

Abstract
Patients with diabetes mellitus can develop cardiac dysfunction in the absence of underlying coronary artery disease or hypertension; a condition defined as diabetic cardiomyopathy. Mice lacking the intracellular protein kinase Akt2 develop a syndrome that is similar to diabetes mellitus type 2. Expression profiling of akt2−/− myocardium revealed that Rab4a, a GTPase involved in glucose transporter 4 translocation and β –adrenergic receptor (β AR) recycling to the plasma membrane, was significantly induced. We therefore hypothesized that Akt2 deficiency increases myocardial β-adrenergic sensitivity. Confirmatory analysis revealed upregulation of Rab4a mRNA and protein in akt2−/− myocardium. In cultured cardiomyocyte experiments, Rab4a was induced by pharmacological inhibition of Akt as well as by specific knockdown of Akt2 with siRNA. Isolated akt2−/− hearts were hypersensitive to isoproterenol (ISO), but exhibited normal sensitivity to forskolin. Prolonged ISO treatment led to increased cardiac hypertrophy in akt2−/− mice compared to wild type mice. In addition, spontaneous hypertrophy was noted in aged akt2−/− hearts that was inhibited by treatment with the β AR blocker propranolol. In agreement with previous results demonstrating increased fatty acid oxidation rates in akt2−/− myocardium, we found increased peroxisome proliferator-activated receptor α (PPARα ) activity in the hearts of these animals. Interestingly, increased myocardial Rab4a expression was present in mice with cardiac-specific overexpression of PPARα and was also observed upon stimulation of PPARα activity in cultured cardiomyocytes. Accordingly, propranolol attenuated the development of cardiac hypertrophy in the PPARα transgenic mice as well. Our results indicate that reduced Akt2 leads to upregulation of Rab4a expression in cardiomyocytes in a cell-autonomous fashion that may involve activation of PPARα . This maladaptive response is associated with hypersensitivity of akt2−/− myocardium to β-adrenergic stimulation.
Keywords: Adrenergic receptors, Insulin resistance, Diabetes mellitus, Cardiac hypertrophy, Cardiac metabolism
INTRODUCTION
Cardiovascular complications are the leading cause of death in patients with diabetes mellitus (DM) [1]. DM patients are at high risk for developing coronary artery disease and hypertensive-related cardiac abnormalities. These patients have a worse prognosis in the setting of heart failure (HF) or myocardial infarction [2]. In addition, DM can promote the development of intrinsic myocardial dysfunction called diabetic cardiomyopathy [3 ].
Insulin signaling is dependent on the activation of a signal transduction pathway that includes the insulin receptor, IRS family members, Phosphatidylinositol-3’ kinase-α , (PI3K) phosphoinositide dependent kinase 1 (PDK1) and Akt family members [4]. Impaired PI3K/Akt signaling has been implicated in the development of insulin resistance and DM type 2 [5, 6]. In cardiomyocytes, the generation of ATP is dependent on the metabolism of both fatty acids and glucose [7]. Cardiomyocytes express the insulin receptor, and insulin treatment leads to translocation of the transmembrane glucose transporter glut4 to the plasma membrane, resulting in increased glucose uptake. The substrates of Akt family members that result in glut4 vesicle translocation to the plasma membrane are not well-established; but may include synip, a syntaxin4 binding partner that regulates glut4 vesicle docking and fusion and AS160, a Rab GAP that is inactivated by Akt-mediated phosphorylation [8, 9]. In addition, Rab4a, a Ras related small GTP binding protein was shown to participate in glut4 translocation to plasma membrane and was found to co-localize with glut4 on the same vesicles [10, 11]. Insulin stimulation also regulates cardiomyocyte physiology by modulating the levels of metabolic enzymes that participate in glucose and fatty acid metabolism [12].
The Akt family of protein serine/threonine kinases is ubiquitously expressed in mammalian tissues. Mice lacking Akt1 exhibit a mild growth defect, but are normal with regard to glucose tolerance and insulin-stimulated disposal of blood glucose [13]. In contrast, mice lacking Akt2 develop a syndrome that is highly similar to type 2 DM in humans. Hyperinsulinemia is present shortly after birth in akt2−/− mice, and animals gradually develop hyperglycemia at about three months of age [14]. Interestingly, a loss-of-function mutation in the akt2 gene was associated with the development of DM and severe insulin resistance in a human pedigree [15]. In addition, analysis of left ventricular (LV) biopsy specimens obtained during coronary artery bypass surgery revealed that Akt activation was significantly reduced in the myocardium of patients with type 2 DM [16].
In previous work, we demonstrated that 2-month-old euglycemic akt2−/− mice have reduced cardiac glucose oxidation and increased fatty acid oxidation rates, but demonstrate normal cardiac function [17]. In addition, we found that akt2−/− mice demonstrate a normal hypertrophic response to transverse aortic constriction, but exhibit increased cardiac remodeling and dysfunction in response to acute myocardial infarction [17]. Since Akt2 plays a key role in insulin signaling we hypothesized that the expression of proteins related to insulin signaling may be altered in akt2−/− cardiac tissue. To address this possibility, a microarray analysis of gene expression was recently performed on 3-month-old akt2−/− and WT ventricular tissue (unpublished data). A prominent finding in the akt2−/− myocardium was an increase in the mRNA encoding Rab4a. While Rab4a is involved in glut4 translocation to the plasma membrane, it was also found to be critical for the recycling of β-adrenergic receptors (β AR) from internal vesicles to the plasma membrane [18, 19]. Since β AR signaling is a critical regulator of cardiac function, our present study was designed to assess the possibility that deficiency of Akt2 can lead to increased myocardial β-adrenergic sensitivity.
RESEARCH DESIGN AND METHODS
Animals
Methods are described in detail in the supplementary material, available in an online appendix. All procedures were approved by the Committee for the Handling and Care of Laboratory Animals (CHCLA) prior to experimentation.
Mice with targeted disruption of the akt2 gene were previously described [14, 17]. Mice overexpressing PPARα in a cardiac-specific manner (MHC-PPARα were a kind gift of Dr. Daniel Kelly, and were described in detail previously [20].
Murine echocardiography
Transthoracic echocardiography was performed in awake mice with an Acuson Sequoia 256 Echocardiography System equipped with a 15 MHz (15L8) transducer as described previously (Acuson, A Siemens Company, Malvern, PA) [17]. The echocardiographer was blinded to the experimental status of the mice.
Cardiac morphometry and histology
Methods are described in detail in the supplementary material. Heart weight to tibia length ratio (HW/T) was calculated to determine cardiac hypertrophy. Histological analysis was performed in paraffin-embedded LV sections. Cardiac myocyte cross-sectional area was calculated in high power fields from the LV lateroanterior region.
Gene expression analysis and protein analysis
Methods are described in detail in the supplementary material. Quantitative real-time RT-PCR analysis was carried out with the Taqman master mix kit or Syber green reagent (Applied Biosystems, Foster City, CA) according to the manufacturer’s specifications. Specific mRNA levels were normalized by the expression of GAPDH [17].
Protein lysates were prepared as previously described [17]. Bands were visualized by use of the ECL system (Amersham).
Ex vivo analysis of cardiac physiology and cAMP levels
Methods are described in detail in the supplementary material. Experiments were conducted with hearts isolated from 8-week-old akt2−/− and wild type C57Bl/6 mice. Hearts were hung on a Langendorff apparatus and perfused with oxygenated, pre-heated buffer (37° C). A fluid-filled balloon was inserted into the LV. Pressure recordings were interfaced with a personal computer for off-line analysis of heart rate, pressure derivatives (+dP/dt, −dP/dt) and developed pressure. For cAMP measurements, hearts were exposed to a solution containing 100 nM isoproterenol and 0.25 mM 3-isobutyl-1-methylxanthine (IBMX) for 5 minutes and were then rapidly freeze clamped. Tissue samples were pulverized on dry ice, suspended in 0.1M HCL and centrifuged for 5 minutes (12000 g). The supernatants were evaluated for cAMP levels using a cAMP Enzyme Immunoassay Kit (Sigma, USA).
In vivo application of drugs
Methods are described in detail in the supplementary material. Long term ISO application (60mg/kg/day) was performed using implanted Alzet osmotic mini-pumps. Propranolol (0.5 g/L), was added directly to the drinking water of akt2−/− mice and matched WT mice and was replaced 3 times a week [21].
Neonatal Rat Ventricular Myocytes
Cardiomyocytes were isolated from 1–2 day old Sprague-Dawley rat pups. Drug application, siRNA treatment and adenoviral infection are described in detail in the supplementary material.
Statistical analysis
Values are expressed as means ± SEM. Student's t-test or one-way ANOVA was used as required (SigmaStat 3.1, Systat Software, Inc, San Jose, CA). Statistical significance was set at p < 0.05.
RESULTS
Rab4a is induced in akt2−/− cardiac tissue
Quantitative real-time PCR confirmed the induction of Rab4a in akt2−/− myocardium that was first uncovered in our preliminary microarray analysis. Rab4a mRNA levels were increased in LV tissue by approximately 2-fold (Figure 1A). Examination of LV protein lysates from animals at different ages (10 days to 9 months-old) revealed that Rab4a protein levels in akt2−/− LV were invariably increased by 4–7-fold when compared to age-matched WT mice (Figure 1B). As akt2−/− mice aged, Rab4a levels declined to some extent from their peak at 2 months of age, but remained markedly higher than age-matched control mice (Figure 1B). We next examined whether complete deficiency of akt2 is required for cardiac Rab4a induction by use of haplo-insufficient mice. Cardiac Rab4a expression was increased in akt2+/− mice although to a lesser extent than in akt2−/− myocardium (Online Figure 1).
Figure 1.
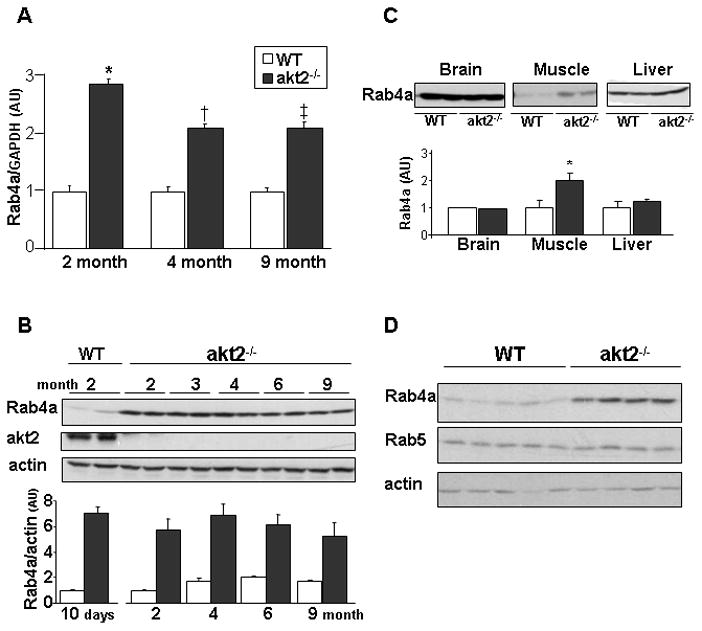
Rab4a expression is increased in akt2−/− myocardium. A. Quantitative real-time RT-PCR analysis of Rab4a mRNA levels in akt2−/− and WT myocardium obtained from 2-month (n=3; *, p=0.0008 akt2−/− vs. WT), 4-month (n=5; *, p=0.0002 akt2−/− vs. WT) and 9-month (n=11; *, p=0.006 akt2−/− vs. WT) old mice. Rab4a mRNA levels were normalized by GAPDH mRNA levels and are presented in arbitrary units. B. Rab4a protein levels in ventricular lysates obtained from 10-day-old mice and from 2-, 3-, 4-, 6- and 9-month-old mice ( WT and akt2−/−). Results were normalized by actin protein levels (lower panel). C. Rab4a protein levels in skeletal muscle, brain and liver lysates obtained from 2–3 month-old WT and akt2−/− mice. Rab4a bands from skeletal muscle were increased by 1.85±0.1 fold in akt2−/− mice (lower panel, n=4–6; *, p=0.01). D. Rab4a and Rab5 protein levels in ventricular lysates obtained from 2-month-old WT (n=5) and akt2−/− (n=4) mice.
To evaluate whether Rab4a up-regulation was ubiquitous in akt2−/− mice, its expression in other tissues was examined. Rab4a expression was unchanged in the brain and the liver of akt2−/− mice (Figure 1C). However, Rab4a protein levels were increased in the akt2−/− skeletal muscle. Rab4a and Rab5 have a reciprocal function in cells. Rab4a promotes the translocation of several receptor types from the early endosome to the plasma membrane while Rab5 regulates internalization from the plasma membrane to the early endosome [22]. We examined the expression of Rab5 in the akt2−/− myocardium and found that Rab5 protein levels were unchanged (Figure 1D).
Rab4a is induced by inhibition of Akt2 signaling in cultured cardiomyocytes
We further investigated the role of Akt2 in Rab4a induction by manipulating Akt2 signaling in cultured neonatal rat ventricular cardiomyocytes. First, cardiomyocytes were treated with the direct Akt1/2 inhibitor A6730 for 48 hours. Treatment with A6730 markedly inhibited insulin-dependent Akt signaling (Figure 2A) and caused upregulation of Rab4a expression in a dose dependent manner (Figure 2B). Next, we inhibited the expression of Akt2 specifically utilizing Accell siRNA (Thermo Scientific, USA) that allows delivery into difficult-to-transfect cells. A 55% decrease of Akt2 protein levels was achieved by use of this siRNA and a significant increase in Rab4A expression was noted (Figure 2C).
Figure 2.
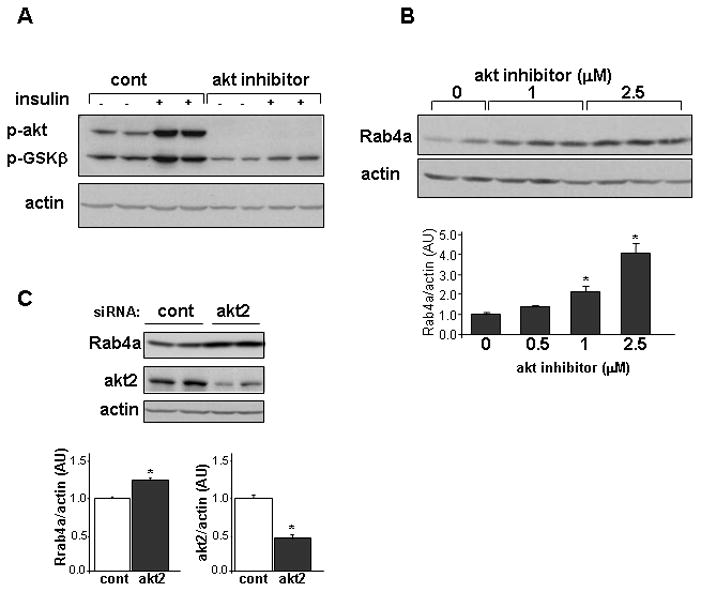
Rab4a expression is induced in cultured rat neonatal ventricular cardiomyocytes by Akt inhibition. A. Analysis of phospho-Akt levels and phospho-GSK3β levels in cultured cardiomyocytes following 10 minutes of stimulation with 10 nM insulin under normal conditions (Cont) and in the presence of the Akt1/2 inhibitor A6730. Cardiomyocytes were exposed to the drug for 24 hours before the activation with insulin. B. Rab4a protein levels following exposure of cardiomyocytes to regular media (0) or to different concentrations of the Akt1/2 inhibitor A6730 (0.5–2.5 μM, n=4–6 wells for each concentration, lower panel *, p< 0.01 compared to control).C. Rab4a protein levels following incubation of cultured cardiomyocytes with 1μM of akt2 siRNA or 1μM of a non–targeting (cont) siRNA in an Accell medium. Rab4a and akt2 bands were normalized by actin protein levels (lower panel *, p< 0.0001 compared to control, n=5 wells in 3 independent experiments)
FoxO family members are important transcription factors that are regulated by Akt signaling. FoxO activity was previously shown to have an inhibitory effect on insulin signaling in cardiomyocytes [23]. We therefore hypothesized that FoxO activity may be involved in Rab4a induction. To evaluate this possibility, cardiomyocytes were infected with an adenoviral expression vector encoding constitutively active FoxO3 (FoxO3A). Infection of cultured cardiomyocytes with this adenoviral vector did not result in increased Rab4a expression (Online Figure 2).
Akt2−/−hearts are sensitized to acute β-adrenergic stimulation
Since Rab4 regulates the recycling of β ARs to the plasma membrane, we hypothesized that the upregulation of Rab4 in akt2−/− myocardium would increase its sensitivity to β-adrenergic stimulation. Indeed, hypersensitivity to β-adrenegic stimulation was previously shown in transgenic mice with cardiac-specific overexpression of Rab4 [18]. Isolated hearts of 2-month-old akt2−/− and WT mice were stimulated with 100 nM isoproterenol (ISO) and the maximal response was recorded and analyzed (Figure 3). The spontaneous heart rate of akt2−/− and WT preparations were similar at baseline and increased to a similar extent following ISO application (Figure 3A). The developed LV pressure, +dP/dT and −dP/dT were also similar at baseline. However, developed LV pressure was higher in akt2−/− hearts following ISO application (Figure 3B). Similarly, +dP/dT and −dP/dT were similar at baseline in both heart preparations, but were affected to a greater extent in akt2−/− hearts following ISO (Figure 3C, D). We further evaluated akt2−/− hearts utilizing the direct adenylate cyclase activatior forskolin. The response of akt2−/− and WT hearts to forskolin was nearly identical for all parameters (Figure 3), indicating that the β –adrenergic hypersensitivity of akt2−/− hearts was upstream of adenylate cyclase activation.
Figure 3.
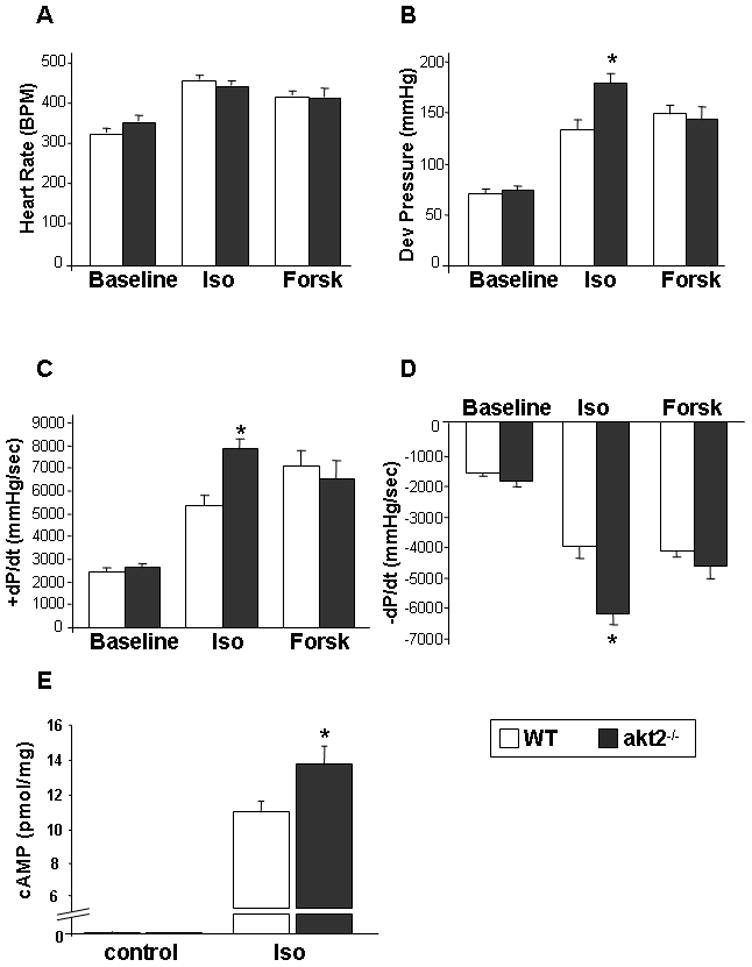
The sensitivity to acute stimulation with ISO is enhanced in akt2−/− hearts. Isolated hearts obtained from 8-week-old akt2−/− and WT mice were perfused with Tyrode's solution containing 100 nM ISO (n=8 and 10 for akt2−/− and WT hearts, respectively) or 1 μM Forskolin (Forsk, n=6 in each group). Parameters at baseline and during the peak pressure response to ISO or Forsk were evaluated. A. Spontaneous heart rate. B. Developed LV pressure (*, p=0.005 akt2−/− ( Iso) vs. WT (Iso)). C. Maximal rate of LV contraction (+dP/dT; *, p=0.0001 akt2−/− ( Iso) vs. WT (Iso)). D. Maximal rate of LV relaxation (−dP/dT; *, p=0.0001 akt2−/− ( Iso) vs. WT (Iso)). E. cAMP levels were measured in WT and akt2−/− hearts at baseline (n=2 in each group) and following stimulation with Iso (100nM) + IBMX (0.25 mM) for 5 min (n=5 and 6 for WT and akt2−/− hearts, respectively. *, p=0.04).
To further confirm the β –adrenergic hypersensitivity of akt2−/− hearts, we evaluated cAMP accumulation following exposure of isolated hearts to 100 nM ISO in the presence of the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (IBMX). Application of ISO + IBMX for 5 minutes markedly increased the tissue levels of cAMP in all tested preparations. However, akt2−/− hearts demonstrated significantly increased accumulation of cAMP in response to this treatment when compared to WT hearts (Figure 3E).
Increased β-adrenergic-mediated cardiac hypertrophy in akt2−/− mice
To determine whether akt2−/− mice were sensitized to the hypertrophic effects of sustained β-adrenergic stimulation, 7-week-old mice were implanted with osmotic Alzet mini-pumps containing ISO or PBS. After 14 days, hearts were isolated and heart weight -to-tibia length ratios were calculated (HW/T). In response to ISO infusion, all mice developed cardiac hypertrophy. However, akt2−/− mice developed significantly more hypertrophy in response to ISO infusion than ISO-treated WT mice (Figure 4). Echocardiographic analysis of ISO-treated animals were performed at day 13 of the infusion to exclude evolving heart failure. The results indicated no difference in LV fractional shortening of akt2−/− compared to WT mice. Histologic examination revealed no foci of fibrosis in WT or akt2−/− mice following ISO treatment. Blood glucose levels obtained at the end of the experiment were similar in akt2−/− and WT mice.
Figure 4.
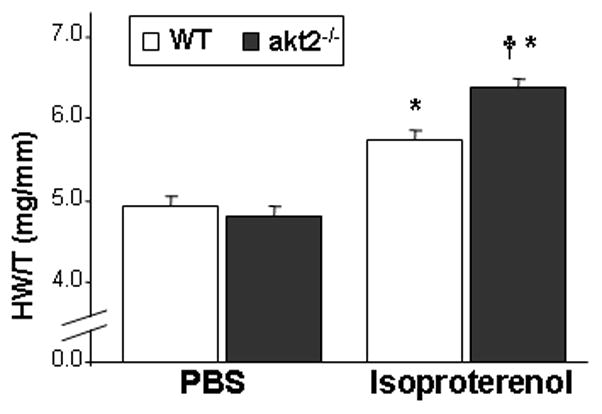
Increased β-adrenergic-mediated cardiac hypertrophy in akt2−/− mice. 7-week-old akt2−/− and WT mice were implanted with Alzet osmotic minipumps to deliver ISO (60 mg/kg/day, n=7–8) or PBS (n=5–6) for 14 days. Note increased ISO-mediated hypertrophy characterized by elevated heart weight-to-tibia length ratio (HW/T) in akt2−/− mice compared with WT mice. *, p<0.001 for WT (iso) vs. WT (PBS) and akt2−/− (iso) vs. akt2−/− (PBS); †, p=0.007 akt2−/− (iso) vs. WT (iso).
Recent studies revealed that Epac, a guanine nucleotide exchange factor directly activated by cAMP, mediates β AR -induced cardiac hypertrophy in a PKA-independent fashion that involved calcineurin and Ca(2+)/calmodulin-dependent protein kinase II (CaMKII) [24]. In addition, Epac expression was previously found to be increased in murine myocardium following chronic ISO infusion [24, 25]. Therefore, we evaluated - the expression- level of Epac in ISO-treated akt2−/− mice as an indicator of β AR stimulation. Indeed, Epac protein levels were increased in ISO-treated akt2−/− compared to ISO-treated WT mice (Online Figure 3). Furthermore, cardiac levels of the active, phosphorylated form of CaMKII were higher in ISO-treated akt2−/− compared to ISO-treated WT mice (Online Figure 3).
Akt2−/− mice develop concentric cardiac hypertrophy over time
Our group previously demonstrated that akt2−/− mice at 8 weeks of age exhibit normal cardiac morphology and Function [17]. Since transgenic overexpression of Rab4a has been shown to cause concentric cardiac hypertrophy at 22 weeks of age, we wished to determine whether the induction of endogenous Rab4a in akt2−/− myocardium would induce a similar hypertrophic phenotype with aging. Analysis of HW/T verified that 2-month-old akt2−/− mice exhibited normal cardiac mass. However, at 3 months of age, akt2−/− mice already demonstrated marked cardiac hypertrophy that persisted thereafter (Figure 5A). To exclude systemic hypertension as a cause of cardiac hypertrophy, noninvasive blood pressure measurements were performed on 2-month-old animals and demonstrated similar systolic and diastolic blood pressure (BP) in the akt2−/− mice and the WT controls (Figure 5B). Random blood glucose measurements of akt2−/− and WT mice indicated that akt2−/− mice were euglycemic at 2 months of age, but exhibited significant hyperglycemia at 3 months of age that was consistently observed thereafter (Online Figure 3).
Figure 5.
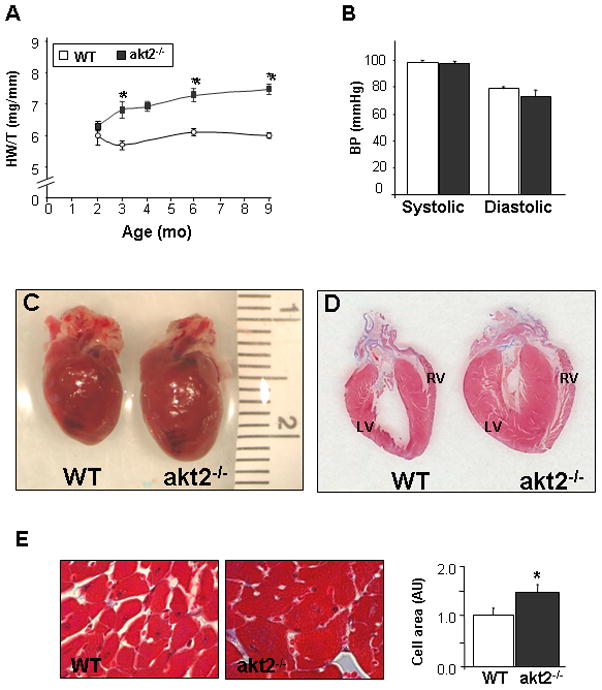
Spontaneous gradual development of cardiac hypertrophy in akt2−/− mice. A. Heart weight to tibial length ratio (HW/T) was determined at various time points. The number of mice analyzed at each time point were: 2 months (3 WT,4 akt2−/−), 3 months (3 WT, 5 akt2−/−), 4 months (6 WT, 3 akt2−/−), 6 months (5 WT, 5 akt2−/−), and 9 months (9 WT, 8 akt2−/−). B. Noninvasive blood pressure measurements obtained from 2 month old akt2−/− and WT mice (n=4 in each group). C. Anatomic appearance of akt2−/− and WT hearts obtained from 9-month-old animals. D. Histological sections from left and right verticals of akt2−/− and WT mice stained with Masson’s trichrome-staining. Notice the absence of intra-myocardial fibrosis in akt2−/− sections. E. Right: representative LV histological sections from 9-month-old akt2−/− and WT mice stained with H&E. Left: Summary of the average cross-sectional area of cardiomyocytes. Results are normalized to WT.
Detailed analysis of the cardiac phenotype of akt2−/− mice at 9 months of age demonstrated significant LV and RV hypertrophy compared to age-matched WT (Online Table 1). Transthoracic echocardiographic analysis also confirmed the development of marked hypertrophy in aged akt2−/− mice (Online Table 2). In addition, fractional shortening (FS) was slightly but significantly reduced in aged akt2−/− mice compared to matched WT mice (Online Table 2). Anatomic examination demonstrated marked concentric cardiac enlargement (Figure 5C) and histologic examination revealed no apparent foci of fibrosis (Figure 5D). Histological analysis also demonstrated that the cross-sectional area of ventricular cardiomyocytes was increased in akt2−/− mice (Figure 5E).
Beta-blocker treatment prevents the development of cardiac hypertrophy in akt2−/− mice
To further evaluate the role of β-adrenergic signaling in the hypertrophy of akt2−/− mice, we treated 2-month-old mice with the β AR blocker propranolol for 6 weeks. Propranolol treatment completely prevented the development of cardiac hypertrophy in akt2−/− mice compared to untreated akt2−/− animals (Figure 6A). The effect of propranolol was also studied in 10-month-old akt2−/− mice where cardiac hypertrophy was already well-established. Treatment of 10-month-old akt2−/− mice with propranolol for 6 weeks reduced the amount of cardiac hypertrophy compared to untreated akt2−/− mice (Figure 6B). Histological analysis verified that the cross-sectional area of ventricular cardiomyocytes was indeed decreased following propranolol treatment (Figure 6C). Importantly, propranolol treatment did not have any effect of the myocardial mass of young or aged WT controls (Figure 6A,B).
Figure 6.
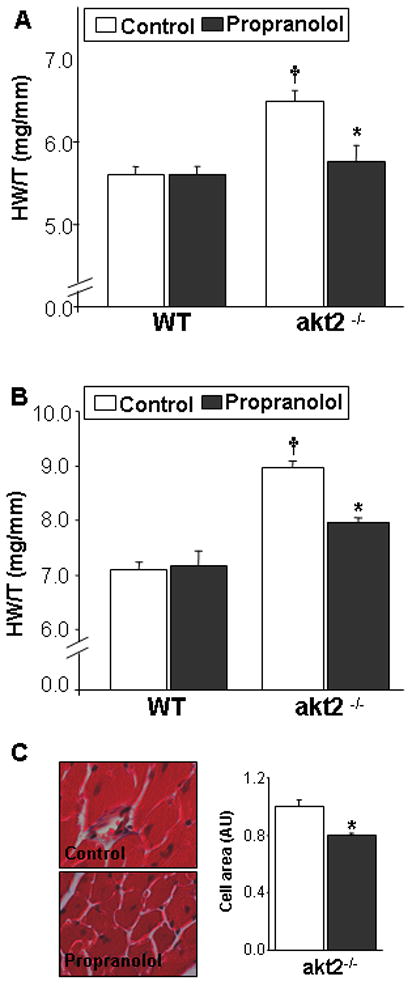
Propranolol treatment of akt2−/− mice inhibits cardiac hypertrophy. A. Reduced cardiac hypertrophy in akt2−/− mice treated with propranolol for 6 weeks from the age of 7-weeks-old, compared to untreated akt2−/− mice (n=4–6; *, p=0.008). Increased cardiac hypertrophy in untreated akt2−/− mice compared to untreated WT mice (n=6; †, p=0.002). B. Decreased cardiac hypertrophy in 10-month-old akt2−/− mice treated with propranolol for 6 weeks (n=3; *, p=0.004) compared to untreated akt2−/− mice. Increased cardiac hypertrophy in untreated akt2−/− mice compared to untreated WT (†, p=0.0004). C. Representative LV histological sections from 10-month old control akt2−/− mice and akt2−/− mice treated with propranolol. Summary of the average cross-sectional area of cardiomyocytes (Right panel)
Increased PPARα activity in Akt2−/− myocardium
A reciprocal relationship may exist between Akt2 signaling and peroxisome proliferator-activated receptor α (PPARα ) activity. Mice overexpressing PPARα in a cardiac-specific manner (MHC-PPARα mice) showed increased myocardial fatty acid oxidation rates as well as cardiac insulin resistance and markedly decreased insulin-stimulated Akt activity [20, 26]. On the other hand, akt2−/− hearts exhibit reduced insulin-stimulated glucose oxidation rates and increased fatty acid oxidation rates [17]. In order to evaluate the role of PPARα activity in akt2−/− myocardium, we examined the mRNA levels of PPARα as well as the PPAR α-regulated genes CPT-1 and MCAD [20]. Our results demonstrated that the mRNA levels for all three genes were significantly increased in akt2−/− compared to WT myocardium (Figure 7). Thus, the presence of increased PPARα activity and decreased insulin-stimulated Akt signaling is a shared finding in the myocardium ofakt2−/− and MHC-PPARα mice.
Figure 7.
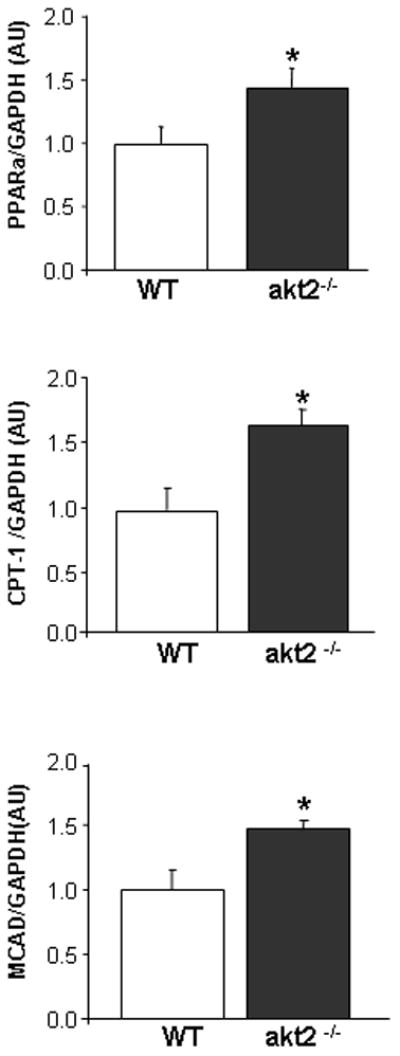
Increase in PPARα and PPARα-target gene expression in akt2−/− myocardium. LV tissue isolated from 9-month-old WT (n=5) and akt2−/− (n=5) mice was used to purify RNA for quantitative RT-PCR analysis. Peroxisome proliferator-activated receptor α (PPARα ), carnitine palmitoyltranferase 1 (CPT-1) and medium-chain acyl-CoA dehydrogenase (MCAD) were normalized to the expression of GAPDH. The data is normalized to WT and is presented in arbitrary units (AU). PPARα (*, p=0.04), mCPT-1 (*, p=0.018) and mCAD (*, p=0.025).
Cardiac Rab4a is induced by activation of PPARα
Current and previous findings suggest a marked similarity in the metabolic state that exists in akt2−/− and MHC-PPARα myocardium. Therefore, we evaluated the possible involvement of PPARα activation in myocardial Rab4a induction. To that end the myocardial levels of Rab4a were evaluated in MHC-PPARα mice. Interestingly, rab4a protein levels were elevated in MHC-PPARα myocardium (Figure 8A). Moreover, adenoviral mediated overexpression of PPARα in combination with the potent PPARα ligand WY-14643, led to a significant increase in Rab4a expression in cultured rat neonatal ventricular cardiomyocytes as well (Figure 8B). Since MHC-PPARα mice develop robust cardiac hypertrophy at an early stage of life we next evaluated whether propranolol treatment would diminish the development of cardiac hypertrophy in this model system. Since overt hypertrophy occurs in this model at a very early age, we treated MHC-PPARα mice and littermates with propranolol from the age of 2 weeks for a period of 6 weeks. Our results demonstrated that propranolol can reduce the development of cardiac hypertrophy in MHC-PPARα mice as well. Propranolol treatment did not affect the myocardial mass of WT controls (Figure 8C)
Figure 8.
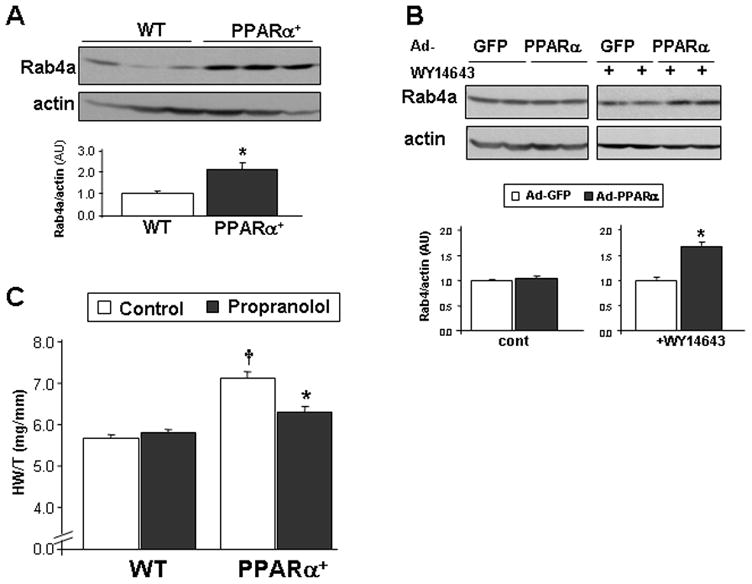
Rab4a is induced in MHC-PPARα myocardium A. Increased Rab4a protein level in transgenic mice with cardiac-specific overexpression of PPARα . Rab4a protein levels in ventricular lysates obtained from 6 week-old MHC-PPARα mice and WT littermates, normalized by actin protein levels (lower panel, n=7–8; *, p= 0.002) B. Rab4a expression following incubation of cardiomyocytes with Ad-GFP-PPARα or Ad-GFP control (MOI of 100). Cell were evaluated with (n=8 wells) or without (n=5 wells) the PPARα agonist WY14643 (1μM). Bands were normalized by actin (lower panel *, p= 0.00005 compared to Ad-GFP+WY). C. Reduced cardiac hypertrophy in MHC-PPARα mice treated with propranolol for 6 weeks from the age of 2-weeks-old, compared to untreated MHC-PPARα (n=9 and 5; respectively *, p=0.0004). Increased cardiac hypertrophy in untreated MHC-PPARα mice compared to untreated WT (n=8; †, p=0.00001).
DISCUSSION
β ARs are critical regulators of cardiac function. In response to acute stress, stimulation of myocardial β ARs by circulating and locally released catecholamines increase cardiac contractility and heart rate and thereby augment blood flow to vital organs [27]. Upon stimulation with agonists, β AR signaling in the heart is negatively regulated by agonist-mediated uncoupling of β AR signaling [28]. In addition, β AR stimulation leads to the β-arrestin-dependent process of transvesicular receptor internalization and trafficking [29].
In two independent studies, the small Ras-like GTPase Rab4a was shown to play an important role in the resensitization of β AR after they are internalized by the action of β-arrestin [18, 19]. As a result, inhibition of Rab4a function by expression of a dominant negative mutant (S27N) resulted in reduced myocardial sensitivity to ISO stimulation [19]. In contrast, mice with cardiac-specific overexpression of Rab4a were found to have an increased concentration of β AR in the plasma membrane and also demonstrated acute hypersensitivity to β AR stimulation as well as concentric cardiac hypertrophy with aging [18]. Our finding that endogenous Rab4a is highly induced by Akt2 loss-of-function, a condition associated with severe insulin resistance and development of type2 DM in mice as well as in humans, is the first clear indication that increased Rab4a protein levels leading to increased β AR signaling may indeed be relevant to a human disease state.
The results of this study suggest that Rab4a is not only increased in concentration in akt2−/− myocardium, but is also functional in terms of promoting β AR recycling and sensitivity. First, isolated akt2−/− myocardium was sensitized to acute ISO stimulation, but was not sensitized to acute forskolin stimulation that targets adenylate cyclase. Of note, while the inotropic effect of ISO was clearly enhanced in akt2−/−hearts, we did not identify a difference in the chronotropic effect of ISO under the same conditions. One possible explanation for these disparate findings is that akt2 deficiency does not induce Rab4a expression in the pacemaker cells of the sinus node. However, since it was not possible to isolate these cells for biochemical analysis, we could not address this possibility in the present study. Second, chronic ISO stimulation led to enhanced cardiac hypertrophy in young euglycemic akt2−/− mice, in association with enhanced Epac expression, which was previously implicated in β-adrenergic stimulation-mediated cardiac hypertrophy [24, 25]. The possibility that changes in systemic blood pressure or vascular resistance contributed to the observed phenotype following ISO treatment cannot be totally excluded. However, this possibility seems unlikely as it was previously shown that changes in systemic blood pressure do not mediate the hypertrophic effect of ISO in mice [30, 31]. Moreover, our group previously demonstrated that akt2−/− mice exhibit a similar hypertrophic response in response to pressure-overload compared to WT controls [17]. Another possibility is that the increased hypertrophy in the akt2−/− mice was secondary to myocardial damage and toxicity, but there were no histological signs of cell damage or fibrosis observed in the akt2−/− hearts after chronic ISO treatment. It should be mentioned that, in comparison to rats, mice in general, and the C57Bl/6 strain in particular, are resistant to ISO-induced cardiomyocyte death and myocardial fibrosis [32, 33]. Third, akt2−/− mice were found to develop a cardiac hypertrophic phenotype over time that was similar to the phenotype of mice with cardiac-specific overexpression of Rab4a. Moreover, β-blocker treatment prevented the spontaneous development of cardiac hypertrophy in akt2−/− mice. The possibility that increased sympathetic tone also contributed to the hypertrophic phenotype of akt2−/− mice was not directly addressed in the present study, but the baseline heart rate and blood pressure of akt2−/− mice was similar to that observed in age-matched WT animals. In addition, elevated sympathetic tone does not explain the increased hypertrophic phenotype in response to ISO infusion, which occurred in an accelerated fashion over a time period of 2 weeks and in an early age in which no spontaneous hypertrophy is normally observed.
Association between cardiac Akt2 and Rab4a expression
Apart from the effect of Rab4a in β AR recycling, Rab4a was previously found to be localized in glut4-containing intracellular vesicles and was shown to participate in glut4 translocation to the plasma membrane following insulin stimulation [10, 11, 34, 35]. The finding that Rab4a up-regulation is not a ubiquitous feature of the akt2−/− mice, but is specifically observed in myocardium and skeletal muscle where glut4-mediated glucose transport is operative, suggests that the up-regulation of Rab4a is a compensatory mechanism to correct the abnormal glucose metabolism in these tissues. Our results in cultured cardiomyocytes with both a pharmacological inhibitor of Akt kinase activity and an siRNA-mediated Akt2 knock-down approach demonstrate that Rab4a expression is promoted by decreased Akt2 signaling in a cell-autonomous manner that is apparently unrelated to extracellular glucose concentration. The increased expression of Rab4a in the hearts of mice haplo-insufficient for Akt2 also supports this model, since these animals exhibit completely normal systemic glucose homeostasis [14]. Further work will be needed to identify the exact downstream effector/s of Akt2 that are involved in Rab4a upregulation. The possibility that FoxO activation is involved in this phenomenon was not supported by our results with adenoviral-mediated FoxO3 overexpression in cultured cardiomyocytes.
Role of PPARα activation in Rab4a induction
Our results indicate that expression of PPARα is increased in akt2−/− myocardium. In addition, we found that the expression of two important PPARα-regulated genes, CPT-1 and MCAD, are increased in akt2−/− myocardium. These findings demonstrate enhanced PPARα activity in the akt2−/− heart and support our previously published finding that there is increased fatty acid oxidation in the akt2−/− heart [17]. Of note, Akt2 may inhibit PPARα activation via phosphorylation and inhibition of the transcriptional coactivator peroxisome proliferator-activated receptor-coactivator-1α [36].
One model to explain our findings is that Akt2 loss-of-function results in PPARα activation that results in Rab4a induction. The detection of increased Rab4a in MHC-PPARα myocardium supports this model. Moreover, overactivation of PPARα in cultured cardiomyocytes also induced Rab4a expression. Interestingly, computerized sequence analysis identified a retinoid X receptor (RXR) binding site upstream to exon 1 in the Rab4a gene.
The hypertrophic phenotype of MHC-PPARα mice is much more robust than the cardiac phenotype of akt2−/− mice, and leads to overt heart failure by 10–12 weeks of-age [20]. In addition, previous reports indicated the Gq activation may be involved in the hepertrophic phenotype of MHC-PPARα mice [37]. Nevertheless, the fact that the hypertrophic phenotype of MHC-PPARα mice could be significantly attenuated by propranolol treatment indicates that myocardial β AR hypersensitivity may be a contributing factor in the development of cardiac hypertrophy in this model. Interestingly, the acute effect of ISO stimulation on cardiac function in PPARα −/− mice was significantly blunted in one study [38]. In addition, β AR receptor expression was found to be reduced in the PPARα −/− myocardium [38].
Possible clinical implications
The therapeutic efficacy of pharmacological β AR blockade has been well-documented in HF and in myocardial ischemia. However, pharmacogenomic studies revealed several genetic and molecular variations in β AR signaling that can affect the clinical outcome of patients with HF and their response to β –blocker treatment [39, 40]. In DM patients, β-blockers were considered harmful for many years due to their metabolic side effects [41]. However, it is currently accepted that DM patients with myocardial ischemia or HF benefit from β-blocker therapy[41–43]. Nevertheless, since β-blocker treatment is not free of side effects and can exacerbate metabolic concerns in DM patients, an ability to predict the responsiveness of these patients to β-blocker treatment would be of high clinical value. Our present findings suggest that a subpopulation of patients in which myocardial Akt2 is significantly inhibited may benefit the most from β-blocker treatment. Furthermore, since type 2 DM is a systemic disease, the cardiac expression of Rab4a and consequent responsiveness to β-blocker therapy may be predicted based on peripheral Akt2 function or PPARα activity., The upregulation of Rab4a in the skeletal muscle of akt2−/− mice might also suggest a way to identify patients with increased cardiac Rab4a by analyzing Rab4a protein levels in skeletal muscle biopsy specimens. Since the activity in circulating cells of the β AR-regulating protein G protein-coupled receptor kinase-2 (GRK2) is a marker of GRK2 alterations in heart [44, 45], it will be interesting to determine whether Rab4a levels and β AR sensitivity in circulating cells is similarly a marker of Akt2 and PPARα activity in heart.
Supplementary Material
Acknowledgments
The authors acknowledge the assistance of the DDRCC Morphology Core Facility at Washington University (NIH DDRCC P30 DK52574). This work was supported by NIH grants HL057278, HL061567, HL 076670, and HL091913 (A.J.M.). S.E. was supported by the Machiah Foundation/Jewish Community Federation Fellowship Program.
Non-standard Abbreviations
- β AR
β adrenergic receptor
- CaMKII
Ca(2+)/calmodulin-dependent protein kinase II
- DM
diabetes mellitus
- HF
heart failure
- HW/T
Heart weight-to-tibia length ratio
- ISO
isoproterenol
- LV
left ventricle
- PDK-1
phosphoinositide dependent kinase 1
- PI3K
Phosphatidylinositol-3’ kinase-α
- PPARα
peroxisome proliferator-activated receptor α
- RV
right ventricle
- WT
wild type
- BP
blood pressure
- PBS
Phospate buffered saline
Footnotes
DISCLOSURES
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mazzone T, Chait A, Plutzky J. Cardiovascular disease risk in type 2 diabetes mellitus: insights from mechanistic studies. Lancet. 2008 May 24;371(9626):1800–9. doi: 10.1016/S0140-6736(08)60768-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen-Solal A, Beauvais F, Logeart D. Heart failure and diabetes mellitus: epidemiology and management of an alarming association. J Card Fail. 2008 Sep;14(7):615–25. doi: 10.1016/j.cardfail.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007 Jun 26;115(25):3213–23. doi: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- 4.Zaid H, Antonescu CN, Randhawa VK, Klip A. Insulin action on glucose transporters through molecular switches, tracks and tethers. Biochem J. 2008 Jul 15;413(2):201–15. doi: 10.1042/BJ20080723. [DOI] [PubMed] [Google Scholar]
- 5.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002 May 31;296(5573):1655–7. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 6.Farese RV, Sajan MP, Standaert ML. Insulin-sensitive protein kinases (atypical protein kinase C and protein kinase B/Akt): actions and defects in obesity and type II diabetes. Exp Biol Med (Maywood) 2005 Oct;230(9):593–605. doi: 10.1177/153537020523000901. [DOI] [PubMed] [Google Scholar]
- 7.Lehman JJ, Kelly DP. Gene regulatory mechanisms governing energy metabolism during cardiac hypertrophic growth. Heart Fail Rev. 2002 Apr;7(2):175–85. doi: 10.1023/a:1015332726303. [DOI] [PubMed] [Google Scholar]
- 8.Yamada E, Okada S, Saito T, Ohshima K, Sato M, Tsuchiya T, et al. Akt2 phosphorylates Synip to regulate docking and fusion of GLUT4-containing vesicles. J Cell Biol. 2005 Mar 14;168(6):921–8. doi: 10.1083/jcb.200408182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sano H, Kane S, Sano E, Miinea CP, Asara JM, Lane WS, et al. Insulin-stimulated phosphorylation of a Rab GTPase–activating protein regulates GLUT4 translocation. J Biol Chem. 2003 Apr 25;278(17):14599–602. doi: 10.1074/jbc.C300063200. [DOI] [PubMed] [Google Scholar]
- 10.Aledo JC, Darakhshan F, Hundal HS. Rab4, but not the transferrin receptor, is colocalized with GLUT4 in an insulin-sensitive intracellular compartment in rat skeletal muscle. Biochem Biophys Res Commun. 1995 Oct 4;215(1):321–8. doi: 10.1006/bbrc.1995.2469. [DOI] [PubMed] [Google Scholar]
- 11.Dransfeld O, Uphues I, Sasson S, Schurmann A, Joost HG, Eckel J. Regulation of subcellular distribution of GLUT4 in cardiomyocytes: Rab4A reduces basal glucose transport and augments insulin responsiveness. Exp Clin Endocrinol Diabetes. 2000;108(1):26–36. doi: 10.1055/s-0032-1329212. [DOI] [PubMed] [Google Scholar]
- 12.Burgering BM. A brief introduction to FOXOlogy. Oncogene. 2008 Apr 7;27(16):2258–62. doi: 10.1038/onc.2008.29. [DOI] [PubMed] [Google Scholar]
- 13.Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001 Oct 19;276(42):38349–52. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- 14.Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB, 3rd, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta) Science. 2001 Jun 1;292(5522):1728–31. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- 15.George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S, Wilson JC, et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science. 2004 May 28;304(5675):1325–8. doi: 10.1126/science.1096706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sasso FC, Torella D, Carbonara O, Ellison GM, Torella M, Scardone M, et al. Increased vascular endothelial growth factor expression but impaired vascular endothelial growth factor receptor signaling in the myocardium of type 2 diabetic patients with chronic coronary heart disease. J Am Coll Cardiol. 2005 Sep 6;46(5):827–34. doi: 10.1016/j.jacc.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 17.DeBosch B, Sambandam N, Weinheimer C, Courtois M, Muslin AJ. Akt2 regulates cardiac metabolism and cardiomyocyte survival. J Biol Chem. 2006 Oct 27;281(43):32841–51. doi: 10.1074/jbc.M513087200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Filipeanu CM, Zhou F, Lam ML, Kerut KE, Claycomb WC, Wu G. Enhancement of the recycling and activation of beta-adrenergic receptor by Rab4 GTPase in cardiac myocytes. J Biol Chem. 2006 Apr 21;281(16):11097–103. doi: 10.1074/jbc.M511460200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Odley A, Hahn HS, Lynch RA, Marreez Y, Osinska H, Robbins J, et al. Regulation of cardiac contractility by Rab4-modulated beta2-adrenergic receptor recycling. Proc Natl Acad Sci U S A. 2004 May 4;101(18):7082–7. doi: 10.1073/pnas.0308335101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest. 2002 Jan;109(1):121–30. doi: 10.1172/JCI14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Asai K, Yang GP, Geng YJ, Takagi G, Bishop S, Ishikawa Y, et al. Beta-adrenergic receptor blockade arrests myocyte damage and preserves cardiac function in the transgenic G(salpha) mouse. J Clin Invest. 1999 Sep;104(5):551–8. doi: 10.1172/JCI7418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seachrist JL, Anborgh PH, Ferguson SS. beta 2-adrenergic receptor internalization, endosomal sorting, and plasma membrane recycling are regulated by rab GTPases. J Biol Chem. 2000 Sep 1;275(35):27221–8. doi: 10.1074/jbc.M003657200. [DOI] [PubMed] [Google Scholar]
- 23.Ni YG, Wang N, Cao DJ, Sachan N, Morris DJ, Gerard RD, et al. FoxO transcription factors activate Akt and attenuate insulin signaling in heart by inhibiting protein phosphatases. Proc Natl Acad Sci U S A. 2007 Dec 18;104(51):20517–22. doi: 10.1073/pnas.0610290104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Metrich M, Lucas A, Gastineau M, Samuel JL, Heymes C, Morel E, et al. Epac mediates beta-adrenergic receptor-induced cardiomyocyte hypertrophy. Circ Res. 2008 Apr 25;102(8):959–65. doi: 10.1161/CIRCRESAHA.107.164947. [DOI] [PubMed] [Google Scholar]
- 25.Ulucan C, Wang X, Baljinnyam E, Bai Y, Okumura S, Sato M, et al. Developmental changes in gene expression of Epac and its upregulation in myocardial hypertrophy. Am J Physiol Heart Circ Physiol. 2007 Sep;293(3):H1662–72. doi: 10.1152/ajpheart.00159.2007. [DOI] [PubMed] [Google Scholar]
- 26.Park SY, Cho YR, Finck BN, Kim HJ, Higashimori T, Hong EG, et al. Cardiac-specific overexpression of peroxisome proliferator-activated receptor-alpha causes insulin resistance in heart and liver. Diabetes. 2005 Sep;54(9):2514–24. doi: 10.2337/diabetes.54.9.2514. [DOI] [PubMed] [Google Scholar]
- 27.Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002 Jan 10;415(6868):206–12. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- 28.Choi DJ, Koch WJ, Hunter JJ, Rockman HA. Mechanism of beta-adrenergic receptor desensitization in cardiac hypertrophy is increased beta-adrenergic receptor kinase. J Biol Chem. 1997 Jul 4;272(27):17223–9. doi: 10.1074/jbc.272.27.17223. [DOI] [PubMed] [Google Scholar]
- 29.Lohse MJ. Molecular mechanisms of membrane receptor desensitization. Biochim Biophys Acta. 1993 Nov 7;1179(2):171–88. doi: 10.1016/0167-4889(93)90139-g. [DOI] [PubMed] [Google Scholar]
- 30.Saadane N, Alpert L, Chalifour LE. Expression of immediate early genes, GATA-4, and Nkx-2.5 in adrenergic-induced cardiac hypertrophy and during regression in adult mice. Br J Pharmacol. 1999 Jul;127(5):1165–76. doi: 10.1038/sj.bjp.0702676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hohimer AR, Davis LE, Hatton DC. Repeated daily injections and osmotic pump infusion of isoproterenol cause similar increases in cardiac mass but have different effects on blood pressure. Can J Physiol Pharmacol. 2005 Feb;83(2):191–7. doi: 10.1139/y04-137. [DOI] [PubMed] [Google Scholar]
- 32.Kudej RK, Iwase M, Uechi M, Vatner DE, Oka N, Ishikawa Y, et al. Effects of chronic beta-adrenergic receptor stimulation in mice. J Mol Cell Cardiol. 1997 Oct;29(10):2735–46. doi: 10.1006/jmcc.1997.0508. [DOI] [PubMed] [Google Scholar]
- 33.Faulx MD, Ernsberger P, Vatner D, Hoffman RD, Lewis W, Strachan R, et al. Strain-dependent beta-adrenergic receptor function influences myocardial responses to isoproterenol stimulation in mice. Am J Physiol Heart Circ Physiol. 2005 Jul;289(1):H30–6. doi: 10.1152/ajpheart.00636.2004. [DOI] [PubMed] [Google Scholar]
- 34.Cormont M, Gautier N, Ilc K, le Marchand-Brustel Y. Expression of a prenylation-deficient Rab4 inhibits the GLUT4 translocation induced by active phosphatidylinositol 3-kinase and protein kinase B. Biochem J. 2001 May 15;356(Pt 1):143–9. doi: 10.1042/0264-6021:3560143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vollenweider P, Martin SS, Haruta T, Morris AJ, Nelson JG, Cormont M, et al. The small guanosine triphosphate-binding protein Rab4 is involved in insulin-induced GLUT4 translocation and actin filament rearrangement in 3T3-L1 cells. Endocrinology. 1997 Nov;138(11):4941–9. doi: 10.1210/endo.138.11.5493. [DOI] [PubMed] [Google Scholar]
- 36.Li X, Monks B, Ge Q, Birnbaum MJ. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature. 2007 Jun 21;447(7147):1012–6. doi: 10.1038/nature05861. [DOI] [PubMed] [Google Scholar]
- 37.Harris IS, Treskov I, Rowley MW, Heximer S, Kaltenbronn K, Finck BN, et al. G-protein signaling participates in the development of diabetic cardiomyopathy. Diabetes. 2004 Dec;53(12):3082–90. doi: 10.2337/diabetes.53.12.3082. [DOI] [PubMed] [Google Scholar]
- 38.Loichot C, Jesel L, Tesse A, Tabernero A, Schoonjans K, Roul G, et al. Deletion of peroxisome proliferator-activated receptor-alpha induces an alteration of cardiac functions. Am J Physiol Heart Circ Physiol. 2006 Jul;291(1):H161–6. doi: 10.1152/ajpheart.01065.2004. [DOI] [PubMed] [Google Scholar]
- 39.Dorn GW, 2nd, Liggett SB. Mechanisms of pharmacogenomic effects of genetic variation within the cardiac adrenergic network in heart failure. Mol Pharmacol. 2009 Sep;76(3):466–80. doi: 10.1124/mol.109.056572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liggett SB, Cresci S, Kelly RJ, Syed FM, Matkovich SJ, Hahn HS, et al. A GRK5 polymorphism that inhibits beta-adrenergic receptor signaling is protective in heart failure. Nat Med. 2008 May;14(5):510–7. doi: 10.1038/nm1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Landray MJ, Toescu V, Kendall MJ. The cardioprotective role of beta-blockers in patients with diabetes mellitus. J Clin Pharm Ther. 2002 Aug;27(4):233–42. doi: 10.1046/j.1365-2710.2002.00419.x. [DOI] [PubMed] [Google Scholar]
- 42.Di Bari M, Marchionni N, Pahor M. Beta-blockers after acute myocardial infarction in elderly patients with diabetes mellitus: time to reassess. Drugs Aging. 2003;20(1):13–22. doi: 10.2165/00002512-200320010-00002. [DOI] [PubMed] [Google Scholar]
- 43.Haas SJ, Vos T, Gilbert RE, Krum H. Are beta-blockers as efficacious in patients with diabetes mellitus as in patients without diabetes mellitus who have chronic heart failure? A meta-analysis of large-scale clinical trials. Am Heart J. 2003 Nov;146(5):848–53. doi: 10.1016/S0002-8703(03)00403-4. [DOI] [PubMed] [Google Scholar]
- 44.Hata JA, Williams ML, Schroder JN, Lima B, Keys JR, Blaxall BC, et al. Lymphocyte levels of GRK2 (betaARK1) mirror changes in the LVAD-supported failing human heart: lower GRK2 associated with improved beta-adrenergic signaling after mechanical unloading. J Card Fail. 2006 Jun;12(5):360–8. doi: 10.1016/j.cardfail.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 45.Iaccarino G, Barbato E, Cipolletta E, De Amicis V, Margulies KB, Leosco D, et al. Elevated myocardial and lymphocyte GRK2 expression and activity in human heart failure. Eur Heart J. 2005 Sep;26(17):1752–8. doi: 10.1093/eurheartj/ehi429. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.