominantly produce SSBs and DSBs and the repair of these lesions is not expected to involve the resynthesis step. If PCNA is required only for the resynthesis step, it would be difficult to explain the formation of PCNA complex in response to DNA strand breaks. Miura et al. (24) have documented the formation of PCNA foci in X-irradiated cells of normal and NER-deficient XP-A cells. It is likely that the lesions generated by X-rays are repaired by PCNA-dependent long patch BER pathway. Although compelling evidence is lacking, the correlation observed in this study between the time course kinetics of PCNA complex and the repair of BLM-induced strand breaks (detected by single gel electrophoresis) suggests a role for PCNA in strand break repair activity in human cells.
We observed a rapid assembly of chromatin-bound PCNA foci soon after γ-irradiation, followed by the disappearance of majority of foci 6 h after treatment. Although the functional significance of this complex remains largely unclear, the time course of PCNA foci formation correlates with well-established kinetics with half-lives of 15–30 min and 1–3 h for SSB and DSB repair, respectively (35,36). The present study raises two possibilities for the potential participation of PCNA in the repair of DNA strand breaks. First, PCNA may scan the damage in the genomic DNA and effectively mediate strand break repair by recruiting the factors involved in both NHEJ and homologous recombination (HR) repair pathways. Secondly, PCNA may help in loading the DNA pol δ and ɛ onto the damaged DNA template and enhancing their processivity. Experimental evidence supports both of these possibilities. The first possibility is supported by the rapid functional transition of PCNA that may participate in a damage-signaling pathway. Recently, PCNA has been demonstrated to recruit chromatin assembly factor to SSBs and gaps (43). This suggests a potential role for PCNA not only during the repair synthesis but also in nucleosome assembly following DNA repair. Alternatively, PCNA may also serve as a loading dock for the catalytic turnover of repair proteins, thereby effectively mediating strand break repair activity in cells. Such a role for PCNA has been proposed for the NER pathway (11). The increased interaction of PCNA with Ku70/80 heterodimer after damage may be supportive of such a role for PCNA in mediating the NHEJ pathway.
The second possibility is strengthened by the time- and dose-dependent formation of PCNA trimeric structures observed in IR- and BLM-treated cells, which reflect DNA polymerizing activity, as PCNA trimeric structures are often associated with active DNA synthesis (44). The association of PCNA foci with the synthetic sites of repair as well as the formation of PCNA trimer observed after DNA damage strongly suggests the involvement of PCNA-dependent polymerization activity during the repair of strand breaks. In corroboration with our results, very long repair patches ranging from 3 to 150 nt were found after DSB repair in IR- and BLM-treated human cells at G1/S border (45). These long repair patches were abolished upon inhibition of pol δ and ɛ by aphidicolin and cytosine arabinoside (45). This study suggests that some of the strand breaks may be processed by DNA pol δ and ɛ, possibly requiring PCNA as an accessory factor for processivity. Whisnant-Hurst and Leadon (45) showed that the long repair patches constituted 20% of the repair label, which is in agreement with the fraction of PCNA trimeric structures observed after IR and BLM treatments in this study. Furthermore, we found that 40–45% of the PCNA foci co-localize with BrdU-labeled repair sites indicating the PCNA-dependent polymerizing activity in γ-treated cells. Also, treatment of cells with the DNA polymerase inhibitors aphidicolin and hydroxyurea after γ-irradiation resulted in the persistence of PCNA foci up to 24 h (data not shown). Budd and Campbell (46) have shown that aphidicolin-sensitive polymerases are involved in the repair of plasmid DNA damaged by γ-rays and BLM. DNA pol δ and ɛ have been found to be involved in the repair of IR-induced DNA damage in yeast (47,48). These studies provide supporting evidence for the role of pol δ and ɛ in the repair of strand breaks.
One of the novel findings of the present study is the demonstration of an efficient PCNA complex in radiation-sensitive AT cells. ATM, the gene mutated in the AT patients, encodes a 370 kDa protein with a phosphatidylinositol kinase (PI-3) domain, a feature shared by several other proteins involved in DNA damage response, cell cycle control and telomere maintenance (49,50). ATM phosphorylates p53 on serine15 and leads to the stabilization of p53 following DNA damage (51,52). Recently, the requirement of ATM and c-Abl has been demonstrated for the IR-induced assembly of a recombination complex comprising Rad51 and 52 proteins (53). However, the DNA damage response of PCNA to IR and BLM occurs independent of both ATM and p53 induction in AT cells. Foray et al. (54) have demonstrated that the repair of strand breaks is faster in AT cells than normal cells during the first 6 h after irradiation. Furthermore, the repair profile was grossly similar in both normal and AT primary fibroblast cells treated either at 4 or at 37°C with 30 Gy of γ-irradiation (35,36). Thus, the rapid PCNA complex formation observed corroborates the efficient strand break repair reported in AT cells. In IR- and BL-treated cells, a punctuated pattern of PCNA distribution is found homogenously throughout the nucleoplasm and the PCNA foci are tightly bound to nuclear substructures. In contrast to the kinetics of PCNA complex formation, human Mre11 and Rad50 form foci 8 h after irradiation in exponentially growing cells (55). It is likely that a small fraction of DSBs are repaired in mammalian cells by the HR pathway involving the Mre11 and Rad50 complex, and this pathway may be much slower than that of the PCNA-dependent repair pathway.
In this study, we have demonstrated efficient PCNA complex formation in a time-dependent manner following DNA damage in normal and AT cells. The rapid formation of a PCNA–chromatin complex, comprising monomer and trimer forms, in response to strand breaks suggests a role for PCNA both in damage recognition and in the rapid repair of SSBs and DSBs. Future studies are required to understand the precise mechanism of PCNA complex formation and its functional significance in the repair of IR- and BLM-induced DNA damage.
Acknowledgments
ACKNOWLEDGEMENTS
The authors wish to thank Dr M.P.Hande for helpful comments on the manuscript. The authors are grateful to Dr T.S.Kumaravel and Mr Althaf Lohani (Gerontology Research Center, NIA, NIH, Baltimore, MD) for help with alkaline single gel electrophoresis. This study was supported by research grants from NIH/NCI (CA75061 and CA49062) and DOE (DEFG02-98ER62667) awarded to C.R.G.
References
- 1.Bravo R., Frank R., Blundell,P.A. and Macdonald-Bravo,H. (1987) Cyclin/PCNA is the auxiliary protein of DNA polymerase-δ. Nature, 326, 515–517. [DOI] [PubMed] [Google Scholar]
- 2.Burgers P.M. (1991) Saccharomyces cerevisiae replication factor C. II. Formation and activity of complexes with the proliferating cell nuclear antigen and with DNA polymerases δ and ɛ. J. Biol. Chem., 266, 22698–22706. [PubMed] [Google Scholar]
- 3.Krishna T.S., Kong,X.P., Gary,S., Burgers,P.M. and Kuriyan,J. (1994) Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA. Cell, 79, 1233–1243. [DOI] [PubMed] [Google Scholar]
- 4.Xiong Y., Zhang,H. and Beach,D. (1992) D type cyclins associate with multiple protein kinases and the DNA replication and repair factor PCNA. Cell, 71, 505–514. [DOI] [PubMed] [Google Scholar]
- 5.Zhang H., Xiong,Y. and Beach,D. (1993) Proliferating cell nuclear antigen and p21 are components of multiple cell cycle kinase complexes. Mol. Biol. Cell, 4, 897–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flores-Rozas H., Kelman,Z., Dean,F.B., Pan,Z.Q., Harper,J.W., Elledge,S.J., O’Donnell,M. and Hurwitz,J. (1994) Cdk-interacting protein 1 directly binds with proliferating cell nuclear antigen and inhibits DNA replication catalyzed by the DNA polymerase δ holoenzyme. Proc. Natl Acad. Sci. USA, 91, 8655–8659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Y., Jenkins,C.W., Nichols,M.A. and Xiong,Y. (1994) Cell cycle expression and p53 regulation of the cyclin-dependent kinase inhibitor p21. Oncogene, 9, 2261–2268. [PubMed] [Google Scholar]
- 8.Podust V.N., Podust,L.M., Goubin,F., Ducommun,B. and Hubscher,U. (1995) Mechanism of inhibition of proliferating cell nuclear antigen-dependent DNA synthesis by the cyclin-dependent kinase inhibitor p21. Biochemistry, 34, 8869–8875. [DOI] [PubMed] [Google Scholar]
- 9.Li X., Li,J., Harrington,J., Lieber,M.R. and Burgers,P.M. (1995) Lagging strand DNA synthesis at the eukaryotic replication fork involves binding and stimulation of FEN-1 by proliferating cell nuclear antigen. J. Biol. Chem., 270, 22109–22112. [DOI] [PubMed] [Google Scholar]
- 10.Wu X., Li,J., Li,X., Hsieh,C.L., Burgers,P.M. and Lieber,M.R. (1996) Processing of branched DNA intermediates by a complex of human FEN-1 and PCNA. Nucleic Acids Res., 24, 2036–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nichols A.F. and Sancar,A. (1992) Purification of PCNA as a nucleotide excision repair protein. Nucleic Acids Res., 20, 2441–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shivji M.K., Podust,V.N., Hubscher,U. and Wood,R.D. (1995) Nucleotide excision repair DNA synthesis by DNA polymerase epsilon in the presence of PCNA, RFC and RPA. Biochemistry, 34, 5011–5017. [DOI] [PubMed] [Google Scholar]
- 13.Aboussekhra A., Biggerstaff,M., Shivji,M.K., Vilpo,J.A., Moncollin,V., Podust,V.N., Protic,M., Hubscher,U., Egly,J.M. and Wood,R.D. (1995) Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell, 80, 859–868. [DOI] [PubMed] [Google Scholar]
- 14.Matsumoto Y., Kim,K. and Bogenhagen,D.F. (1994) Proliferating cell nuclear antigen-dependent abasic site repair in Xenopus laevis oocytes: an alternative pathway of base excision DNA repair. Mol. Cell. Biol., 14, 6187–6197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsumoto Y., Kim,K., Hurwitz,J., Gary,R., Levin,D.S., Tomkinson,A.E. and Park,M.S. (1999) Reconstitution of proliferating cell nuclear antigen-dependent repair of apurinic/apyrimidinic sites with purified human proteins. J. Biol. Chem., 274, 33703–33708. [DOI] [PubMed] [Google Scholar]
- 16.Fortini P., Pascucci,B., Parlanti,E., Sobol,R.W., Wilson,S.H. and Dogliotti,E. (1998) Different DNA polymerases are involved in the short- and long-patch base excision repair in mammalian cells. Biochemistry, 37, 3575–3580. [DOI] [PubMed] [Google Scholar]
- 17.Gary R., Ludwig,D.L., Cornelius,H.L., MacInnes,M.A. and Park,M.S. (1997) The DNA repair endonuclease XPG binds to proliferating cell nuclear antigen (PCNA) and shares sequence elements with the PCNA-binding regions of FEN-1 and cyclin-dependent kinase inhibitor p21. J. Biol. Chem., 272, 24522–24529. [DOI] [PubMed] [Google Scholar]
- 18.Gary R., Kim,K., Cornelius,H.L., Park,M.S. and Matsumoto,Y. (1999) Proliferating cell nuclear antigen facilitates excision in long-patch base excision repair. J. Biol. Chem., 274, 4354–4363. [DOI] [PubMed] [Google Scholar]
- 19.Umar A., Buermeyer,A.B., Simon,J.A., Thomas,D.C., Clark,A.B., Liskay,R.M. and Kunkel,T.A. (1996) Requirement for PCNA in DNA mismatch repair at a step preceding DNA resynthesis. Cell, 87, 65–73. [DOI] [PubMed] [Google Scholar]
- 20.Tsurimoto T. (1999) PCNA binding proteins. Front. Biosci., 4, D849–D858. [DOI] [PubMed] [Google Scholar]
- 21.Miura M., Domon,M., Sasaki,T. and Takasaki,Y. (1992) Induction of proliferating cell nuclear antigen (PCNA) complex formation in quiescent fibroblasts from a xeroderma pigmentosum patient. J. Cell. Physiol., 150, 370–376. [DOI] [PubMed] [Google Scholar]
- 22.Aboussekhra A. and Wood,R.D. (1995) Detection of nucleotide excision repair incisions in human fibroblasts by immunostaining for PCNA. Exp. Cell. Res., 221, 326–332. [DOI] [PubMed] [Google Scholar]
- 23.Balajee A.S., May,A. and Bohr,V.A. (1998) Fine structural analysis of DNA repair in mammalian cells. Mutat. Res., 404, 3–11. [DOI] [PubMed] [Google Scholar]
- 24.Miura M., Sasaki,T. and Takasaki,Y. (1996) Characterization of X-ray-induced immunostaining of proliferating cell nuclear antigen in human diploid fibroblasts. Radiat. Res., 145, 75–80. [PubMed] [Google Scholar]
- 25.Savio M., Stivala,L.A., Bianchi,L., Vannini,V. and Prosperi,E. (1998) Involvement of the proliferating cell nuclear antigen (PCNA) in DNA repair induced by alkylating agents and oxidative damage in human fibroblasts. Carcinogenesis, 19, 591–596. [DOI] [PubMed] [Google Scholar]
- 26.Balajee A.S., Dianova,I. and Bohr,V.A. (1999) Oxidative damage-induced PCNA complex formation is efficient in xeroderma pigmentosum group A but reduced in Cockayne syndrome group B cells. Nucleic Acids Res., 27, 4476–4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miura M., Nakamura,S., Sasaki,T., Takasaki,Y., Shiomi,T. and Yamaizumi,M. (1996) Roles of XPG and XPF/ERCC1 endonucleases in UV-induced immunostaining of PCNA in fibroblasts. Exp. Cell Res., 226, 126–132. [DOI] [PubMed] [Google Scholar]
- 28.McAlear M.A., Howell,E.A., Espenshade,K.K. and Holm,C. (1994) Proliferating cell nuclear antigen (pol30) mutations suppress cdc44 mutations and identify potential regions of interaction between the two encoded proteins. Mol. Cell. Biol., 14, 4390–4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ayyagari R., Impellizzeri,K.J., Yoder,B.L., Gary,S.L. and Burgers,P.M. (1995) A mutational analysis of the yeast proliferating cell nuclear antigen indicates distinct roles in DNA replication and DNA repair. Mol. Cell. Biol., 15, 4420–4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arroyo M.P. and Wang,T.S. (1998) Mutant PCNA alleles are associated with cdc phenotypes and sensitivity to DNA damage in fission yeast. Mol. Gen. Genet., 257, 505–518. [DOI] [PubMed] [Google Scholar]
- 31.Eissenberg J.C., Ayyagari,R., Gomes,X.V. and Burgers,P.M. (1997) Mutations in yeast proliferating cell nuclear antigen define distinct sites for interaction with DNA polymerase δ and DNA polymerase ɛ. Mol. Cell. Biol., 17, 6367–6378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prosperi E., Stivala,L.A., Sala,E., Scovassi,A.I. and Bianchi,L. (1993) Proliferating cell nuclear antigen complex formation induced by ultraviolet irradiation in human quiescent fibroblasts as detected by immunostaining and flow cytometry. Exp. Cell Res., 205, 320–325. [DOI] [PubMed] [Google Scholar]
- 33.Maruyama E. (1996) Biochemical characterization of mouse brain necdin. Biochem. J., 314, 895–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Angelis K.J., Dusinska,M. and Collins,A.R. (1999) Single cell gel electrophoresis: detection of DNA damage at different levels of sensitivity. Electrophoresis, 20, 2133–2138. [DOI] [PubMed] [Google Scholar]
- 35.Foray N., Monroco,C., Marples,B., Hendry,J.H., Fertil,B., Goodhead,D.T., Arlett,C.F. and Malaise,E.P. (1998) Repair of radiation-induced DNA double-strand breaks in human fibroblasts is consistent with a continuous spectrum of repair probability. Int. J. Radiat. Biol., 74, 551–560. [DOI] [PubMed] [Google Scholar]
- 36.Foray N., Arlett,C.F. and Malaise,E.P. (1999) Underestimation of the small residual damage when measuring DNA double-strand breaks (DSB): is the repair of radiation-induced DSB complete? Int. J. Radiat. Biol., 75, 1589–1595. [DOI] [PubMed] [Google Scholar]
- 37.Cheong N. and Iliakis,G. (1997) In vitro rejoining of double strand breaks induced in cellular DNA by bleomycin and restriction endonucleases. Int. J. Radiat. Biol., 71, 365–375. [DOI] [PubMed] [Google Scholar]
- 38.Xu J. and Morris,G.F. (1999) p53-mediated regulation of proliferating cell nuclear antigen expression in cells exposed to ionizing radiation. Mol. Cell. Biol., 19, 12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Balajee A.S., May,A., Dianova,I. and Bohr,V.A. (1998) Efficient PCNA complex formation is dependent upon both transcription coupled repair and genome overall repair. Mutat. Res., 409, 135–146. [DOI] [PubMed] [Google Scholar]
- 40.Li L., Peterson,C.A., Zhang,X. and Legerski,R.J. (2000) Requirement for PCNA and RPA in interstrand crosslink-induced DNA synthesis. Nucleic Acids Res., 28, 1424–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stucki M., Pascucci,B., Parlanti,E., Fortini,P., Wilson,S.H., Hubscher,U. and Dogliotti,E. (1998) Mammalian base excision repair by DNA polymerases δ and ɛ. Oncogene, 17, 835–843. [DOI] [PubMed] [Google Scholar]
- 42.Araujo S.J., Tirode,F., Coin,F., Pospiech,H., Syvaoja,J.E., Stucki,M., Hubscher,U., Egly,J.M. and Wood,R.D. (2000) Nucleotide excision repair of DNA with recombinant human proteins: definition of the minimal set of factors, active forms of TFIIH and modulation by CAK. Genes Dev., 14, 349–359. [PMC free article] [PubMed] [Google Scholar]
- 43.Moggs J.G., Grandi,P., Quivy,J.P., Jonsson,Z.O., Hubscher,U., Becker,P.B. and Almouzni,G. (2000) A CAF-1-PCNA-mediated chromatin assembly pathway triggered by sensing DNA damage. Mol. Cell. Biol., 20, 1206–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jonsson Z.O., Podust,V.N., Podust,L.M. and Hubscher,U. (1995) Tyrosine 114 is essential for the trimeric structure and the functional activities of human proliferating cell nuclear antigen. EMBO J., 14, 5745–5751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Whisnant-Hurst N. and Leadon,S.A. (1999) Induced repair of DNA double-strand breaks at the G1/S-phase border. Radiat. Res., 151, 257–262. [PubMed] [Google Scholar]
- 46.Budd M.E. and Campbell,J.L. (1997) The roles of the eukaryotic DNA polymerases in DNA repair synthesis. Mutat. Res., 384, 157–167. [DOI] [PubMed] [Google Scholar]
- 47.Wang Z., Wu,X. and Friedberg,E.C. (1993) DNA repair synthesis during base excision repair in vitro is catalyzed by DNA polymerase ɛ and is influenced by DNA polymerases α and δ in Saccharomyces cerevisiae. Mol. Cell. Biol., 13, 1051–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blank A., Kim,B. and Loeb,L.A. (1994) DNA polymerase δ is required for base excision repair of DNA methylation damage in Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA, 91, 9047–9051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Savitsky K., Sfez,S., Tagle,D.A., Ziv,Y., Sartiel,A., Collins,F.S., Shiloh,Y. and Rotman,G. (1995) The complete sequence of the coding region of the ATM gene reveals similarity to cell cycle regulators in different species. Hum. Mol. Genet., 4, 2025–2032. [DOI] [PubMed] [Google Scholar]
- 50.Savitsky K., Bar-Shira,A., Gilad,S., Rotman,G., Ziv,Y., Vanagaite,L., Tagle,D.A., Smith,S., Uziel,T., Sfez,S. et al. (1995) A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science, 268, 1749–1753. [DOI] [PubMed] [Google Scholar]
- 51.Banin S., Moyal,L., Shieh,S., Taya,Y., Anderson,C.W., Chessa,L., Smorodinsky,N.I., Prives,C., Reiss,Y., Shiloh,Y. and Ziv,Y. (1998) Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science, 281, 1674–1677. [DOI] [PubMed] [Google Scholar]
- 52.Canman C.E., Lim,D.S., Cimprich,K.A., Taya,Y., Tamai,K., Sakaguchi,K., Appella,E., Kastan,M.B. and Siliciano,J.D. (1998) Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science, 281, 1677–1679. [DOI] [PubMed] [Google Scholar]
- 53.Chen G., Yuan,S.S., Liu,W., Xu,Y., Trujillo,K., Song,B., Cong,F., Goff,S.P., Wu,Y., Arlinghaus,R., Baltimore,D., Gasser,P.J., Park,M.S., Sung,P. and Lee,E.Y. (1999) Radiation-induced assembly of Rad51 and Rad52 recombination complex requires ATM and c-Abl. J. Biol. Chem., 274, 12748–12752. [DOI] [PubMed] [Google Scholar]
- 54.Foray N., Priestley,A., Alsbeih,G., Badie,C., Capulas,E.P., Arlett,C.F. and Malaise,E.P. (1997) Hypersensitivity of ataxia telangiectasia fibroblasts to ionizing radiation is associated with a repair deficiency of DNA double-strand breaks. Int. J. Radiat. Biol., 72, 271–283. [DOI] [PubMed] [Google Scholar]
- 55.Maser R.S., Monsen,K.J., Nelms,B.E. and Petrini,J.H. (1997) hMre11 and hRad50 nuclear foci are induced during the normal cellular response to DNA double-strand breaks. Mol. Cell. Biol., 17, 6087–6096. [DOI] [PMC free article] [PubMed] [Google Scholar]