

SUMMARY
A prominent feature of the promoters of Bordetella pertussis fimbrial subunit genes fim2, fim3, and fimX is the presence of a “C-stretch”, a monotonic run of C residues. The C-stretch renders these genes capable of phase-variation, through spontaneous variations in its length. For each of these we determined the length of the C-stretch that gave maximal transcriptional activity, and found that the three optimized promoters align perfectly, with identical distances between conserved upstream sequences and the downstream −10 elements and transcriptional start sites. We also demonstrated, for Pfim3, that the conserved sequence corresponds to BvgA-binding sites. The more upstream of the two binding sites is predicted to be high affinity, by comparison to a functionally-derived consensus BvgA-binding sequence. The other binding site is a fairly poor match to this consensus, with 10 of 14 bp belonging to the C-stretch. Interestingly, the center of this downstream site of BvgA binding coincides exactly with the center of the expected typical location of a −35 sequence. However, the lack of a recognizable −35 element (CCCCCC vs. TTGACA), and the occupation of this site by BvgA~P suggest that activation of the fim promoters involves unusual interactions among BvgA, RNA polymerase, and promoter DNA.
Keywords: Bordetella, BvgA, sigma factor, fimbriae, phase-variation
INTRODUCTION
Like many bacterial pathogens, Bordetella pertussis, the causative agent of the human disease whooping cough, exhibits global regulation of genes involved in its pathogenic lifestyle (Stibitz, 2007). In B. pertussis, and the related species B. parapertussis and B. bronchiseptica, the bvgASR locus is the central player in virulence gene regulation. This locus embodies many features currently recognized as typical of bacterial virulence gene regulation systems. For one, it encodes a two-component system, consisting of the membrane sensor histidine-kinase BvgS and the phosphate-accepting response regulator BvgA, allowing for modulation of virulence potential in response to environmental cues. Although the actual environmental signals that modulate BvgS activity “in the wild” have not been identified, compounds such as MgSO4 or nicotinic acid have been used to study this phenomenon in the laboratory (Lacey, 1960; Scarlato and Rappuoli,1991). Expression of typical virulence genes, such as those encoding toxins and adhesins, is activated through the activities of BvgA and BvgS, while a separate set of genes, the vrgs, or Bvg-repressed genes, is repressed through the action of BvgR, itself encoded by a BvgA-activated gene. Although phosphorylated BvgA (BvgA~P) has been shown to be both necessary and sufficient for in vitro activation of several virulence genes (Steffen et al., 1996; Boucher et al., 1997; Kinnear et al. 1999; Merkel et al., 2003; Williams et al., 2005), the responses of individual promoters vary in terms of the levels of BvgA~P required for full activation (Scarlato et al., 1991; Steffen et al., 1996). Furthermore, in the case of the bipA gene, low to moderate levels activate expression while high levels repress expression (Williams et al., 2005). These individual responses can be understood in terms of promoter architecture, specifically, the number, affinity, and location, of BvgA-binding sites in the regions upstream of the core promoter elements.
Bordetella pertussis is capable of synthesizing two different serotypes of fimbriae, known as Fim2 and Fim3, that belong to the chaperone-usher family of fimbriae (Nuccio and Baumler, 2007). Three proteins, the usher, the chaperone, and an adhesin located at the fimbrial tip, are common to the biogenesis of both serotype fimbriae, and are encoded within the operon for filamentous hemagglutinin (Locht et al., 1992; Willems et al., 1994). However, the major fimbrial subunit genes, fim2, fim3, and a silent locus, fimX, are located at unlinked chromosomal locations (Livey et al., 1987; Mooi et al., 1990, Pedroni et al., 1988). Interestingly, fimbrial subunit genes are capable of switching expression off or on at frequencies several orders of magnitude higher than that predicted by random mutation. Ground-breaking work of Mooi and coworkers showed that this fimbrial phase-variation was due to changes in the DNA sequence of the promoters for the subunit genes; specifically, changes in the length of a long run of C residues (Willems et al., 1990). For example, in the fim3 promoter, a shift from on to off was associated with a change in the length of this “C-stretch” from 14 to 13 residues. The fimX promoter appeared to be silent due to degradation in the length of the C-stretch to only 7 residues, thus rendering it essentially incapable of returning spontaneously to an active state. Alignment of the promoter sequences of these three promoters identified a short region of conserved sequence upstream of the predicted RNA polymerase (RNAP) binding region and the C-stretch. The authors hypothesized that this represented a binding site for an as yet unidentified activator protein. Although it was known that the fim genes were ultimately regulated by the bvgAS locus, it was not known if this activation was direct, i.e. due to the binding of BvgA~P to fim promoters. Also not fully understood was the means by which addition or deletion of a single nucleotide base pair could so dramatically affect promoter activity.
In this study we addressed these and related questions in order to more completely understand fim promoter structure/function. Our findings indicate that BvgA~P binds directly to fim promoters and that it is sufficient for their transcriptional activation. Unexpectedly, they also reveal that the spatial relationship of BvgA~P and RNAP bound to these promoters is indicative of a novel configuration that implies a novel mechanism of transcriptional activation. We hypothesize that phase-variation is a consequence of the requirements for correct spacing and axial alignment of BvgA~P with respect to RNAP when bound to the promoter, requirements that are either met, or not met, depending on the length of the C-stretch. Interestingly, although DNA binding by BvgA~P and crucial interactions with RNAP appear to take place within the region demarcated by the C-stretch, specific DNA sequences required for their interaction do not appear to be encoded within it.
RESULTS
The optimal length of the C-stretch in fim promoters aligns conserved upstream sequences with −10 elements and transcriptional start sites
Previous studies examining the mechanism of fimbrial phase-variation detailed instances in which the switch, for a given fim gene, from fim expression “off” to fim expression “on” was due to a lengthening of the C-stretch from a transcription-nonpermissive to a transcription-permissive length (Willems et al., 1990). Thus, these studies defined the lower limit of this parameter. In order to determine if an upper limit to the permissible length of the C-stretch existed, and what the optimal length might be, length variants of the fim2, fim3, and fimX promoters were constructed and tested for activity in vivo. This was accomplished through the use of transcriptional gene fusions with the luxCDABE operon of Photorhabdus luminescens, which encodes a bacterial luciferase enzyme together with enzymes responsible for luciferase substrate synthesis. Light output from Bordetella pertussis strains harboring these fusions inserted into the chromosome in single-copy revealed that lengthening of the C-stretch beyond a certain point was detrimental in each case (Fig. 1A&B). Except in the case of fimX, a single optimal sequence was observed, while the activity of longer or shorter variants decreased with increasing difference in length from the optimum. For fimX, it was observed that, while the 17C variant was most active, and the 16C derivative was significantly reduced, the 15C derivative was increased relative to the 16C version. This pattern is consistent with the presence of an alternative −10 sequence that exists in the fimX promoter, offset by 2 bp, as shown in Fig. 1C. In the 15C derivative, the distance from upstream conserved sequences to this alternative −10 sequence is the same as to the native −10 region for the 17C variant. To verify whether the transcriptional activities seen in the promoter variants were BvgA dependent, we tested activities for all the promoters under modulating conditions, i.e. in the presence of 50mM MgSO4. Under these conditions the BvgAS system is not active. For all variants of Pfim2, Pfim3 and PfimX, the promoter activities were completely off (data not shown), suggesting that these promoters remain under the control of the BvgAS locus..
Fig. 1. fim promoter comparison. A&B) C-stretch optimization.

Variants of Pfim3, Pfim2, and PfimX that differed in the length of the C-stretch were constructed, cloned into the luxCDABE fusion vector pSS3967, and conjugated into B. pertussis strain BP536. Growth of the resulting chromosomal integrants and quantitation of light production is described in Materials and Methods. C) Alignment of optimal-length fim promoters. DNA sequences of fim promoters aligned by sequence identity, with identical nucleotides depicted by colons between juxtaposed sequences. Inverted arrows indicate the positions of BvgA-binding sites. Boxed regions correspond to the start of transcription (+1), the −10 region, and the expected position of a −35 element were one present (“−35 region”). Nucleotides in bold type within the boxed −10 regions correspond to those that match the consensus sequence TATAAT. Brackets indicate the positions of an extended −10 TG dinucleotide in the fim3 promoter, and an alternative −10 sequence in the fimX promoter. Numbers in parentheses above the inverted arrows indicate the predicted score for BvgA-binding half-sites in the fim3 promoter according to the algorithm represented in Fig. 2C. D) Transcription start site determination for fim3, fim2, and fimX promoters. Primer extension products and sequencing ladders are shown for each promoter. Sequencing ladders correspond to the sequence of the complementary (template) strand. In the fim2 and fimX panels, some of the sequence ladders (denoted by asterisk) were doped with primer extension product to unambiguously identify the starting nucleotide.
Fig. 1C presents a DNA sequence alignment of the optimal length variants for each of the three promoter sequences. From this it can be seen that the superficially disparate lengths of the C-stretches in the three different promoters actually correspond to identical spacing between the previously noted conserved sequences upstream (Willems et al., 1990), and core promoter features, such as predicted −10 regions and transcriptional start sites, downstream. Since accurate determination of a transcriptional start is crucial to elucidating promoter architecture, we determined the start sites by primer extension as shown in Fig. 1D. With the exception of Pfim3, in which our determination differed by one bp from that previously reported (Willems et al., 1990), the start sites were as expected. It should be noted that the fim3 start site we identified is in a more favorable position relative to the predicted −10 sequence and initiates with a more favorable nucleotide than that previously identified (reviewed in Hook-Barnard & Hinton, 2007).
Although each of the three C-stretches extends to a different position downstream, their upstream boundaries are identical. Interestingly, the sequence adjacent to, and just upstream of, the C-stretches is also identical. It should be noted that the expected position of a −35 sequence, i.e. separated by 17 bp from the −10 sequence, corresponds to the leftmost 6 bp of the C-stretch in all three promoters. These promoters therefore lack a properly positioned sequence with any similarity to the consensus −35 element, −35TTGACA-30, except for the C at position −31. Since the major features of these promoters are similar, we chose one promoter, Pfim3, for further detailed analyses.
Upstream conserved sequences correspond to BvgA-binding sites
To address whether BvgA-activation of fim promoters might be due to direct interaction, we sought to determine if BvgA~P was able to bind specifically to the fim3 promoter. Fragments containing Pfim3 were subjected to BvgA-binding analysis using FeBABE-BvgA, as previously reported for the fha and bipA promoters (Boucher et al., 2003, Williams et al., 2005). The V148C and T194C proteins used for FeBABE conjugation are two BvgA variants derived from cysteine-free BvgA (C93A and C103A double mutant), and are as active as wild type in transcriptional activation both in vivo and in vitro (Boucher et al. 2003). As shown in Fig. 2A, clear cleavage signals were obtained with BvgA in which FeBABE was conjugated to V148C, that cleaves at the outer boundaries of a DNA-bound BvgA dimer, as well as with the T194C-conjugated derivative, that cleaves near the dimer interface, as depicted in Fig. 2B. Minor cleavages produced by the V148C-conjugated derivative at approximately −36 and −58 on the template strand, and −28 and −50 on the non-template strand have been observed previously, but inconsistently, and are not taken to represent the true position of the V148C-conjugated FeBABE moiety (Boucher et al. 2003). The geometry of these residues relative to the predicted structure of the BvgA C-terminal domain bound to DNA is shown in Fig. 2B. These data clearly show that two head-to-head dimers of BvgA~P bind to the fim3 promoter. The spacing of the centers of binding for the two dimers, 22 bp, is typical for that observed at several other BvgA-regulated promoters, such as fha, bipA, ptx, cya, prn, bvgAS, and bvgR (Boucher et al., 2003; Williams et al., 2005; Boucher and Stibitz, unpublished results), and corresponds to approximately two helical turns, thus indicating that the BvgA molecules bind to the same face of the DNA helix.
Fig. 2. BvgA-FeBABE cleavage of fim3 promoter DNA.

A) Patterns resulting from cleavage by BvgA-FeBABE of DNA fragments containing the fim3 promoter. BvgA was derivatized with FeBABE at a cysteine residue, introduced by site-directed mutagenesis into the gene encoding BvgA C93A C103A, at either position 148, corresponding to the outer boundaries of a BvgA dimer, or at position 194, corresponding to the dimer interface (see Fig. 2B). Pfim3 promoter fragments were 32P-labeled at the 5′ end of the template strand (left) or the 3′ end of the non-template strand (right) as previously described (Boucher et al., 2003). The same fragments were used in the A+G sequencing reaction described by Maxam and Gilbert (1977) to allow determination of cleavage coordinates, numbered relative to the start of transcription. Closed and open arrowheads indicate the positions of maximum cleavage, for each FeBABE moiety, by BvgA derivatized with FeBABE at position 148, and 194, respectively. B) Predicted structure of the BvgA C-terminal domain bound to DNA, derived by modeling of the BvgA protein sequence on the crystal structure of the homologous NarL C-terminal domain bound to its operator site (Maris et al., 2002). Positions of the V148 and T194 residues substituted with cysteine for the purpose of FeBABE-labeling are indicated. Side and top views are provided. Figure from Boucher et al. (2003). C) Table representing an algorithm for assessing functionality of DNA sequences bound by BvgA. Overall penalty scores for a given seven bp sequence are derived by summing the individual penalty scores at each position. These penalty scores were derived from data collected through systematic mutagenesis of the primary binding site of the fha promoter (Boucher et al., 2001a). Original figure from Merkel et al. (2003). An optimal sequence, such as the fha primary binding site, scores 0 for both half sites. More negative scores indicate lower predicted affinity for BvgA~P. D) Schematic illustration of the positions of cleavage of each strand of fim3 promoter DNA by BvgA-FeBABE-V148 (filled arrow heads) and by BvgA-FeBABE-T194 (open arrow heads), as shown in panel A. Nucleotide coordinates are given relative to the transcription start site. Nucleotides in bold type represent the transcriptional start site as well as those within the −10 region that match the consensus −10 sequence −12 TATAAT - 7. Also depicted are the sites of cleavage by KMnO4 of the template strand in a tertiary complex with BvgA~P and RNAP. Thickness of these arrows corresponds to intensity of cleavage, as shown in Fig. 3B.
Interestingly, the positions of binding correspond, in part, to the upstream conserved sequences previously noted by Willems et al. (1990). The predicted strength of these binding sites can be evaluated using an algorithm, represented by the table in Fig. 2C. This table is based on systematic mutational studies of an isolated BvgA primary-binding site, tested in in vitro binding assays. Mutants were also tested in vivo, in the context of an fha promoter derivative, for their ability to mediate activation of transcription by BvgA (Boucher et al., 2001a). This algorithm accurately identified the positions of binding of BvgA at the bvgR and bipA promoters (data not shown), and was used successfully to analyze BvgA binding strength in these promoters (Merkel, et al. 2003; Williams, et al. 2005). Scores for the binding sites identified at the fim3 promoter by BvgA-FeBABE analysis are presented on the Pfim3 sequence (Fig. 1C), and indicate that the upstream conserved sequence noted previously corresponds to a nearly perfect BvgA-binding half-site. Its less-conserved partner half-site in the most upstream location scores only moderately well. Surprisingly, the downstream BvgA dimer binds largely within the C-stretch itself. The conserved four bp directly upstream of the C-stretch in each promoter, together with the first 10 bp of the C-stretch, constitute the binding site for this dimer, and its sequence is therefore nearly identical in the three promoters. As indicated in Fig. 1C, the algorithm-determined scores for the two fim3 half-sites in this, the secondary binding site, are relatively low, but it is worth noting that for each half-site the most crucial nucleotide, i.e. a C at position 4, is present. Of particular interest is the location of the secondary binding site relative to the core promoter elements. If the optimal, 17 bp, spacing between −10 and −35 sequences is invoked, the BvgA secondary binding site is centered at exactly the same position as the center of a typically-positioned 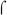 70-dependent −35 sequence.
70-dependent −35 sequence.
BvgA~P is necessary and sufficient for transcriptional activation of the fim3 promoter in vitro
To further address the role of BvgA in activation of fim promoters, we asked whether purified BvgA~P activated transcription of fim3 RNA in vitro. As shown in Fig. 3A, in the presence of a supercoiled template containing the fim3-15C promoter and E. coli RNAP, addition of BvgA~P resulted in transcription from the fim3 promoter in a dose-dependent manner. As expected, the BvgA~P-dependent fhaB promoter was also active under these conditions, while the fim3 promoter in the off configuration, here represented by the 13C version, retained only a basal level of transcription. In addition, transcription from the 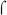 70-dependent promoters PRNAI (Morita & Oka, 1979), present on the plasmid DNAs, and PuvsX-sigma (Hinton & Vuthoori, 2000), added as a linear template, was observed in all lanes. In transcriptions containing the Pfha template, we also observed an RNA corresponding to the fha* promoter, in the absence of BvgA~P, and its disappearance, in the presence of BvgA~P. This result agrees with previously published findings of a BvgA-independent promoter (Pfha*) initiating at approximately 60bp upstream of the Pfha transcription start site (Boucher et al., 1997).
70-dependent promoters PRNAI (Morita & Oka, 1979), present on the plasmid DNAs, and PuvsX-sigma (Hinton & Vuthoori, 2000), added as a linear template, was observed in all lanes. In transcriptions containing the Pfha template, we also observed an RNA corresponding to the fha* promoter, in the absence of BvgA~P, and its disappearance, in the presence of BvgA~P. This result agrees with previously published findings of a BvgA-independent promoter (Pfha*) initiating at approximately 60bp upstream of the Pfha transcription start site (Boucher et al., 1997).
Fig. 3. BvgA~P activation of Pfim3 and Pfha and identification of Pfim3 transcription bubble in vitro.

A) Denaturing gels showing the products of in vitro transcription reactions, assembled as follows. Left panel: 0.25 pmol template DNA (0.2 pmol Pfha or Pfim3 plus 0.05 pmol PuvsX-sigma) was mixed with unphosphorylated or phosphorylated BvgA (0, 2, or 4 pmol) in 5.9 ul protein buffer I on ice. A solution containing 1.5 pmol of reconstituted RNAP in 4.1 ul protein buffer II was then added. Right panel: 0.1 pmol template DNA (0.05 pmol Pfha or Pfim3-15C plus 0.05 pmol Pre#) was mixed with phosphorylated BvgA (0, 1, 2pmol) in 5 ul protein buffer III on ice. A solution containing 0.75 pmol reconstituted RNAP in 5 ul protein buffer IV was then added. In each case, reactions were incubated for 15 min at 37° C, and transcription was initiated by adding 1 ul of NTP mix containing heparin to limit transcription to a single round. After incubation at 37° C for 10 min, reactions were collected on dry ice and treated as described in Materials and Methods. Sigma70-dependent promoters PRNAI (Morita & Oka, 1979), present on the supercoiled plasmid template, and Puvsx-sigma (Hinton & Vuthoori, 2000), added as a linear template, served as BvgA-independent controls. B) Denaturing gel showing the products of potassium permanganate footprinting of Pfim3 DNA that had been 32P-labeled at the 5′ end of the template (bottom) strand. The DNA was incubated with the indicated proteins with (lanes 1, 2) or without (lane 3) potassium permanganate or was used in an A+G sequencing reaction (lane 4) (Maxam & Gilbert, 1980) to allow determination of cleavage coordinates, numbered relative to the start of transcription.
E. coli 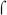 70 and B. pertussis
70 and B. pertussis 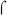 are highly similar in sequence (Gruber & Gross, 2003) and have been shown to be interchangeable in other in vitro transcription systems using promoters with and without activators (Boucher et al., 1997, Steffen & Ullmann, 1998, Baxter et al., 2006). The same result was observed here with BvgA~P activation of the fim3-15C and fha promoters (Fig. 3A, right), demonstrating that our system faithfully recapitulates BvgA~P activation with the B. pertussis
are highly similar in sequence (Gruber & Gross, 2003) and have been shown to be interchangeable in other in vitro transcription systems using promoters with and without activators (Boucher et al., 1997, Steffen & Ullmann, 1998, Baxter et al., 2006). The same result was observed here with BvgA~P activation of the fim3-15C and fha promoters (Fig. 3A, right), demonstrating that our system faithfully recapitulates BvgA~P activation with the B. pertussis 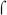 factor. As expected, transcription from the
factor. As expected, transcription from the 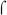 70-dependent promoters, Pre# (Kumar et al., 1993) and PRNAI was observed with polymerase reconstituted with either
70-dependent promoters, Pre# (Kumar et al., 1993) and PRNAI was observed with polymerase reconstituted with either  factor.
factor.
Having an in vitro system also allowed us to definitively and functionally identify the fim3 −10 element using permanganate footprinting. As shown in Fig. 3B, in the presence of BvgA~P, RNAP, and a DNA fragment containing the fim3-15C promoter, we observed pronounced cleavage signals for template (bottom) - strand thymidines at positions −5 and −11 and a less intense signal at position +2. These results are consistent with the upstream edge of the single-stranded “transcription bubble” lying at the predicted −11 position and agrees with the fim3 −10 sequence of −12TATTCT-7 (Fig. 2D).
Mutagenesis studies of the fim3 promoter C-stretch
It is intriguing that the C-stretch, lacking any clearly meaningful DNA sequence information, apparently plays host to crucial interactions between BvgA~P and RNAP. We therefore sought to examine the effect of introducing sequence information expected to enhance the interaction of either of these two proteins with the C-stretch DNA. To this end mutant derivatives of the fim3 promoter were constructed and their effect was assessed in vivo. The sequences of these derivatives are shown in Fig. 4A. Pfim3-mut1, harboring a nearly perfect −35 sequence at the appropriate location, displayed dramatically reduced activity in vivo. A minimal level of BvgA-independent activity tested was observed in Pfim3-mut1 under modulating conditions (data not shown) and in a bvgA-deletion strain (Figure 4B&C). Two other variants, in which known BvgA-binding sites from other BvgA-regulated promoters were introduced at the same location as the secondary binding site in Pfim3, were also constructed. One of these, Pfim3-mut2 harbors the primary binding site from the fhaB promoter, one of the strongest sites known, with each of the half-sites scoring a perfect 0 according to the algorithm in Fig. 2C. The other, Pfim3-mut4, harbors a binding site from the ptx promoter that is weaker, with each half-site scoring −5. For each of these derivatives a control promoter was constructed in which the fim3 primary binding site was destroyed by mutation of each base to a least productive alternative, according to the algorithm in Fig. 2C. These control promoters are Pfim3-mut3 and Pfim3-mut5. The in vivo expression from these promoters indicated that introduction of a strong BvgA-binding site, from Pfha, at the position of secondary binding, had a strong negative effect on Pfim3 activity. An only moderately strong binding site, from ptx, also inhibited activity, although to a lesser degree. In each case the remaining activity was dependent on the integrity of the upstream primary binding site, indicating its crucial role, even when the secondary binding site presumably had increased binding potential for BvgA~P.
Fig. 4. Effects of DNA sequence substitutions in the fim3 promoter.

A) The sequences of the fim3 wild type promoter and derivatives substituted with specific sequences at the secondary BvgA-binding site that are predicted to mediate interaction with region 4.2 of the sigma subunit of RNA polymerase (Pfim3-mut1) or increased binding of BvgA~P (Pfim3-mut2 – Pfim3-mut5). Pfim3-mut1 contains a near-consensus −35 region. Pfim3-mut2 and Pfim3-mut3 contain the primary binding site of the fha promoter in place of the secondary binding site of Pfim3. Pfim3-mut4 and Pfim3-mut5 contain a binding site from the ptx promoter in place of the secondary binding site of Pfim3. In Pfim3-mut3 and Pfim3-mut5, the primary binding site has been destroyed by substitution with a sequence of identical length in which every nucleotide has been changed to a least-productive alternative, according to the table in Fig. 2C. Constructs with Pfim3-15C and Pfim3-13C were included as positive and negative controls, respectively. B&C) Activities of wild type and substituted promoters were analyzed for their abilities to promote lux transcription in B. pertussis strains containing the wildtype bvg locus (BP536) or a bvgA deletion (BP1526). Intensity of light production after growth on BG agar is depicted in a false-color image (B) and by quantitative data derived from the analysis of several such images (C).
To more directly assess the potential DNA-sequence specific contribution of the C-stretch to fim3 promoter activity, site-directed mutagenesis studies were performed in which every bp in the 15 bp C-stretch was changed to A, G, or T. As shown in Fig. 5A, no single residue appeared crucial, as all substituted promoters had activity comparable to the wildtype. Even the one C residue for which an effect might plausibly be predicted, the −31 C, which is in the correct position to serve as the C in the −35 region consensus sequence (−35TTGACA-30), was not observed to be crucial, nor was any adjacent or nearby C residue.
Fig. 5. Site specific mutagenesis of the C-stretch of Pfim3-15C.

A) B. pertussis strains BP536 harboring chromosomally-integrated pSS3967 containing no insert (vector), negative control promoter (Pfim3-13C), positive control promoter (Pfim3-15C), or mutant derivatives of Pfim3-15C substituted at single residues in the C-stretch, were constructed and streaked in sectors on Bordet-Gengou agar. The three substitutions at each position were streaked in the same quadrant of each plate, with the position of the mutated C residue indicated (e.g. −33C). Following incubation, light production was visualized in the IVIS instrument, with false color rendering to indicate intensity. B) Graph showing distribution of activities of Pfim3-15C derivatives in which the C-stretch was substituted with random DNA sequence. Sixty independent, DNA-sequence- verified derivatives were assayed. The X-axis delineates a number of ranges of relative promoter activity (% of wild type) equal or more than the lower limit and less than the higher limit. The Y-axis is the % of population calculated by the number of mutants in each range against the total sixty mutants used in this assay.
To assess the potential existence of DNA-sequence-specific contributions of the C-stretch by a different experimental approach, a pool of mutant promoters was constructed in which the entire C-stretch was replaced by random DNA sequences. Individual variants from this pool were isolated at random, their fim3 promoter sequences were determined, and they were assessed for in vivo activity. Sixty such derivatives were examined. As shown in the distribution graph in Fig. 5B, ~12% of these variants (7 mutants) had activity that was greater than 50% of that of the wild type promoter. No sequence features, such as a discernible BvgA binding site, were apparent in the substituted region of these 7 mutants, although all remained BvgA-regulated as indicated by their down-regulation in the presence of MgSO4 (data not shown). Taken together these results indicate that the primary contribution of C-stretch DNA to promoter activity is to act as a spacer, and does not depend to a major degree on specific sequences contained within.
DISCUSSION
We report here a molecular genetic analysis of the fimbrial subunit promoters of Bordetella pertussis. Since these promoters represent additional examples of BvgA-mediated transcriptional activation, it is perhaps not surprising that BvgA interacts with these promoters in much the same way that it does at other BvgA-activated promoters that have been more intensively studied. For example, at the fhaB and bipA promoters, BvgA has been demonstrated to bind as a head-to-head dimer, with a periodicity of 22 bp, or two helical turns (Boucher et al, 2003, Williams et al., 2005). Thus the three dimers that bind at fhaB, and the two that bind at bipA, occupy one face of the DNA helix. We have shown here that two BvgA dimers bind to the fim3 promoter, and presumably the fim2 and fimX promoters, in a similar configuration.
Another aspect of promoter activation by BvgA that is echoed in the fim promoters is the theme of primary and secondary binding. For example, at fhaB and bipA, the most upstream binding position of BvgA corresponds to the presence of a high affinity site that is in perfect agreement with a consensus binding sequence derived from mutational studies (Boucher et al., 2001a). This is termed primary binding. Occupation of the adjacent downstream positions, i.e. secondary binding, is apparently assisted by cooperative dimer-dimer interactions with BvgA bound to this primary site. Studies on the fhaB promoter have indicated that, while the DNA sequence of the primary site is crucial, that of the secondary sites is much less so. The latter conclusion comes from the observation that substitution of the secondary binding region with random DNA sequences gave rise, in an unexpectedly high percentage of cases, to wild type levels of Bvg-regulated promoter activity (Boucher et al., 2001b). Interestingly, the natural example of the fim promoters demonstrates the same point, and in essentially the same way. In each of these promoters, rather than randomized sequences, introduced experimentally, the secondary binding region is composed primarily of the C-stretch, which is apparently devoid of sequence information that could direct the binding of BvgA or RNAP. The apparent lack of contribution of DNA sequence in this region is consistent with non-specific, lower affinity binding of BvgA~P. In fact, our accumulated experience suggests that secondary binding must be relatively weak in order for transcriptional activation to occur. For example, in the case of the fhaB promoter, multiple attempts to introduce higher affinity binding sites at the most promoter-proximal secondary binding position all failed to produce active promoters (Boucher and Stibitz, unpublished results). In the case of the fim3 promoter variants reported here, introduction of the fha high affinity BvgA-binding site in place of the secondary binding site that overlaps the C-stretch also severely reduced promoter activity, while a BvgA-binding site of lower affinity, from the ptx promoter, had a less severe effect. The reason for this requirement is unclear, but it may be that lower affinity of binding by BvgA~P molecules in this region provides them with greater flexibility, perhaps necessary to adopt a conformation or configuration that allows productive contacts to be made with RNAP. Alternatively, weaker interactions may be necessary to allow RNAP to escape the promoter and begin elongation (promoter clearance).
Sequence analysis reveals that all three fim promoters lack a consensus  70-dependent −35 sequence, yet they all display a −10 region that exhibits a good match to the canonical
70-dependent −35 sequence, yet they all display a −10 region that exhibits a good match to the canonical 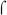 70-dependent −10 region, −12-TATAAT-7. In addition, Pfim3 contains the extended −10 sequence, TG at positions −15/−14. Previous work with E. coli
70-dependent −10 region, −12-TATAAT-7. In addition, Pfim3 contains the extended −10 sequence, TG at positions −15/−14. Previous work with E. coli 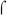 70-dependent extended −10 promoters has shown that this sequence, when present with a good −10 element, can confer independence from the requirement for a good −35 element (reviewed in (Hook-Barnard & Hinton, 2007)). However, in the absence of BvgA~P, Pfim3 is not active with RNAP in vivo and only slightly active in vitro (Fig. 3A, and data not shown). Thus, like many promoters that require activators, Pfim3, Pfim2, and PfimX have non-ideal sequences within the core promoter, insufficient by themselves to direct transcriptional initiation without assistance from the activator.
70-dependent extended −10 promoters has shown that this sequence, when present with a good −10 element, can confer independence from the requirement for a good −35 element (reviewed in (Hook-Barnard & Hinton, 2007)). However, in the absence of BvgA~P, Pfim3 is not active with RNAP in vivo and only slightly active in vitro (Fig. 3A, and data not shown). Thus, like many promoters that require activators, Pfim3, Pfim2, and PfimX have non-ideal sequences within the core promoter, insufficient by themselves to direct transcriptional initiation without assistance from the activator.
Despite these various similarities between the fim promoters and other promoters that require activation, the most striking aspect of BvgA-binding to the fim promoters is the atypical spatial relationship of BvgA-binding sites to the sequence motifs making up the core promoter elements with which RNAP is predicted to interact. At the fhaB and bipA promoters, the most downstream BvgA~P binding site abuts the predicted −35 region (Boucher et al., 2003, Williams et al., 2005). Such a configuration is typical of bacterial promoters regulated by class II activators (reviewed in (Barnard et al., 2004)), and should allow both BvgA~P and RNAP to occupy the promoter simultaneously. In fact, binding of BvgA~P at these promoters is apparently stimulated by RNAP binding, and vice versa (Boucher et al., 2001b). In contrast, the configuration at the fim promoters is strikingly different. The experimental determination of the transcriptional start sites, together with the presence, in each promoter, of an appropriately spaced −10 region, allows a prediction, with high confidence, of the position of RNAP binding. In fact, at the fim3 promoter, functionality of the predicted −10 region was confirmed by permanganate footprinting. Use of FeBABE-BvgA~P gives us an equally reliable indication of the positions of BvgA~P binding to the fim3, and by extension, the fim2 and fimX promoter. However, in the context of conventional paradigms for interactions between RNAP and transcriptional activator proteins, these two findings appear to be in conflict. The expected position of the −35 region is centered exactly on the center of binding of the downstream, secondary, BvgA~P dimer, i.e. at −32.5. Recognition of the −35 element by 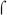 70 occurs in the major groove, mediated by a helix-turn-helix domain in
70 occurs in the major groove, mediated by a helix-turn-helix domain in 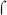 70 region 4.2 ((Campbell et al., 2002) and references therein), and each monomer within the BvgA~P dimer is also thought to bind via a helix-turn-helix motif in the major groove. Interestingly, −32.5 is within the C-stretch and thus neither interaction is expected to be stabilized by typical sequence-specific interactions with the promoter DNA in this region, but rather to be dependent on protein-protein and protein-DNA interactions outside the C-stretch. In the case of BvgA~P this involves presumed cooperative dimer-dimer interactions with a BvgA~P dimer bound to the upstream primary site via higher affinity, sequence-specific interactions, as well as the upstream 4 bp of the 14 bp secondary binding site. In the case of RNAP, we hypothesize that interactions with the downstream −10 region are crucial.
70 region 4.2 ((Campbell et al., 2002) and references therein), and each monomer within the BvgA~P dimer is also thought to bind via a helix-turn-helix motif in the major groove. Interestingly, −32.5 is within the C-stretch and thus neither interaction is expected to be stabilized by typical sequence-specific interactions with the promoter DNA in this region, but rather to be dependent on protein-protein and protein-DNA interactions outside the C-stretch. In the case of BvgA~P this involves presumed cooperative dimer-dimer interactions with a BvgA~P dimer bound to the upstream primary site via higher affinity, sequence-specific interactions, as well as the upstream 4 bp of the 14 bp secondary binding site. In the case of RNAP, we hypothesize that interactions with the downstream −10 region are crucial.
It appears then that BvgA~P and 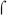 region 4 must either compete or cooperate in binding to the −35 region of the fim3 promoter DNA. One precedent for a competition mechanism is
region 4 must either compete or cooperate in binding to the −35 region of the fim3 promoter DNA. One precedent for a competition mechanism is 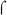 -appropriation at bacteriophage T4 middle promoters, in which the T4 transcriptional activator, MotA, binds to a site in the T4 middle promoters that overlaps the
-appropriation at bacteriophage T4 middle promoters, in which the T4 transcriptional activator, MotA, binds to a site in the T4 middle promoters that overlaps the 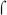 70-dependent −35 element (reviewed in (Hinton et al., 2005)). MotA activation depends on the action of another T4 protein, AsiA, which remodels
70-dependent −35 element (reviewed in (Hinton et al., 2005)). MotA activation depends on the action of another T4 protein, AsiA, which remodels 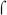 Region 4, preventing its interaction with the −35 DNA, but also allowing MotA to bind to the resulting σ-AsiA complex. BvgA~P activation at the fim3 promoter cannot be completely analogous to this situation since it does not require additional proteins to act as co-activators. Nonetheless, our results suggest that at the fim promoters, transcriptional activation by BvgA~P involves unusual interactions between RNAP, the activator, and the promoter DNA.
Region 4, preventing its interaction with the −35 DNA, but also allowing MotA to bind to the resulting σ-AsiA complex. BvgA~P activation at the fim3 promoter cannot be completely analogous to this situation since it does not require additional proteins to act as co-activators. Nonetheless, our results suggest that at the fim promoters, transcriptional activation by BvgA~P involves unusual interactions between RNAP, the activator, and the promoter DNA.
The aspect of the fim promoters that originally attracted the most interest, the C-stretch, with its clearly demonstrated involvement in phase-variation, can now be understood in somewhat more molecular detail. We now see that BvgA~P binds specifically to these promoters, and is responsible for activating transcription. This appears to occur by a mechanism that is perhaps novel but incompletely understood at this point. However, it is expected, and experimental results indicate, that the tertiary complex formed via this mechanism is sensitive to the relative spatial positioning of BvgA~P and RNAP bound to the promoter. Alteration of the length of the C-stretch alters this crucial parameter with the result that a change from a permissive to a nonpermissive state, or vice versa, can occur by the insertion or deletion of a single residue. The nature of the DNA helix dictates that changing the length of a spacer region between two bound moieties alters their relative spatial relationship in two ways: by the linear distance between them, and by their relative angles with respect to the axis of the DNA helix. Apart from determining this crucial spacing between bound BvgA~P and RNAP, the C-stretch appears to play a relatively passive role in the mechanisms of promoter activation. Our mutagenesis studies have shown that no one specific C residue is crucial for Bvg-regulated promoter activity. Furthermore, a surprisingly high percentage of random DNA sequences of the same length can serve the same function. On the other hand, the unique and clearly non-random nature of the C-stretch suggests that this structure has evolved to fill a specific purpose. We are therefore left to conclude that the primary role of the C-stretch is to provide a mechanism for phase-variation, as originally observed by Willems et al, (1980). The ability to switch between fimbriated and non-fimbriated phenotypes, or to change the immunological profile of these surface-exposed structures, presumably provides a survival advantage to Bordetellae as they vie against mammalian host defenses. An interesting question that remains is whether the unique mode of BvgA-RNAP interaction implied by this unique promoter architecture is also related to the phenomenon of phase-variation.
EXPERIMENTAL PROCEDURES
Bacterial strains and media
Bacterial strains and plasmids used in this study are shown in Table 1. Escherichia coli strains were grown on Luria-Bertani (LB) agar or in LB broth (Miller, 1972). Bordetella pertussis strains were grown on Bordet Gengou (BG) agar containing 15% defibrinated sheep blood and supplemented with 1% proteose peptone (Becton, Dickinson and Company). To repress the activity of BvgAS, modulating conditions were achieved by the inclusion of 50 mM MgSO4. Unless otherwise indicated, antibiotics (Sigma) were used at the following concentrations in LB agar: ampicillin, 200 g/ml; gentamicin sulfate, 5
g/ml; gentamicin sulfate, 5  g/ml; kanamycin sulfate, 10
g/ml; kanamycin sulfate, 10  g/ml. Concentrations used in BG agar were: gentamicin sulfate, 10
g/ml. Concentrations used in BG agar were: gentamicin sulfate, 10  g/ml; streptomycin sulfate, 100
g/ml; streptomycin sulfate, 100  g/ml.
g/ml.
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant features | Source or reference |
|---|---|---|
| E. coli | ||
| DH5⟨ | High-efficiency transformation | Bethesda Research Laboratories |
| SM10 | Conjugation proficient | Simon et al. (1983) |
| B. pertussis | ||
| Tohama I | Patient isolate | Kasuga et al. (1954) |
| BP536 | StrR, NalR Tohama I | Stibitz and Yang (1991) |
| BP953 | BP536, fha-lacZ, ptx-phoA | Stibitz (1994) |
| BP1526 | BP953, ΔbvgA–1215 | This study |
| Plasm ids | ||
| pSS3110 | lacZYA promoter assay vector | Veal-Carr and Stibitz (2005) |
| pSS3967 | luxCDABE promoter assay vector | This study |
| pQC1025 | pSS3967::Pfim2* | This study |
| pSS4159 | pSS3967::Pfim3** | This study |
| pQC1030_ | pSS3967::PfimX*** | This study |
| pPuvsX-sigma | T4 uvsX promoter transcription vector | Hinton and Vuthoori (2000) |
| pPre# | PPre# transcription vector | This study |
| pTE103 | in vitro transcription vector | Elliott and Geiduscheck (1984) |
| pfha | pTE103::Pfha | Boucher et al. (1997) |
| pPfim3-13C | pTE103::Pfim3-13C | This study |
| pPfim3-15C | pTE103::Pfim3-15C | This study |
| pETsigmaBp |
B. pertussis 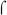 subunit expression subunit expression |
This study |
cloned fim2 fragment contains nucleotides 1166483 to 1176644 of the B. pertussis Tohama I genome and extends, relative to the start of transcription, from −130 to +32.
cloned fim3 fragment contains nucleotides 1647430 to 1647592 of the B. pertussis Tohama I genome and extends, relative to the start of transcription, from −130 to +33.
cloned fimX fragment contains nucleotides 2839614-2839776 of the B. pertussis Tohama I genome and extends, relative to the start of transcription, from −123 to +40.
DNA, cloning, mutagenesis, and strain construction
For determination of promoter activity in vivo we constructed the plasmid vector pSS3967. This plasmid is a derivative of pSS3110 in which the lacZYA operon of E. coli has been replaced by the luxCDABE operon of Photorhabdus luminescens. Like pSS3110, pSS3967 contains the oriT of plasmid R6K and can therefore be transferred from appropriate E. coli donor strains to B. pertussis recipients via conjugation. Also like pSS3110, pSS3967 is incapable of replication in B. pertussis and therefore selection for its maintenance (by gentamicin resistance) allows the isolation of B. pertussis exconjugants in which this plasmid has inserted by homologous recombination at a specific location (Veal-Carr and Stibitz, 2005). Promoters to be assayed can be cloned between the EcoRI and SalI restriction sites located upstream of the luxCDABE operon, and downstream of tandem transcriptional terminators. In this way PCR fragments containing the fim2, fim3, or fimX promoters, flanked by an upstream EcoRI site and a downstream SalI site, were cloned into pSS3967 to create pQC1069, pSS4228, and pQC1058, respectively (Table 1). Derivatives of these promoters and plasmids were created using site-directed mutagenesis methods that take advantage of the characteristics of the type IIS restriction enzyme BsaI, as described by Stemmer and Morris (Stemmer and Morris, 1992), although inverse PCR was not performed. These DNA cloning procedures used E. coli DH5⟨ as an initial transformation host. Following verification by DNA sequencing, pSS3967 derivatives were transformed into SM10 to permit conjugation into BP536 or BP1526. BP1526 was derived by the introduction of a deletion of the bvgA gene, from the EcoRI site upstream to the SphI site downstream into BP953 (Stibitz, 1994). Matings were performed by swabbing the E. coli donor strain together with the B. pertussis recipient strain on BG agar plus 10mM MgCl2. After incubation at 37° for 3 hours, bacteria were recovered by swabbing and re-swabbed onto BG agar plus gentamicin and streptomycin. Exconjugants arose after 3-4 days incubation at 37°.
An expression vector for B. pertussis sigmaA, pETsigmaBp, was constructed as follows. B. pertussis chromosomal DNA was isolated from cells using the MasterPure kit (Epicentre Biotechnologies). The rpoD gene was PCR-amplified from the chromosomal DNA template with oligos containing either an NdeI site (upstream of coding region) or EcoRI site (downstream of coding region). The resulting fragment was digested with NdeI and EcoRI and then ligated into pET28a(+) (Novagen) that had been digested with the same enzymes, to generate a plasmid that expressed His6-tagged sigmaBp protein..
Templates for in vitro transcription were as follows. pPuvsx-sigma (Hinton and Vuthoori, 2000) and pfha (Boucher et al., 1997) have been described previously. Plasmids pPfim3-15C and pPfim3-13C were constructed similarly to pfha. PCR fragments containing the fim3 promoter 13C and 15C derivatives, each flanked by a BamHI site upstream of position −130, and a SalI site downstream of the +33 position, were cloned between the BamHI and SalI sites of pTE103 (Elliott and Geiduscheck, 1984). Transcription of these plasmids in vitro yields transcripts 260 nucleotides in length. The transcription template plasmid pPre# was constructed by inserting oligonucleotides encoding the Pre# promoter sequence from −55 to +5 (Kumar et al., 1993) and flanked by restriction sites for BamHI and EcoRI into similarly-digested pDKT90 (March-Amegdazie & Hinton, 1995). Digestion of the resulting plasmid with NaeI generated a linear template that yields a Pre# RNA of 296 nucleotides in length.
Visualization and quantitation of luciferase activity
Light production by B. pertussis strains streaked in sectors on BG plus gentamicin agar plates, and incubated for 48 hours at 37°C was revealed by imaging with the refrigerated CCD camera of an IVIS-50 instrument (Caliper Life Sciences). All such images presented in this study were adjusted to have the same range and scale for false-color visualization of light intensity. For quantitative determination, the total flux (photons/second) of a circular region of interest (ROI) 0.5 cm diameter in the middle region of each sector was derived using Living Image software v2.6.1 (Caliper Life Sciences) running in Igor Pro Carbon (WaveMetrics). Absolute levels of light production can vary significantly from assay to assay, however we have found it to be a very reliable measure of the relative light production of strains streaked and incubated as sectors on the same plate. For this reason, data from each strain was expressed as a percentage relative to the most luminescent strain on a given plate. Each assay was performed a minimum of three times, on different days, and from independent streaks, and the results are presented as the mean, with error bars indicating the standard deviation.
Primer extension
B. pertussis BP536 harboring chromosomally integrated plasmids pQC1069, pSS4228, and pQC1058, were grown for 2 days on Bordet Gengou agar with gentamicin. These strains contain the luxCDABE operon driven by the fim2, fim3, or fimX, promoter, respectively. RNA was isolated by Method II described in Hinton, 1989. Primer extension by AMV reverse transcriptase (Life Sciences, Inc) was performed using a protocol based on that of Guild et al., 1988. The priming oligonucleotide was oligonucleotide 1054 (5′-GAGAGTCATTCAATATTGGCAGGTA-3′), which had been labeled with 32P at the 5′ end using the Optikinase Kit from USB, and which primes within the luxB gene that is fused to each promoter in these strains. The same labeled primer was used for dideoxy-sequencing (Sanger et al., 1977), using cognate plasmid or PCR-generated templates, to generate sequencing ladders that were electrophoresed alongside primer extension reactions to allow precise location of the initiation nucleotide. In some cases, sequencing ladders were doped with primer extension product to allow unambiguous identification. Electrophoresis was performed on a 7M urea/5% acrylamide sequencing gel, and gels were autoradiographed at −80°C.
Proteins
E. coli RNAP core was purchased from Epicentre Technologies.  70 was purified as described (Gerber and Hinton, 1996). His6-tagged B. pertussis
70 was purified as described (Gerber and Hinton, 1996). His6-tagged B. pertussis  was purified as described for His6-tagged
was purified as described for His6-tagged  70 (Vuthoori et al., 2001) from BL21(DE3)[pLysE] cultures (Studier, 1991) containing pETsigmaBp that had been grown at 37°C in LB broth supplemented with 50 μg/ml kanamycin and 25 μg/ml chloramphenicol.
70 (Vuthoori et al., 2001) from BL21(DE3)[pLysE] cultures (Studier, 1991) containing pETsigmaBp that had been grown at 37°C in LB broth supplemented with 50 μg/ml kanamycin and 25 μg/ml chloramphenicol.
BvgA used in in vitro transcription was purified following the method of Boucher et al. (1997). BL21(DE3)/pLysE/pBvgA cells were grown to mid-log phase at 37°C in LB broth supplemented with 100ug/ml of ampicillin and 25 ug/ml of chloramphenicol. After addition of isopropyl-B-D-thiogalactopyranoside (IPTG) to 1 mM, cells were grown for 3 additional hr and then harvested by centrifugation at 15,300 × g for 10 min. The cell pellet was washed with cold 1X PBS, then lysed by sonication in lysis buffer pH 7.5 (40 mM HEPES-NaOH, 50 mM KCl, 5 mM EDTA, 1 mM DTT, 1 mM phenylmethylsulfonyl fluoride, 0.25 mg/ml lysozyme). After centrifugation, the pellet was washed in 20 mM HEPES (pH 7.5), 0.5 M NaCl, 5 mM EDTA, 1 mM DTT, 0.1% TritonX−100, 0.25mg/ml lysozyme and then washed in 20 mM HEPES (pH 7.5) 1 mM EDTA, 1 mM DTT. The pellet was resuspended in 6 mL of column buffer (50 mM imidazole (pH 6.8), 8 M de-ionized urea, 1 mM DTT), and insoluble material was removed by centrifugation at 12,100 × g for 30 min. The supernatant was loaded onto a 20-mL Hi-Prep 16/10 Q Sepharose Fast Flow (GE Healthcare) column pre-equilibrated with column buffer. The flowthrough fraction containing purified BvgA was collected and dialyzed against 10 mM HEPES (pH 7.4), 1 mM EDTA, 0.1 mM DTT. The white precipitate that formed was collected by centrifugation at 10,000 × g for 10 min. This precipitate was resuspended to a final concentration of 0.15 mg/ml in 50 mM Na2HPO4 (pH 6.8), 6 M guanidinium-HCl, 2 mM MgCl2, 1 mM DTT. Insoluble material was removed by centrifugation at 16,000 × g for 30 min. The sample was then dialyzed against 2 changes of storage buffer 1 (20 mM HEPES (pH 7.4), 0.3 M KCl, 10 mM MgCl2, 1 mM DTT, 45% glycerol) then 2 changes of storage buffer 2 (20 mM HEPES (pH 7.4), 50 mM KCl, 1 mM MgCl2, 0.1 mM DTT, 45% glycerol) over 48 hr at 4°C. Aliquots of BvgA in this buffer (1.3 pmol/ul) were stored at −20°C. The purity of BvgA in our preparation was confirmed by SDS-PAGE.
BvgA-FeBABE cleavage
BvgA-C93A,C103A,V148C, and BvgA-C93A,C103A,T194C proteins were purified, covalently labeled with Fe(III) (S)-1-(p-Bromoacetamido-benzyl) ethylene diamine tetra-acetic acid (Fe-BABE), and used to reveal binding to the fim3 promoter essentially as previously described for the fha promoter (Boucher et al., 2003). Cleavage was performed in the presence of RNAP. FeBABE was obtained from Dojindo Corporation, Japan.
Potassium permanganate footprinting
DNA-protein complexes were treated with potassium permanganate following the method of Sasse-Dwight & Gralla (1989). A 248 bp 32P end-labeled Pfim3 DNA fragment, extending from −138 to +111, was made by PCR with pPfim3-15C as a template and primers that annealed upstream and downstream, with the latter 32P-labeled using the Optikinase kit from USB. The labeled fragment (0.025 pmol) was incubated for 10 minutes at 37° C in a solution (10ul) containing 51.8 mM Tris, 37.3 mM NaCl, 20 mM acetyl phosphate, 7.7 mM KCl, 3.08 mM HEPES, 3.0 mM MgOAc, 2.0 mM CaCl2, 1.54 mM MgCl2, 0.11 mM EDTA, 12% glycerol, 25 ug/ml BSA, and, as indicated, 2.0 pmol of phosphorylated BvgA and 0.75 pmol of RNA polymerase. Potassium permanganate (0.5 ul of 50 mM solution) was added, and the mixture was incubated for 2.5 minutes at 37° C. After the addition of 5 ul stop solution (1.5 M sodium acetate, 1.0 M 2-mercaptoethanol, 100 ug/ml tRNA), the DNA was precipitated with ethanol and dried. DNA pellets were resuspended in 0.1 M piperidine (100 ul) and incubated at 90° C for 30 minutes. After precipitation with butanol, DNA products were separated on 8% polyacrylamide, 7M urea denaturing gels.
In vitro transcription
BvgA (1.3 pmol/ul) was phosphorylated by incubating the protein with a solution of 20 mM acetyl phosphate, 20 mM Tris-HCl pH 8 for 30 min at room temperature. Non-phosphorylated BvgA was treated similarly, but without acetyl phosphate. Transcription reactions were assembled as described in the Figure legend. Once reactions were collected, 25 ul gel loading solution (TBE, 7 M urea, 0.1% (w/v) bromophenol blue, 0.1% (w/v) xylene cyanol FF) were added, and the samples were heated at 95°C for two minutes prior to electrophoresis on 7 M urea, 4% polyacrylamide denaturing gels run in 0.5 × TBE. After autoradiography, films were scanned on a Powerlook 2100XL densitometer.
Transcription buffers contained the following. Protein Buffer I: 45.8 mM Tris-Cl pH 8, 42.4 mM NaCl, 11.8 mM KCl, 4.72 mM HEPES-NaOH, 2.55 mM magnesium acetate, 0.24 mM MgCl2, 0.11 mM DTT, 0.08 mM EDTA, 21.2 ug/mL BSA, 10.6% glycerol, +/− 34 mM acetyl phosphate for phosphorylated or non-phosphorylated BvgA reactions, respectively. Protein Buffer II: 72 mM Tris-Cl pH 8, 148.8 mM NaCl, 3.65 mM magnesium acetate, 0.15 mM EDTA, 0.34 mM DTT, 30.4 ug/mL BSA, 58% glycerol. Protein Buffer III: 62 mM Tris-Cl pH 8, 48 mM acetyl phosphate, 20 mM KCl, 8 mM HEPES-NaOH pH 7.4, 4.0 mM MgCl2, 3.0 mM MgOAc, 0.5 mM DTT, 0.1 mM EDTA, 25 mg/ml BSA, 20% glycerol. Protein Buffer IV: 63 mM Tris-Cl pH 8, 22 mM NaCL, 3.0 mM MgOAc, 0.3 mM EDTA, 0.17 mM DTT, 25 mg/ml BSA, 13% glycerol. NTP mix was 2.5 mM each ATP, GTP, CTP; 0.25 mM UTP at a specific activity of 4.4×103 dpm/pmol; and 2 mg/ml heparin.
ACKNOWLEDGEMENTS
The authors would like to thank M. Schmitt, C. Jones, R. Bonocora, L.Knipling, T. James, and M. Hsieh for helpful discussions. K. Decker is a graduate student in the Graduate Partnership Program, Johns Hopkins University-National Institutes of Health. This research was supported in part by the Intramural Research Program of the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases.
REFERENCES
- Barnard A, Wolfe A, Busby S. Regulation at complex bacterial promoters: how bacteria use different promoter organizations to produce different regulatory outcomes. Curr. Opin. Microbiol. 2004;7:102–108. doi: 10.1016/j.mib.2004.02.011. [DOI] [PubMed] [Google Scholar]
- Baxter K, Lee J, Minakhin L, Severinov K, Hinton DM. Mutational analysis of sigma70 region 4 needed for appropriation by the bacteriophage T4 transcription factors AsiA and MotA. J. Mol. Biol. 2006;363:931–944. doi: 10.1016/j.jmb.2006.08.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher PE, Murakami K, Ishihama A, Stibitz S. Nature of DNA binding and RNA polymerase interaction of the Bordetella pertussis BvgA transcriptional activator at the fha promoter. J. Bacteriol. 1997;179:1755–1763. doi: 10.1128/jb.179.5.1755-1763.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher PE, Yang M-S, Stibitz S. Mutational analysis of the high-afinity binding site in the fha promoter of Bordetella pertussis. Mol. Microbiol. 2001a;40:991–999. doi: 10.1046/j.1365-2958.2001.02442.x. [DOI] [PubMed] [Google Scholar]
- Boucher PE, Yang M-S, Schmidt DM, Stibitz S. Genetic and biochemical analyses of BvgA interaction with the secondary binding region of the fha promoter of Bordetella pertussis. J. Bacteriol. 2001b;183:536–544. doi: 10.1128/JB.183.2.536-544.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher PE, Maris AE, Yang M-S, Stibitz S. The response regulator BvgA and RNA polymerase alpha subunit C-terminal domain bind simultaneously to different faces of the same segment of promoter DNA. Mol. Cell. 2003;11:163–173. doi: 10.1016/s1097-2765(03)00007-8. [DOI] [PubMed] [Google Scholar]
- Campbell EA, Muzzin O, Chlenov JL, Sun JL, Olson CA, Weoinman O, Trester-Zedlitz ML, Darst SA. Structure of the bacterial RNA polymerase promoter specificity sigma subunit. Mol. Cell. 2002;9:527–539. doi: 10.1016/s1097-2765(02)00470-7. [DOI] [PubMed] [Google Scholar]
- Elliott T, Geiduscheck EP. Defining a bacteriophage T4 late promoter: absence of a “−35” region. Cell. 1984;36:211–219. doi: 10.1016/0092-8674(84)90091-6. [DOI] [PubMed] [Google Scholar]
- Gerber JS, Hinton DM. An N-terminal muation in the bacteriophage T4 motA gene yields a protein that binds DNA but is defective for activation of transcription. J. Bacteriol. 1996;178:6133–6139. doi: 10.1128/jb.178.21.6133-6139.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber TM, Gross CA. Multiple sigma subunits and the partitioning of bacterial transcription space. Ann. Rev. Microbiol. 2003;57:441–466. doi: 10.1146/annurev.micro.57.030502.090913. [DOI] [PubMed] [Google Scholar]
- Guild N, Gayle M, Sweeney R, Hollingsworth T, Modeer L, Gold L. Transcriptional activation of bacteriophage T4 middle promoters by the MotA protein. J. Mol. Biol. 1988;199:241–258. doi: 10.1016/0022-2836(88)90311-7. [DOI] [PubMed] [Google Scholar]
- Hinton DM. Transcript analyses of the uvsX-40-41 region of bacteriophage T4. Changes in the RNA as infection proceeds. J. Biol. Chem. 1989;264:14432–14439. [PubMed] [Google Scholar]
- Hinton DM, Vuthoori S. Efficient inhibition of Escherichia coli TNA polymerase by the bacteriophage T4 AsiA protein requires that AsiA binds first to free sigma70. J. Mol. Biol. 2000;304:731–739. doi: 10.1006/jmbi.2000.4113. [DOI] [PubMed] [Google Scholar]
- Hinton DM, Pande S, Wais N, Johnson XB, Vuthoori M, Makela A, Hook-Barnard I. Transcription takeover by sigma appropriation: remodeling of the sigma70 subunit of Escherichia coli RNA polymerase by the bacteriophage T4 activator MotA and co-activator AsiA. Microbiology. 2005;151:1729–1740. doi: 10.1099/mic.0.27972-0. [DOI] [PubMed] [Google Scholar]
- Hook-Barnard IG, Hinton DM. Transcription initiation by mix and match elements: flexibility for polymerase binding to bacterial promoters. Gene Regulation and Systems Biology. 2007:275–293. http://la-press.com/article.php?article_id=481. [PMC free article] [PubMed] [Google Scholar]
- Kasuga T, Nakase Y, Ukishima K, Takatsu K. Studies on Haemophilus pertussis. V. Relation between the phase of bacilli and the progress of the whooping-cough. Kitasato Arch. Exp. Med. 1954;27:57–62. [PubMed] [Google Scholar]
- Kinnear SM, Boucher PE, Stibitz S, Carbonetti NH. Analysis of BvgA activation of the pertactin gene promoter in Bordetella pertussis. J. Bacteriol. 1999;181:5234–5241. doi: 10.1128/jb.181.17.5234-5241.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Malloch RA, Fujita N, Smillie DA, Ishihama A, Hayward RS. The minus 35-recognition region of Escherichia coli sigma 70 is inessential for initiation of transcription at an “extended minus 10” promoter. J. Mol. Biol. 1993;232:406–418. doi: 10.1006/jmbi.1993.1400. [DOI] [PubMed] [Google Scholar]
- Livey I, Duggleby CJ, Robinson A. Cloning and nucleotide sequence analysis of the serotype 2 fimbrial subunit gene of Bordetella pertussis. Mol. Microbiol. 1987;1:203–209. doi: 10.1111/j.1365-2958.1987.tb00513.x. [DOI] [PubMed] [Google Scholar]
- Locht C, Geoffroy MC, Renauld G. Common accessory genes for the Bordetella pertussis filamentous hemagglutinin and fimbriae share sequence similarities with the papC and papD gene families. EMBO J. 1992;11:3175–3183. doi: 10.1002/j.1460-2075.1992.tb05394.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- March-Amegadzie R, Hinton DM. The bacteriophage T4 middle promoter PuvsX: analysis of regions important for binding of the T4 transcriptional activator MotA and for activation of transcription. Mol. Microbiol. 1995;15:649–660. doi: 10.1111/j.1365-2958.1995.tb02374.x. [DOI] [PubMed] [Google Scholar]
- Maris AE, Sawaya MR, Kaczor-Grzeskowiak M, Jarvis MR, Brearson SM, Kopka ML, Schröder I, Gunsalus RP, Dickerson RE. Dimerization allows DNA target site recognition by the NarL response regulator. Nat. Struct. Biol. 2002;9:771–8. doi: 10.1038/nsb845. [DOI] [PubMed] [Google Scholar]
- Maxam AM, Gilbert W. A new method for sequencing DNA. Proc. Natl. Acad. Sci. USA. 1977;74:560–564. doi: 10.1073/pnas.74.2.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkel TJ, Boucher PE, Stibitz S, Grippe VK. Analysis of bvgR expression in Bordetella pertussis. J. Bacteriol. 2003;185:6902–6912. doi: 10.1128/JB.185.23.6902-6912.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JH. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1972. [Google Scholar]
- Mooi FR, ter Avest A, van der Heide HG. Structure of the Bordetella pertussis gene coding for the serotype 3 fimbrial subunit. FEMS Microbiol. Lett. 1990;54:327–331. doi: 10.1016/0378-1097(90)90307-c. [DOI] [PubMed] [Google Scholar]
- Morita M, Oka A. The structure of a transcriptional unit on colicin E1 plasmid. Eur. J. Biochem. 1979;97:435–443. doi: 10.1111/j.1432-1033.1979.tb13131.x. [DOI] [PubMed] [Google Scholar]
- Nuccio S-P, Bäumler AJ. Evolution of the chaperone/usher assembly pathway: fimbrial classification goes Greek. Microbiol. Mol. Biol. Rev. 2007;71:551–575. doi: 10.1128/MMBR.00014-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedroni P, Riboli B, de Ferra F, Grandi G, Toma S, Aricò B, Rappuoli R. Cloning of a novel pilin-like gene from Bordetella pertussis: homology to the fim2 gene. Mol. Microbiol. 1988;2:539–543. doi: 10.1111/j.1365-2958.1988.tb00061.x. [DOI] [PubMed] [Google Scholar]
- Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasse-Dwight S, Gralla JD. KMnO4 as a probe for lac promoter DNA melting and mechanism in vivo. J. Biol. Chem. 1989;264:8074–8081. [PubMed] [Google Scholar]
- Scarlato V, Arico B, Prugnola A, Rappuoli R. Sequential activation and environmental regulation of virulence genes in Bordetella pertussis. EMBO J. 1991;10:3971–3975. doi: 10.1002/j.1460-2075.1991.tb04967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon R, Priefer U, Puhler A. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram-negative bacteria. Bio/Technology. 1983;1:784–789. [Google Scholar]
- Steffen P, Goyard S, Ullmann A. Phosphorylated BvgA is sufficient for transcriptional activation of virulence-regulated genes in Bordetella pertussis. EMBO J. 1996;15:102–109. [PMC free article] [PubMed] [Google Scholar]
- Steffen P, Ullmann A. Hybrid Bordetella pertussis-Escherichia coli RNA polymerases: selectivity of promoter activation. J. Bacteriol. 1998;180:1567–1569. doi: 10.1128/jb.180.6.1567-1569.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmer WP, Morris SK. Enzymatic inverse PCR: a restriction site independent, single-fragment method for high-efficiency, site directed mutagenesis. Biotechniques. 1992;13:214–220. [PubMed] [Google Scholar]
- Stibitz S. The bvg regulon. In: Locht Camille., editor. Bordetella Molecular Microbiology. Horizon Bioscience; Norfolk, UK: 2007. pp. 47–67. [Google Scholar]
- Stibitz S, Yang M-S. Subcellular localization and immunological detection of proteins encoded by the vir locus of Bordetella pertussis. J. Bacteriol. 1991;173:4288–4296. doi: 10.1128/jb.173.14.4288-4296.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stibitz S. Mutations in the bvgA gene of Bordetella pertussis that differentially affect regulation of virulence determinants. J. Bacteriol. 1994;176:5615–5621. doi: 10.1128/jb.176.18.5615-5621.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier FW. Use of bacteriophage T7 lysozyme to improve an inducible T7 expression system. J. Mol. Biol. 1991;219:37–44. doi: 10.1016/0022-2836(91)90855-z. [DOI] [PubMed] [Google Scholar]
- Veal-Carr WL, Stibitz S. Demonstration of differential virulence gene promoter activation in vivo in Bordetella pertussis using RIVET. Mol. Microbiol. 2005;55:788–798. doi: 10.1111/j.1365-2958.2004.04418.x. [DOI] [PubMed] [Google Scholar]
- Vuthoori S, Bowers CW, McCracken A, Dombrowski AJ, Hinton DM. Domain 1.1 of the sigma (70) subunit of Escherichia coli RNA polymerase modulates the formation of stable polymerase/promoter complexes. J. Mol. Biol. 2001;309:561–572. doi: 10.1006/jmbi.2001.4690. [DOI] [PubMed] [Google Scholar]
- Willems R, Paul A, van der Heide HG, ter Avest AR, Mooi FR. Fimbrial phase variation in Bordetella pertussis: a novel mechanism for transcriptional regulation. EMBO J. 1990;9:2803–2809. doi: 10.1002/j.1460-2075.1990.tb07468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems RJ, Geuijen C, van der Heide HG, Renauld G, Bertin P, van den Akker WM, Locht C, Mooi FR. Mutational analysis of the Bordetella pertussis fim/fha gene cluster: identification of a gene with sequence similarities to haemolysin accessory genes involved in export of FHA. Mol. Microbiol. 1994;11:337–347. doi: 10.1111/j.1365-2958.1994.tb00314.x. [DOI] [PubMed] [Google Scholar]
- Williams CL, Boucher PE, Stibitz S, Cotter PA. BvgA functions as both an activator and a repressor to control Bvg phase expression of bipA in Bordetella pertussis. Mol. Microbiol. 2005;56:175–188. doi: 10.1111/j.1365-2958.2004.04526.x. [DOI] [PubMed] [Google Scholar]