

Abstract
Human ether-á-go-go related gene (hERG) potassium (K+) channels play a critical role in cardiac action potential repolarization. This is due, in large part, to the unique gating properties of these channels, which are characterized by relatively slow activation and an unusually fast and voltage-dependent inactivation. A large number of structurally diverse compounds bind to hERG and carry an unacceptably high risk of causing arrhythmias. On the other hand, drugs that increase hERG current may, at least in principle, prove useful for treatment of long QT syndrome. A few blockers have been shown to increase hERG current at potentials close to the threshold for channel activation – a process referred to as facilitation. More recently, a novel group of hERG channel activators have been identified that slow deactivation and/or attenuate inactivation. Structural determinants for the action of two different types of activators have been identified. These compounds bind at sites that are distinct from each other and also separate from the binding site of high affinity blockers. They reveal not only novel ways of chemically manipulating hERG channel function, but also interactions between structural domains that are critical to normal activation and inactivation gating.
John Mitcheson (left) did his PhD in Bristol and was first introduced to IKr and hERG as a post-doc with Jules Hancox. In 1998 he was awarded a Fellowship from the Wellcome Trust to work with Mike Sanguinetti on hERG channel gating and pharmacology. Attracted by the prospect of working alongside Peter Stanfield and Nick Standen, he moved to Leicester in 2000 and currently holds the post of Reader in the Department of Cell Physiology and Pharmacology. Matt Perry (centre) gained his PhD in Leeds, worked for 3 years with John Mitcheson in Leicester and 5 years in Salt Lake City with Mike Sanguinetti. He is currently pursuing his interests in hERG channel gating at the Victor Chang Cardiac Research Institute in Sydney with Jamie Vandenberg. Mike Sanguinetti (right) is Professor of Physiology and a member of the Nora Eccles Harrison Cardiovascular Research and Training Institute at the University of Utah. His current research interests include the molecular pharmacology of cardiac K+ channels, the mechanisms of K+ channel gating and why he can no longer ride fast on a bicycle.

Introduction
There has been a great deal of interest in manipulating human ether-á-go-go related gene (hERG) potassium channel function since the important role of these channels in the heart was discovered. hERG (Kv11.1) channels conduct the rapid delayed rectifier K+ current (IKr), a critical current in the phase 3 repolarization of the cardiac action potential (Sanguinetti et al. 1995; Trudeau et al. 1995). Inherited mutations of hERG were found to cause type 2 long QT syndrome, a disorder of cardiac action potential repolarization that predisposes affected patients to arrhythmias and sudden death (Splawski et al. 2000). Patients with long QT syndrome have abnormal ECG traces characterized by a prolonged Q–T interval, which represents the time between initial depolarization and final repolarization of the ventricles of the heart. Type 2 long QT syndrome results from a decrease of hERG current, which reduces the total amount of K+ current and slows repolarization of the ventricular action potential (Sanguinetti et al. 1995). Even before the genetic condition was identified it was known that a similar phenomenon could be caused by some commonly prescribed medications. Indeed, it rapidly became evident that a very large number of drugs (most of which were not even designed to treat cardiac disease) had the potential to inhibit IKr (or hERG current) and cause this potentially lethal side effect (Roden, 1998). In most cases, these compounds had a much higher affinity for hERG than other cardiac ion channels, suggesting there was something quite distinct about the molecular characteristics of hERG channels that caused them to bind to such a large number of structurally diverse compounds. It also became normal practice for pharmaceutical and biotechnology companies to test large numbers of compounds for hERG block early in preclinical safety evaluation. In the process, a small number of compounds have been discovered that had the opposite activity of blockers – they increased hERG channel currents (Grunnet et al. 2008). These compounds can be separated into facilitators (blockers, which paradoxically increase hERG currents at membrane potentials close to the threshold for channel activation; Carmeliet, 1993; Jiang et al. 1999; Hosaka et al. 2007) and activators (or agonists, which increase hERG currents through several different mechanisms; e.g. Kang et al. 2005; Zhou et al. 2005; Hansen et al. 2006), which will be covered later in this review. hERG activators shorten cardiac action potentials and thus could be developed to treat patients with long QT syndrome (Grunnet et al. 2008).
Recent studies have revealed the molecular determinants for block, facilitation and activation of hERG channels. These studies suggest that compounds bind at distinct sites to alter single channel conductance, or activation and inactivation gating. These compounds bind to structural domains critical for hERG channel function.
hERG channel gating
hERG channel gating is distinctive, in large part due to its unusual inactivation properties (Schonherr & Heinemann, 1996; Smith et al. 1996; Spector et al. 1996b). Unlike most other voltage gated K+ (Kv) channels, hERG inactivation is strongly voltage dependent and has much faster time dependent kinetics than activation gating. At a holding potential of −80 mV, the activation gate is closed and the channels are in a stable non-conducting state. The channels activate relatively slowly with depolarizations to potentials positive to −60 mV and the resulting outward currents reach a peak amplitude at ∼−10 mV (Fig. 1). With progressive depolarization, channels rapidly enter the inactivated state and become non-conducting. This rapid and voltage-dependent inactivation is the cause of the characteristic decrease in amplitude of hERG currents at positive potentials (see Fig. 1B). Repolarization causes the channels to rapidly recover from inactivation within a few milliseconds and this gives rise to the large tail currents, which then slowly decay as the activation gate closes (deactivation). The amplitude of the tail currents measured at a single potential reflect the number of channels activated by the previous depolarization and thus can be used to determine the voltage dependence of activation (Fig. 1C).
Figure 1. Schematic representation of block, facilitation and activation of hERG channel currents.
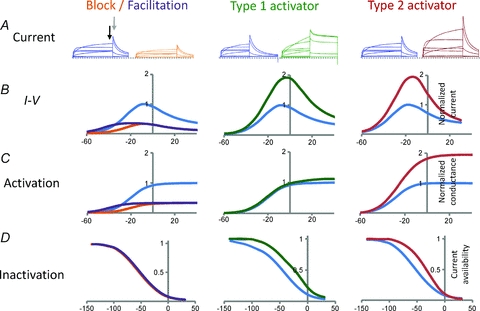
Current traces and current–voltage relationships have been colour coded according to recording conditions – blue, control; orange, block; purple, facilitation; green, type 1 activator; red, type 2 activator. To facilitate comparison, all currents in the presence of the modulator are shown normalized relative to peak control value. A, hERG currents obtained by depolarization of the membrane potential to voltages between −60 mV and +40 mV (in 20 mV increments). Following each test pulse the membrane potential was stepped to −70 mV in order to invoke tail currents. Initially the tail current rapidly increases to a peak (grey arrow) due to the rapid recovery from inactivation and then more slowly decreases due to closure of the activation gate (deactivation). B, end pulse current–voltage (I–V) relationships. Current measured at the end of each test pulse (indicated by black arrow in A) was plotted against voltage. Current increases progressively with potentials up to −10 mV, but then declines at more positive voltages due to voltage-dependent inactivation. C, the voltage dependence of activation. Peak tail current amplitude (indicated by grey arrow in A) plotted against the preceding test pulse potential. D, the voltage dependence of inactivation (or current availability). Currents are maximally activated with a strong prepulse and current availability measured from extrapolated peak currents with voltage pulses to potentials between −160 and +40 mV. At negative potentials there is no channel inactivation and so current availability is close to 1, whereas at positive potentials channels inactivate and current availability declines toward 0. hERG channel blockers (left panels, orange lines) reduce hERG current without effecting channel gating properties. Facilitators (left panels, purple lines) also reduce hERG current, except at voltages negative to −30 mV, where a small increase in current is observed (adapted from Hosaka et al. 2007). This increase is due to a small hyperpolarizing shift in the voltage dependence of activation (see C). Type 1 hERG activators such as RPR260243 (centre, green traces) enhance hERG current by a dual mechanism of slowing channel deactivation (see tail current in A) and reducing inactivation by shifting the voltage dependence to more positive potentials (D). The predominant mechanism of action for type 2 hERG activators such as PD118057 (right, red traces) is to reduce channel inactivation by shifting the voltage dependence to more depolarized potentials (D), without affecting activation or deactivation properties.
hERG channel structure
Recently solved crystal structures of bacterial and mammalian K+ channels have provided tremendous insight into the structural basis of K+ channel gating (Swartz, 2004). Like other Kv channels, hERG is formed by coassembly of four α-subunits, each of which has six transmembrane spanning α-helical segments (S1–S6). Within each hERG subunit, the S1–S4 helices form a voltage sensor domain (VSD) that senses transmembrane potential and is coupled to a central K+-selective pore domain (e.g. Smith & Yellen, 2002; Liu et al. 2003; Piper et al. 2003, 2005; Zhang et al. 2004; Subbiah et al. 2004, 2005). Each pore domain is composed of an outer helix (S5) and inner helix (S6) that together coordinate the pore helix and selectivity filter (Fig. 2B–D). The carboxy end of the pore helix and selectivity filter contain the highly conserved K+ channel signature sequence, which in hERG is Thr-Ser-Val-Gly-Phe-Gly. This sequence forms a narrow conduction pathway at the extracellular end of the pore in which K+ ions are coordinated by the backbone carbonyl oxygen atoms of the signature sequence residues. Inactivation gating in hERG and other channels is not fully understood, but is likely to involve subtle conformational changes to the backbone of the selectivity filter (e.g. Stansfeld et al. 2008) that impair K+ ion coordination and block conduction. hERG inactivation is sensitive to point mutations in the selectivity filter (Smith et al. 1996), pore helix (Ficker et al. 1998) and outer mouth of the pore (Fan et al. 1999; Jiang et al. 2005; Ju et al. 2009) that are likely to influence the stability of the conducting state of the selectivity filter. Mutations at more remote regions, such as S4, that perturb inactivation may influence coupling between the voltage sensor and pore (Piper et al. 2005).
Figure 2. Structural determinants of hERG channel modulators.
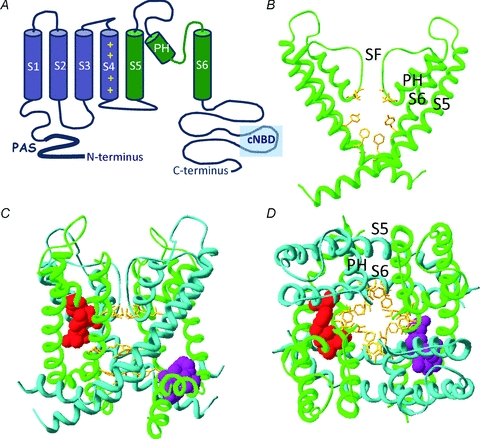
A, cartoon representation of the secondary structure of a single hERG channel subunit. The six transmembrane spanning α-helices (S1–S6) are shown along with the intracellularly located N- and C-termini. The first four helices (S1–S4 shown in blue) form a voltage sensing domain (VSD) which detects changes to the membrane potential. A short linker (S4–S5 linker) connects the VSD to the pore domain, which is composed of the S5 through S6 segments (in green). Within the pore domain are a short α-helix (pore helix, PH) and the selectivity filter (SF), four of which (one from each subunit) come together to coordinate movement of K+ ions through the central pore of the channel. B, homology model of hERG pore domain based upon crystal structure of Kv1.2. Only two of the four subunits are shown for clarity. High-affinity hERG channel blockers bind to amino acid residues (yellow sticks) that line the inner cavity. Two aromatic residues on the S6 helix (Phe656, Tyr652) are critical for all high-affinity blockers. By contrast, the two polar residues on the pore helix (Thr623 and Ser624) are important determinants for some, but not all, high-affinity drugs. C and D, homology model highlighting the putative binding sites for type 1 and type 2 hERG channel activators viewed from the side (C) and extracellular end (D) of the channel. The pore domains from all four subunits are shown by alternating green or blue ribbons. The purple side chains interact with the type 1 activator RPR260243 and are located at the intracellular ends of the S5 helix (Leu553, Phe557) and S6 helix (Asp658, Val659) of a single subunit. In contrast, side chains for the type 2 hERG activator PD118057 are coloured red and are located towards the extracellular end of the S6 helix of one subunit (Leu646) and on the pore helix of a neighbouring subunit (Phe619, Leu622).
Below the selectivity filter the pore widens to form a water-filled central cavity that is lined by residues from the S6 helices. As with other Kv channels, the cytoplasmic ends of the inner (S6) helices of hERG are thought to act as a barrier (gate) to movement of ions across the pore (Mitcheson et al. 2000b). Upon depolarization, conformational changes in the voltage sensor are coupled to the pore so that the S6 helices splay apart to create a wide aperture at the interface with the cytoplasm, which ions and drugs can move through to enter the inner cavity. Opening of Kv channels requires the S6 helices to bend. A number of studies suggest two motifs can confer the necessary flexibility. One is a Gly residue (referred to as a glycine hinge) that is highly conserved across all K+ channel families (Jiang et al. 2002). Mutating this glycine locks many different channels in the closed state. The second putative hinge region found in many Kv channels (but not hERG) is a Pro-X-Pro motif located two helical turns below (towards cytoplasm) the glycine hinge (Labro et al. 2003; Webster et al. 2004; Long et al. 2005). In the crystal structure of Kv1.2 there is a significant swivel and kink at this position (Long et al. 2005) and mutagenesis studies also indicate it is important for activation gating (Labro et al. 2003). hERG channels have two glycines on S6 – one at the conserved glycine hinge position and a second within an Ile-Phe-Gly sequence in place of the Pro-X-Pro motif. However, mutation of either hERG S6 glycine fails to reduce the flexibility of the helix and activation is actually facilitated rather than hindered (Hardman et al. 2007). In hERG, it seems likely that glycines are required not for their ability to introduce flexibility but because the small side chains in these positions permit close packing with neighbouring helices.
Structural basis of drug induced long QT syndrome
Many medications have been shown to prolong QT interval and induce arrhythmias such as torsade de pointes (Recanatini et al. 2005). In most cases these drugs exert these effects by reducing hERG current during the cardiac action potential. Most frequently this is due to drugs binding directly to the channel and blocking conduction (Redfern et al. 2003; Hancox et al. 2008), but it can also occur by interference with trafficking of hERG channels to the surface membrane (Ficker et al. 2005; Rajamani et al. 2006). One of the most striking features of drug induced QT prolongation is the structural and therapeutic diversity of compounds that block hERG (see Fig. 3) and the selectivity they exhibit for hERG relative to other ion channels. High affinity compounds exhibit an open-state-dependent block suggesting that the activation gate acts as a barrier to drugs gaining access to their receptor (Spector et al. 1996a; Mitcheson et al. 2000b). This finding and abundant evidence from a variety of other ion channels pointed to the central cavity forming an important binding site for drugs. An Ala-scanning mutagenesis approach was used to identify amino acid resides in hERG that could interact with several different drugs (Mitcheson et al. 2000a). Residues on S6 and the pore helices that were predicted to line the central cavity were mutated to Ala and tested to see if the mutation reduced the sensitivity to drug block. Mutation of two S6 aromatic residues Tyr652 and Phe656 was found to substantially decrease the potency of MK-499, a compound originally developed as a class III antiarrhythmic (Mitcheson et al. 2000a; see Fig. 2B). These residues have now been found to be critical for interactions with a large number of different hERG channel blockers (e.g. Lees-Miller et al. 2000; Kamiya et al. 2001, 2006; Sanchez-Chapula et al. 2002; Ridley et al. 2004; Milnes et al. 2006). A study in which the Tyr652 and Phe656 were systematically mutated to different residues suggested that a hydrophobic residue at position 656 is sufficient for high affinity binding of terfenadine, cisapride and MK-499, whereas an aromatic residue is required at position 652 (Fernandez et al. 2004). Interestingly, these aromatic residues are not conserved in most other Kv channels that instead have an Ile or Val in the analogous positions. Aromatic residues are not only hydrophobic, but the π-electron orbitals of the cyclic rings also permit a variety of electrostatic interactions with drug groups – including π–π and π–cation interactions. Tandem dimer studies, in which mutations are made in only two diametrically opposite subunits of the tetrameric channel, suggest that some compounds span the pore to interact with Tyr residues on more than one subunit (Myokai et al. 2008; Imai et al. 2009). EAG (Kv10) channels have Tyr and Phe residues in analoguous positions to hERG and yet most blockers have a much lower potency for this closely related channel (Ficker et al. 1998, 2001; Herzberg et al. 1998). The explanation is likely to relate to differences in the orientation of these residues relative to the central cavity. Mutation of EAG, so that the Tyr or Phe residues are repositioned towards the C-terminal ends of S6 by just one residue (which reorientates the side chain on the helix by ∼ 110 deg), increased the sensitivity to cisapride so that it was similar to hERG (Chen et al. 2002). Repositioning the residues in hERG reduced cisapride sensitivity, suggesting that the aromatic residues are already optimally positioned for blocker interactions. EAG channels do not inactivate. One intriguing hypothesis is that inactivation gating of hERG channels includes conformational changes that position the aromatic residues for interactions with compounds in the central cavity (Chen et al. 2002). This may explain the widely reported decrease of drug potency in inactivation-deficient hERG channel constructs (e.g. Ficker et al. 1998, 2001; Milnes et al. 2003; McPate et al. 2008; Perrin et al. 2008). Conformational changes to the cavity may also underlie voltage dependent changes in onset and dissociation observed with some hERG blockers (Sanchez-Chapula et al. 2002, 2003; Stork et al. 2007).
Figure 3. Chemical structure of hERG channel blockers.
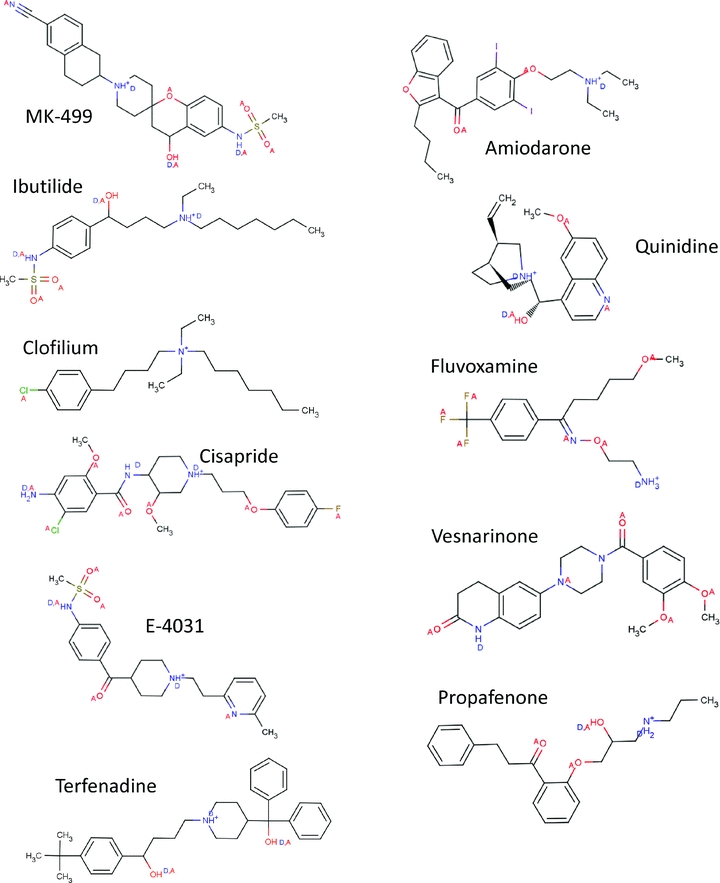
These ligands illustrate some aspects of the structural diversity of hERG blockers and have been shown to block hERG by binding to sites within the inner cavity of the channel. The structures illustrate the protonated states of nitrogen atoms at physiological pH, and the hydrogen bond acceptor (A) and donor (D) atoms. The structures of all ligands were obtained from ChemIDplus website, redrawn using Marvin Beans 4.0.5 (Chemaxon) and pKA calculations for all ligands were measured at pH 7.2 using the plug-in within MarvinSketch. The authors are grateful to Dr Phill Stansfeld, University of Oxford, for the illustrations of chemical structure in this figure.
Mutation of two polar residues (Thr623 and Ser624) on the pore helices also reduce the sensitivity of hERG channels to block, but in a more compound specific manner (Mitcheson et al. 2000a; Perry et al. 2004, 2006; Kamiya et al. 2006, 2008; see Fig. 2B). For example, in studies of close analogues of ibutilide and clofilium, the T623A:S624A hERG double mutant changed the potency of compounds in the range of 30- to 1000-fold depending on the nature of a para-substituent on the phenyl ring in one part of the molecule (Perry et al. 2006). Polar residues are conserved in these positions in most Kv channels, and thus can't explain the extraordinary pharmacological properties of hERG. Nevertheless these residues are important components of the blocker binding site. With a total of eight aromatic residues arranged in two rings of four within the central cavity, and another eight polar residues close to the selectivity filter, it is perhaps not surprising that this channel can interact with such a large number of different compounds (Fig. 2B).
Mechanisms of action of facilitators and activators of hERG current
In contrast to high affinity blockers which share a common binding region within the aqueous inner cavity of the pore, activators of hERG channels may differ in their binding location depending on the mechanism by which they enhance current.
A small number of class III anti-arrhythmic drugs (almokalant, azimilide and nifekalant), which act primarily as hERG channel blockers, can paradoxically enhance current when hERG channels are activated by weak depolarizations that are preceded by a strong depolarizing prepulse step (Carmeliet, 1993; Jiang et al. 1999; Hosaka et al. 2007). Activation in this manner is termed ‘facilitation’ and is due to a modification of channel gating that shifts the voltage dependence of activation to more hyperpolarized membrane potentials (Fig. 1) (Hosaka et al. 2007). Although this mechanism would enhance current throughout the voltage range, it is likely to be obscured at potentials positive to −30 mV by the voltage-dependent block that also occurs with these drugs. Mutations of residues that are important for block (Thr623, Tyr652 and Phe656) by nifekalant also abolish facilitation suggesting that these processes are intrinsically linked (Hosaka et al. 2007). However, other point mutations within the S6 helix either enhance or attenuate facilitation without affecting block (Hosaka et al. 2007). Thus, it appears that binding of nifekalant within the pore, together with a strong depolarizing prepulse, is a prerequisite for facilitation. It may be that strong depolarization allows the already bound blocker access to a second binding site that mediates facilitation. Alternatively, depolarization may alter the conformation of the channel–blocker complex such that activation is allosterically modulated through the S6 helix. Further studies are required to differentiate between these possibilities and to determine if facilitation reduces the pro-arrhythmic potential of hERG blockers.
Several small molecule activators of hERG channels have recently been identified (Kang et al. 2005; Zhou et al. 2005; Casis et al. 2006; Su et al. 2009; Gerlach et al. 2010; Gessner et al. 2010). Many of these target an important feature of hERG channel gating, which is the rapid, voltage-dependent inactivation process that mediates inward rectification (Zhou et al. 2005; Casis et al. 2006; Perry et al. 2007; Xu et al. 2008; Su et al. 2009; Gerlach et al. 2010). The apparent complexity of this mechanism raises the possibility that activators, despite sharing – at least in part – a common mechanism, can modulate inactivation through distinct interactions with the channel protein. Based on our current understanding we have separated these hERG channel activators into two distinct classes.
Type 1 activators
RPR260243 has been designated as a type 1 hERG channel activator (Perry et al. 2009). This small molecule enhances current by attenuating inactivation and severely slowing the rate of channel closure (deactivation) (Kang et al. 2005; Perry et al. 2007). Although slowed deactivation has been observed with other hERG channel activators (Casis et al. 2006; Su et al. 2009; Gerlach et al. 2010), this affect is much more profound with RPR260243 (Perry et al. 2007). A scanning mutagenesis technique was used to identify a putative binding site for this activator (Perry et al. 2007). Mutation to alanine of four residues located at the cytoplasmic ends of the S5 domain (Leu553, Phe557) and S6 domain (Asp658, Val659) of the channel completely abolished activation by RPR260243. When viewed on a homology model of hERG based on the crystal structure of Kv1.2, the side chains of these key residues form a binding region at the interface of S5 and S6 on the cytoplasmic end of the pore, facing away from inner cavity and conduction pathway (Fig. 2C and D). The cytoplasmic ends of the four S6 domains form the activation gate that controls K+ conduction through the channel. Deactivation involves conformational changes to the S6 segments that close the cytoplasmic aperture to the inner cavity and prevent conduction. Binding of RPR260243 to a single subunit may directly constrain movement of the S6 domains to slow the rate of channel closure. In contrast, any alterations to inactivation gating by RPR260243 are likely to occur allosterically through restricted coupling of the voltage sensor to the selectivity filter via the S5 or S6 domains (Perry et al. 2007).
Type 2 activators
Most other hERG channel activators, such as PD118057 (Zhou et al. 2005), its analogue PD307243 (Xu et al. 2008), NS1643 (Casis et al. 2006), A935142 (Su et al. 2009) and ICA-105574 (Gerlach et al. 2010), act primarily to attenuate inactivation and are designated as type 2 activators. Impaired inactivation occurs through a dual mechanism involving both a shift in the voltage dependence of inactivation to more depolarized membrane potentials and a slowing of the onset rate (Xu et al. 2008; Perry et al. 2009; Su et al. 2009). In addition, a secondary increase in conductance is likely to occur through another mechanism, tentatively identified as enhanced single channel open probability (Perry et al. 2009). This secondary mechanism is most evident in conditions where inactivation is largely absent, such as at negative membrane potentials and in certain mutant hERG channels (Xu et al. 2008; Perry et al. 2009). Other minor affects such as alterations in the rate and voltage dependence of activation and/or a minor slowing of the rate of channel deactivation are activator specific.
Scanning mutagenesis has identified a putative binding site for the designated type 2 hERG channel activator, PD118057 (Perry et al. 2009). Mutation of Leu646 in the S6 helix or Phe619 at the base of the pore helix abolishes current activation by this compound. Other mutations such as L622A in the pore helix also substantially reduce PD118057-dependent modulation. Molecular modelling suggests that PD118057 binding is coordinated by hydrophobic residues on two different channel subunits – the compound interacts with Leu646 in the S6 domain of one subunit and Leu622 and Phe619 in the pore helix of the adjacent subunit. This type 2 binding site is located much closer to the selectivity filter than the type 1 binding site (see Fig. 2C and D). Binding at this position may impede coupling between the voltage sensor and selectivity filter to alter inactivation, or may simply stabilize the selectivity filter in the open conformation (Perry et al. 2009). In either case, a direct interaction of PD118057 with the pore helix to attenuate inactivation contrasts with the allosteric mechanism proposed for the type 1 activator RPR260243.
Whether other type 2 activators share the same, or at least closely related, binding sites with PD118057 remains uncertain. It has been proposed that NS1643 and PD306243 act on residues located on the outer mouth of the pore (Xu et al. 2008), a region of the channel that is highly important for inactivation (Liu et al. 2002). A substantial part of the evidence for this proposal is derived from the failure of these molecules to activate hERG channels when applied to the intracellular side of the channels by injecting them into Xenopus oocytes (Xu et al. 2008). Although this could indicate binding to the extracellular face of the channel protein, it is also important to consider that the lipophilic yolk inside oocytes may impair diffusion of these molecules to the intracellular surface of the membrane. In fact, PD307243 produced a substantial increase in hERG current when applied directly to the intracellular face of excised membrane patches (Gordon et al. 2008). Further clarification of the ‘sidedness’ of these activators, using inside-out and outside-out patch clamp recordings, is therefore required.
Point mutations at the extracellular mouth of the pore, which impair inactivation, completely abolish the current enhancing affects of NS1643 but not of PD306243 (Xu et al. 2008). One interpretation is that NS1643 acts solely to impair inactivation whereas PD306243 may have an additional secondary mechanism, such as the enhanced single channel open probability proposed for its closely related structural analogue PD118057 (Perry et al. 2009). It is also likely that the binding sites differ for NS1643 and PD306243. Further studies on a variety of type 2 activators are required to determine if they share a common binding site and to determine the basis for the activator-specific changes to hERG channel activation and deactivation.
Impaired inactivation appears to be a common mechanism for most hERG channel activators. Two notable exceptions to this are mallotoxin (Zeng et al. 2006) and the amiodarone derivative KB130015 (Gessner et al. 2010). Neither of these molecules affects inactivation, but instead they enhance hERG channel current by accelerating the rate of activation and shifting its voltage dependence to more negative potentials. These affects are distinct from the mechanism of facilitation as they do not require a strong depolarizing prepulse (Gessner et al. 2010). Despite their similar modes of action, mallotoxin and KB130015 have synergistic effects on hERG channel activation and thus likely occupy distinct, but functionally coupled, binding sites on hERG (Gessner et al. 2010). The location of the binding residues for either of these molecules remains to be elucidated.
Conclusions and future perspectives
Drug binding sites have now been identified for hERG channel blockers and activators. These ligands are providing useful insights into the molecular and structural basis for the unusual gating properties of hERG. Most high affinity blockers have little influence on channel gating and simply block K+ conduction by binding within the inner cavity, close to the selectivity filter. In contrast, hERG activators primarily exert their effects by modulating channel gating. Although there is much less information on the structural basis of hERG binding with activators than for blockers, initial findings are intriguing. RPR260243 and PD118057 have distinct binding sites both from each other and from blockers. The type 1 activator RPR260243 binds to a site at the interface between the pore and the voltage sensor and slows deactivation by hindering conformational changes to S6 as the pore closes. In addition, it shifts the voltage dependence of inactivation by an allosteric mechanism that has yet to be determined. By contrast, PD118057 binds closer to the selectivity filter, forming intersubunit interactions between the pore helix and S6 that shifts the voltage dependence of inactivation to more depolarized potentials. Whether this decrease of inactivation is due to stabilizing the conducting state of the selectivity filter or uncoupling of the pore to the voltage sensor remains to be determined. Further studies are also required to determine if the binding sites for RPR260243 and PD118057 are shared with other activators of the same type.
In principle, hERG activators have significant therapeutic potential. They could be used to enhance hERG currents in patients with LQTS or developed as a novel class of antiarrhythmic drugs (reviewed by Grunnet et al. 2008). In practice, because of the history of drug induced long QT syndrome, drug regulatory agencies and the pharmaceutical industry as a whole are likely to be wary of developing drugs that are targeted against hERG. The current generation of hERG activators are not very potent (active in the micromolar range) and this raises concerns about their specificity and potential for cardiac and non-cardiac side effects. Nevertheless, hERG activators are an attractive proposition since they increase repolarizing K+ current, stabilize the repolarization phase of the cardiac action potential and may protect against unsynchronized depolarizations triggered by delayed or early afterdepolarizations (Grunnet et al. 2008). The characterization of the binding sites of activators may facilitate development of more potent and specific agents that could be developed as a novel alternative to current antiarrhythmic treatments.
References
- Carmeliet E. Use-dependent block and use-dependent unblock of the delayed rectifier K+ current by almokalant in rabbit ventricular myocytes. Circ Res. 1993;73:857–868. doi: 10.1161/01.res.73.5.857. [DOI] [PubMed] [Google Scholar]
- Casis O, Olesen SP, Sanguinetti MC. Mechanism of action of a novel human ether-a-go-go-related gene channel activator. Mol Pharmacol. 2006;69:658–665. doi: 10.1124/mol.105.019943. [DOI] [PubMed] [Google Scholar]
- Chen J, Seebohm G, Sanguinetti MC. Position of aromatic residues in the S6 domain, not inactivation, dictates cisapride sensitivity of HERG and eag potassium channels. Proc Natl Acad Sci U S A. 2002;99:12461–12466. doi: 10.1073/pnas.192367299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan JS, Jiang M, Dun W, Mcdonald TV, Tseng GC. Effects of outer mouth mutations on hERG channel function: a comparison with similar mutations in the Shaker channel. Biophys J. 1999;76:3128–3140. doi: 10.1016/S0006-3495(99)77464-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez D, Ghanta A, Kauffman GW, Sanguinetti MC. Physicochemical features of the HERG channel drug binding site. J Biol Chem. 2004;279:10120–10127. doi: 10.1074/jbc.M310683200. [DOI] [PubMed] [Google Scholar]
- Ficker E, Dennis A, Kuryshev Y, Wible BA, Brown AM. HERG channel trafficking. Novartis Found Symp. 2005;266:57–69. discussion 70–54, 95–59. [PubMed] [Google Scholar]
- Ficker E, Jarolimek W, Brown AM. Molecular determinants of inactivation and dofetilide block in ether a-go-go (EAG) channels and EAG-related K+ channels. Mol Pharmacol. 2001;60:1343–1348. doi: 10.1124/mol.60.6.1343. [DOI] [PubMed] [Google Scholar]
- Ficker E, Jarolimek W, Kiehn J, Baumann A, Brown AM. Molecular determinants of dofetilide block of HERG K+ channels. Circ Res. 1998;82:386–395. doi: 10.1161/01.res.82.3.386. [DOI] [PubMed] [Google Scholar]
- Gerlach AC, Stoehr SJ, Castle NA. Pharmacological removal of human ether-a-go-go-related gene potassium channel inactivation by 3-nitro-N-(4-phenoxyphenyl) benzamide (ICA-105574) Mol Pharmacol. 2010;77:58–68. doi: 10.1124/mol.109.059543. [DOI] [PubMed] [Google Scholar]
- Gessner G, Macianskiene R, Starkus JG, Schonherr R, Heinemann SH. The amiodarone derivative KB130015 activates hERG1 potassium channels via a novel mechanism. Eur J Pharmacol. 2010;632:52–59. doi: 10.1016/j.ejphar.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon E, Lozinskaya IM, Lin Z, Semus SF, Blaney FE, Willette RN, Xu X. 2-[2-(3,4-Dichloro-phenyl)-2,3-dihydro-1H-isoindol-5-ylamino]-nicotinic acid (PD-307243) causes instantaneous current through human ether-a-go-go-related gene potassium channels. Mol Pharmacol. 2008;73:639–651. doi: 10.1124/mol.107.041152. [DOI] [PubMed] [Google Scholar]
- Grunnet M, Hansen RS, Olesen SP. hERG1 channel activators: a new anti-arrhythmic principle. Prog Biophys Mol Biol. 2008;98:347–362. doi: 10.1016/j.pbiomolbio.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Hancox JC, Mcpate MJ, El Harchi A, Zhang YH. The hERG potassium channel and hERG screening for drug-induced torsades de pointes. Pharmacol Ther. 2008;119:118–132. doi: 10.1016/j.pharmthera.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Hansen RS, Diness TG, Christ T, Wettwer E, Ravens U, Olesen SP, Grunnet M. Biophysical characterization of the new human ether-a-go-go-related gene channel opener NS3623 [N-(4-bromo-2-(1H-tetrazol-5-yl)-phenyl)-N′-(3′-trifluoromethylphenyl)urea] Mol Pharmacol. 2006;70:1319–1329. doi: 10.1124/mol.106.026492. [DOI] [PubMed] [Google Scholar]
- Hardman RM, Stansfeld PJ, Dalibalta S, Sutcliffe MJ, Mitcheson JS. Activation gating of hERG potassium channels: S6 glycines are not required as gating hinges. J Biol Chem. 2007;282:31972–31981. doi: 10.1074/jbc.M705835200. [DOI] [PubMed] [Google Scholar]
- Herzberg IM, Trudeau MC, Robertson GA. Transfer of rapid inactivation and sensitivity to the class III antiarrhythmic drug E-4031 from HERG to M-eag channels. J Physiol. 1998;511:3–14. doi: 10.1111/j.1469-7793.1998.003bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosaka Y, Iwata M, Kamiya N, Yamada M, Kinoshita K, Fukunishi Y, Tsujimae K, Hibino H, Aizawa Y, Inanobe A, Nakamura H, Kurachi Y. Mutational analysis of block and facilitation of HERG current by a class III anti-arrhythmic agent, nifekalant. Channels (Austin) 2007;1:198–208. doi: 10.4161/chan.4691. [DOI] [PubMed] [Google Scholar]
- Imai YN, Ryu S, Oiki S. Docking model of drug binding to the human ether-a-go-go potassium channel guided by tandem dimer mutant patch-clamp data: a synergic approach. J Med Chem. 2009;52:1630–1638. doi: 10.1021/jm801236n. [DOI] [PubMed] [Google Scholar]
- Jiang M, Dun W, Fan JS, Tseng GN. Use-dependent ‘agonist’ effect of azimilide on the HERG channel. J Pharmacol Exp Ther. 1999;291:1324–1336. [PubMed] [Google Scholar]
- Jiang M, Zhang M, Maslennikov IV, Liu J, Wu DM, Korolkova YV, Arseniev AS, Grishin EV, Tseng GN. Dynamic conformational changes of extracellular S5-P linkers in the hERG channel. J Physiol. 2005;569:75–89. doi: 10.1113/jphysiol.2005.093682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YX, Lee A, Chen JY, Cadene M, Chait BT, Mackinnon R. The open pore conformation of potassium channels. Nature. 2002;417:523–526. doi: 10.1038/417523a. [DOI] [PubMed] [Google Scholar]
- Ju P, Pages G, Riek RP, Chen PC, Torres AM, Bansal PS, Kuyucak S, Kuchel PW, Vandenberg JI. The pore domain outer helix contributes to both activation and inactivation of the HERG K+ channel. J Biol Chem. 2009;284:1000–1008. doi: 10.1074/jbc.M806400200. [DOI] [PubMed] [Google Scholar]
- Kamiya K, Mitcheson JS, Yasui K, Kodama I, Sanguinetti MC. Open channel block of HERG K+ channels by vesnarinone. Mol Pharmacol. 2001;60:244–253. doi: 10.1124/mol.60.2.244. [DOI] [PubMed] [Google Scholar]
- Kamiya K, Niwa R, Mitcheson JS, Sanguinetti MC. Molecular determinants of HERG channel block. Mol Pharmacol. 2006;69:1709–1716. doi: 10.1124/mol.105.020990. [DOI] [PubMed] [Google Scholar]
- Kamiya K, Niwa R, Morishima M, Honjo H, Sanguinetti MC. Molecular determinants of hERG channel block by terfenadine and cisapride. J Pharmacol Sci. 2008;108:301–307. doi: 10.1254/jphs.08102fp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, Chen Xl, Wang H, Ji J, Cheng H, Incardona J, Reynolds W, Viviani F, Tabart M, Rampe D. Discovery of a small molecule activator of the human ether-a-go-go-related gene (HERG) cardiac K+ channel. Mol Pharmacol. 2005;67:827–836. doi: 10.1124/mol.104.006577. [DOI] [PubMed] [Google Scholar]
- Labro AJ, Raes AL, Bellens I, Ottschytsch N, Snyders DJ. Gating of shaker-type channels requires the flexibility of S6 caused by prolines. J Biol Chem. 2003;278:50724–50731. doi: 10.1074/jbc.M306097200. [DOI] [PubMed] [Google Scholar]
- Lees-Miller JP, Duan Y, Teng GQ, Duff HJ. Molecular determinant of high-affinity dofetilide binding to HERG1 expressed in Xenopus oocytes: involvement of S6 sites. Mol Pharmacol. 2000;57:367–374. [PubMed] [Google Scholar]
- Liu J, Zhang M, Jiang M, Tseng GN. Structural and functional role of the extracellular S5-P linker in the HERG potassium channel. J Gen Physiol. 2002;120:723–737. doi: 10.1085/jgp.20028687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Zhang M, Jiang M, Tseng GN. Negative charges in the transmembrane domains of the HERG K channel are involved in the activation- and deactivation-gating processes. J Gen Physiol. 2003;121:599–614. doi: 10.1085/jgp.200308788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- McPate MJ, Duncan RS, Hancox JC, Witchel HJ. Pharmacology of the short QT syndrome N588K-hERG K+ channel mutation: differential impact on selected class I and class III antiarrhythmic drugs. Br J Pharmacol. 2008;155:957–966. doi: 10.1038/bjp.2008.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milnes JT, Crociani O, Arcangeli A, Hancox JC, Witchel HJ. Blockade of HERG potassium currents by fluvoxamine: incomplete attenuation by S6 mutations at F656 or Y652. Br J Pharmacol. 2003;139:887–898. doi: 10.1038/sj.bjp.0705335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milnes JT, Witchel HJ, Leaney JL, Leishman DJ, Hancox JC. hERG K+ channel blockade by the antipsychotic drug thioridazine: An obligatory role for the S6 helix residue F656. Biochem Biophys Res Commun. 2006;351:273–280. doi: 10.1016/j.bbrc.2006.10.039. [DOI] [PubMed] [Google Scholar]
- Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. A structural basis for drug-induced long QT syndrome. Proc Natl Acad Sci U S A. 2000a;97:12329–12333. doi: 10.1073/pnas.210244497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitcheson JS, Chen J, Sanguinetti MC. Trapping of a methanesulfonanilide by closure of the HERG potassium channel activation gate. J Gen Physiol. 2000b;115:229–240. doi: 10.1085/jgp.115.3.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myokai T, Ryu S, Shimizu H, Oiki S. Topological mapping of the asymmetric drug binding to the human ether-a-go-go-related gene product (HERG) potassium channel by use of tandem dimers. Mol Pharmacol. 2008;73:1643–1651. doi: 10.1124/mol.107.042085. [DOI] [PubMed] [Google Scholar]
- Perrin MJ, Kuchel PW, Campbell TJ, Vandenberg JI. Drug binding to the inactivated state is necessary but not sufficient for high-affinity binding to human ether-a-go-go-related gene channels. Mol Pharmacol. 2008;74:1443–1452. doi: 10.1124/mol.108.049056. [DOI] [PubMed] [Google Scholar]
- Perry M, De Groot MJ, Helliwell R, Leishman D, Tristani-Firouzi M, Sanguinetti MC, Mitcheson J. Structural determinants of HERG channel block by clofilium and ibutilide. Mol Pharmacol. 2004;66:240–249. doi: 10.1124/mol.104.000117. [DOI] [PubMed] [Google Scholar]
- Perry M, Sachse FB, Abbruzzese J, Sanguinetti MC. PD-118057 contacts the pore helix of hERG1 channels to attenuate inactivation and enhance K+ conductance. Proc Natl Acad Sci U S A. 2009;106:20075–20080. doi: 10.1073/pnas.0906597106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry M, Sachse FB, Sanguinetti MC. Structural basis of action for a human ether-a-go-go-related gene 1 potassium channel activator. Proc Natl Acad Sci U S A. 2007;104:13827–13832. doi: 10.1073/pnas.0703934104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry M, Stansfeld PJ, Leaney J, Wood C, De Groot MJ, Leishman D, Sutcliffe MJ, Mitcheson JS. Drug binding interactions in the inner cavity of HERG channels: molecular insights from structure-activity relationships of clofilium and ibutilide analogs. Mol Pharmacol. 2006;69:509–519. doi: 10.1124/mol.105.016741. [DOI] [PubMed] [Google Scholar]
- Piper DR, Varghese A, Sanguinetti MC, Tristani-Firouzi M. Gating currents associated with intramembrane charge displacement in HERG potassium channels. Proc Natl Acad Sci U S A. 2003;100:10534–10539. doi: 10.1073/pnas.1832721100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper DR, Hinz WA, Tallurri CK, Sanguinetti MC, Tristani-Firouzi M. Regional specificity of hERG channel activation and inactivation gating. J Biol Chem. 2005;280:7206–7217. doi: 10.1074/jbc.M411042200. [DOI] [PubMed] [Google Scholar]
- Rajamani S, Eckhardt LL, Valdivia CR, Klemens CA, Gillman BM, Anderson CL, Holzem KM, Delisle BP, Anson BD, Makielski JC, January CT. Drug-induced long QT syndrome: hERG K+ channel block and disruption of protein trafficking by fluoxetine and norfluoxetine. Br J Pharmacol. 2006;149:481–489. doi: 10.1038/sj.bjp.0706892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recanatini M, Poluzzi E, Masetti M, Cavalli A, De Ponti F. QT prolongation through hERG K+ channel blockade: current knowledge and strategies for the early prediction during drug development. Med Res Rev. 2005;25:133–166. doi: 10.1002/med.20019. [DOI] [PubMed] [Google Scholar]
- Redfern WS, Carlsson L, Davis AS, Lynch WG, Mackenzie I, Palethorpe S, Siegl PK, Strang I, Sullivan AT, Wallis R, Camm AJ, Hammond TG. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc Res. 2003;58:32–45. doi: 10.1016/s0008-6363(02)00846-5. [DOI] [PubMed] [Google Scholar]
- Ridley JM, Dooley PC, Milnes JT, Witchel HJ, Hancox JC. Lidoflazine is a high affinity blocker of the HERG K+ channel. J Mol Cell Cardiol. 2004;36:701–705. doi: 10.1016/j.yjmcc.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Roden DM. Mechanisms and management of proarrhythmia. Am J Cardiol. 1998;82:49I–57I. doi: 10.1016/s0002-9149(98)00472-x. [DOI] [PubMed] [Google Scholar]
- Sanchez-Chapula JA, Ferrer T, Navarro-Polanco RA, Sanguinetti MC. Voltage-dependent profile of human ether-a-go-go-related gene channel block is influenced by a single residue in the S6 transmembrane domain. Mol Pharmacol. 2003;63:1051–1058. doi: 10.1124/mol.63.5.1051. [DOI] [PubMed] [Google Scholar]
- Sanchez-Chapula JA, Navarro-Polanco RA, Culberson C, Chen J, Sanguinetti MC. Molecular determinants of voltage-dependent human ether-a-go-go related gene (HERG) K+ channel block. J Biol Chem. 2002;277:23587–23595. doi: 10.1074/jbc.M200448200. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- Schonherr R, Heinemann SH. Molecular determinants for activation and inactivation of HERG, a human inward rectifier potassium channel. J Physiol. 1996;493:635–642. doi: 10.1113/jphysiol.1996.sp021410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PL, Baukrowitz T, Yellen G. The inward rectification mechanism of the HERG cardiac potassium channel. Nature. 1996;379:833–836. doi: 10.1038/379833a0. [DOI] [PubMed] [Google Scholar]
- Smith PL, Yellen G. Fast and slow voltage sensor movements in HERG potassium channels. J Gen Physiol. 2002;119:275–293. doi: 10.1085/jgp.20028534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector PS, Curran ME, Keating MT, Sanguinetti MC. Class III antiarrhythmic drugs block HERG, a human cardiac delayed rectifier K+ channel. Open-channel block by methanesulfonanilides. Circ Res. 1996a;78:499–503. doi: 10.1161/01.res.78.3.499. [DOI] [PubMed] [Google Scholar]
- Spector PS, Curran ME, Zou A, Keating MT, Sanguinetti MC. Fast inactivation causes rectification of the IKr channel. J Gen Physiol. 1996b;107:611–619. doi: 10.1085/jgp.107.5.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, Moss AJ, Schwartz PJ, Towbin JA, Vincent GM, Keating MT. Spectrum of mutations in long-QT syndrome genes: KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178–1185. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- Stansfeld PJ, Grottesi A, Sands ZA, Sansom MS, Gedeck P, Gosling M, Cox B, Stanfield PR, Mitcheson JS, Sutcliffe MJ. Insight into the mechanism of inactivation and pH sensitivity in potassium channels from molecular dynamics simulations. Biochemistry. 2008;47:7414–7422. doi: 10.1021/bi800475j. [DOI] [PubMed] [Google Scholar]
- Stork D, Timin EN, Berjukow S, Huber C, Hohaus A, Auer M, Hering S. State dependent dissociation of HERG channel inhibitors. Br J Pharmacol. 2007;151:1368–1376. doi: 10.1038/sj.bjp.0707356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbiah RN, Clarke CE, Smith DJ, Zhao J, Campbell TJ, Vandenberg JI. Molecular basis of slow activation of the human ether-á-go-go related gene potassium channel. J Physiol. 2004;558:417–431. doi: 10.1113/jphysiol.2004.062588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbiah RN, Kondo M, Campbell TJ, Vandenberg JI. Tryptophan scanning mutagenesis of the HERG K+ channel: the S4 domain is loosely packed and likely to be lipid exposed. J Physiol. 2005;569:367–379. doi: 10.1113/jphysiol.2005.097386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z, Limberis J, Souers A, Kym P, Mikhail A, Houseman K, Diaz G, Liu X, Martin RL, Cox BF, Gintant GA. Electrophysiologic characterization of a novel hERG channel activator. Biochem Pharmacol. 2009;77:1383–1390. doi: 10.1016/j.bcp.2009.01.015. [DOI] [PubMed] [Google Scholar]
- Swartz KJ. Towards a structural view of gating in potassium channels. Nat Rev Neurosci. 2004;5:905–916. doi: 10.1038/nrn1559. [DOI] [PubMed] [Google Scholar]
- Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995;269:92–95. doi: 10.1126/science.7604285. [DOI] [PubMed] [Google Scholar]
- Webster SM, Del Camino D, Dekker JP, Yellen G. Intracellular gate opening in Shaker K+ channels defined by high-affinity metal bridges. Nature. 2004;428:864–868. doi: 10.1038/nature02468. [DOI] [PubMed] [Google Scholar]
- Xu X, Recanatini M, Roberti M, Tseng GN. Probing the binding sites and mechanisms of action of two human ether-a-go-go-related gene channel activators, 1,3-bis-(2-hydroxy-5-trifluoromethyl-phenyl)-urea (NS1643) and 2-[2-(3,4-dichloro-phenyl)-2,3-dihydro-1H-isoindol-5-ylamino]-nicotinic acid (PD307243) Mol Pharmacol. 2008;73:1709–1721. doi: 10.1124/mol.108.045591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Lozinskaya IM, Lin Z, Willette RN, Brooks DP, Xu X. Mallotoxin is a novel human ether-a-go-go-related gene (hERG) potassium channel activator. J Pharmacol Exp Ther. 2006;319:957–962. doi: 10.1124/jpet.106.110593. [DOI] [PubMed] [Google Scholar]
- Zhang M, Liu J, Tseng GN. Gating charges in the activation and inactivation processes of the HERG channel. J Gen Physiol. 2004;124:703–718. doi: 10.1085/jgp.200409119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Augelli-Szafran CE, Bradley JA, Chen X, Koci BJ, Volberg WA, Sun Z, Cordes JS. Novel potent human ether-a-go-go-related gene (hERG) potassium channel enhancers and their in vitro antiarrhythmic activity. Mol Pharmacol. 2005;68:876–884. doi: 10.1124/mol.105.014035. [DOI] [PubMed] [Google Scholar]