

Abstract
The postsynaptic muscle-specific kinase (MuSK) coordinates formation of the neuromuscular junction (NMJ) during embryonic development. Here we have studied the effects of MuSK autoantibodies upon the NMJ in adult mice. Daily injections of IgG from four MuSK autoantibody-positive myasthenia gravis patients (MuSK IgG; 45 mg day−1i.p. for 14 days) caused reductions in postsynaptic ACh receptor (AChR) packing as assessed by fluorescence resonance energy transfer (FRET). IgG from the patients with the highest titres of MuSK autoantibodies caused large (51–73%) reductions in postsynaptic MuSK staining (cf. control mice; P < 0.01) and muscle weakness. Among mice injected for 14 days with control and MuSK patient IgGs, the residual level of MuSK correlated with the degree of impairment of postsynaptic AChR packing. However, the loss of postsynaptic MuSK preceded this impairment of postsynaptic AChR. When added to cultured C2 muscle cells the MuSK autoantibodies caused tyrosine phosphorylation of MuSK and the AChR β-subunit, and internalization of MuSK from the plasma membrane. The results suggest a pathogenic mechanism in which MuSK autoantibodies rapidly deplete MuSK from the postsynaptic membrane leading to progressive dispersal of postsynaptic AChRs. Moreover, maintenance of postsynaptic AChR packing at the adult NMJ would appear to depend upon physical engagement of MuSK with the AChR scaffold, notwithstanding activation of the MuSK-rapsyn system of AChR clustering.
Introduction
Myasthenia gravis (MG) is the archetypal autoimmune disease of the synapse. In classical MG, autoantibodies against the postsynaptic AChR reduce the efficacy of neuromuscular transmission by hindering AChR channel function, accelerating AChR degradation and activating complement attack upon the postsynaptic membrane (Conti-Fine et al. 2006). However, some MG patients display plasma autoantibodies against MuSK instead of classical AChR autoantibodies (Hoch et al. 2001). Experimental animal models involving either active immunization with MuSK, or passive transfer of patient IgG have recently confirmed that MuSK autoantibodies can cause MG, but the precise actions of the antibodies upon the endplate remain uncertain (Jha et al. 2006; Shigemoto et al. 2006; Cole et al. 2008; ter Beek et al. 2009).
MuSK is a postsynaptic receptor tyrosine kinase needed for development of NMJs (Glass et al. 1996; Sanes & Lichtman, 2001). MuSK forms a protein complex together with its agrin-binding co-receptor, the low density lipoprotein receptor-related protein 4 (LRP4; Kim et al. 2008; Zhang et al. 2008), and several cytoplasmic proteins that collectively reinforce MuSK phosphorylation and convey the signals needed for AChR clustering (Luo et al. 2002; Linnoila et al. 2008; Inoue et al. 2009). While the signalling pathways remain to be fully defined, activation of the MuSK complex initiates reorganization of the cortical actin cytoskeleton and may help regulate sub-synaptic transcription of MuSK, AChR and other synaptic genes (Weston et al. 2000; Lacazette et al. 2003; Madhavan & Peng, 2005). The MuSK–rapsyn system is perhaps the best-understood effector of AChR clustering. Activation of the MuSK complex (by agrin) leads to tyrosine phosphorylation of the AChR β-subunit (Y390), thereby recruiting the adaptor protein, rapsyn (Borges et al. 2008). Binding of extra rapsyn to each AChR helps pack the postsynaptic AChRs tightly together in an AChR scaffold and slows the metabolic turnover of the AChRs (Bezakova et al. 2001; Moransard et al. 2003; Gervásio & Phillips, 2005; Gervásio et al. 2007; Borges et al. 2008; Brockhausen et al. 2008). This tight packing of postsynaptic AChRs (approximately 104μm−2) is required for the fast, effective response to ACh (Land et al. 1980). Apart from signalling functions, MuSK may interact with other proteins in the postsynaptic membrane (Fuhrer et al. 1999; Zhou et al. 1999; Antolik et al. 2007). Thus, MuSK may also play a structural role by helping to hold various components of the AChR scaffold together.
It remains unclear whether MuSK autoantibodies cause synaptic failure at the NMJ by interfering in some way with the expression or function of MuSK. Here we show that injection of IgG from MuSK autoantibody-positive MG patients into mice depleted MuSK from the postsynaptic membrane. The reduction in postsynaptic MuSK was followed by a delayed loosening of postsynaptic AChR packing, suggesting that the presence of MuSK in the membrane supports the long-term stability of the postsynaptic AChR scaffold at the adult NMJ.
Methods
Ethical approval
The article by Drummond (2009) was read carefully to ensure that our experiments complied with the policies and regulations regarding animal experimentation and other ethical matters. Mouse experiments were conducted at the University of Sydney and were approved by the University of Sydney Animal Ethics Committee in accordance with the New South Wales Government Animal Research Act 1985, associated regulations (2005) and the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes, 7th edn (National Health and Medical Research Council, 2004). Consent was obtained from the patients involved in this study in accordance with the Declaration of Helsinki (5th revision, 2004). The project was approved by the Human Research Ethics Committee of the Sydney South West Area Health Service.
Passive transfer of human IgG
Four MuSK autoantibody-positive MG patients who undertook therapeutic plasmapheresis provided the IgG for this study (MuSK1 to MuSK4). Their plasma was screened positive (by immunoprecipitation) for MuSK autoantibodies and negative for anti-AChR by immunoprecipitation. Pooled control plasma was screened negative for antibodies against both MuSK and AChR (Cole et al. 2008). Supplementary data S1 summarizes clinical details. Enzyme-linked immunosorbant assay revealed differences in the MuSK autoantibody titres among the four patients (Supplementary data S2). The rank order for the titres was MuSK2 > MuSK3 > MuSK4 ≈ MuSK1. Passive IgG transfer to mice was conducted as previously described (Cole et al. 2008). Briefly, 6-week-old female C57BL/6J mice (Animal Resources Centre, Western Australia) were injected daily i.p. with 45 mg filter-sterilized human IgG dissolved in PBS. For experiments in which the mouse was to be injected for more than 5 days, a single i.p. injection of cyclophosphamide (300 mg kg−1) was given 24 h after the first IgG injection to suppress any active immune response to the human protein (Toyka et al. 1977). Mice were inspected daily and were killed after 14 days, or when they displayed weight loss and developed muscle weakness (whichever came first). Mice were graded for muscle weakness according to their posture before and after a brief exercise regime (Stacy et al. 2002; Cole et al. 2008). Mice were first rendered unconscious by anaesthesia (2.5 mg ketamine and 0.5 mg Xylazine per 20 g body wt, i.p.) then killed by cervical dislocation after the foot-withdrawal reflex was completely suppressed. A total of 54 mice were used in this study.
Immunofluorescence
Monoclonal mouse anti-rapsyn (mab1234; Peng & Froehner, 1985) hybridoma supernatant was diluted 1:2. Anti-AChR β-subunit antibody (mab124; a gift from Dr Jon Lindstrom) was used for immunoblotting at 1 nm. Affinity-purified fluorescent secondary antibodies were diluted as follows: 1:200 tetramethyl-rhodamine isothiocyanate-conjugated donkey anti-sheep IgG (H+L; Jackson Labs, Baltimore, PA, USA) and 1:500 Alexafluor 488-conjugated donkey anti-mouse IgG (Molecular Probes, Carlsbad, CA, USA). Alexafluor 488–α-bungarotoxin (Alexa488–α-bungarotoxin) and Alexafluor 555–α-bungarotoxin (Molecular Probes) were diluted to a final concentration of 588 nm. All antibodies were diluted in 1% bovine serum albumin (BSA) in PBS.
Antibodies were raised against the recombinant extracellular domain of human MuSK (RSR Limited, Cardiff, UK) by immunizing a sheep (IMVS, South Australia). The antibodies were affinity purified on a column of the immunogen protein and recognized a single band of approximately 100 kD on immunoblots of mouse C2 myoblasts (Fig. 6B). To reduce non-specific binding of the affinity-purified sheep anti-MuSK IgG the following procedure was employed. A strip of PVDF membrane (GE Healthcare, Amersham Hybond-P, PVDF Transfer Membrane, Buckinghamshire, UK) was washed once in methanol (90%, 1 min), twice in water (1 min each) then dot blotted with BSA–PBS (5% w/v). The membrane was dried and the methanol and water washes repeated. The membrane was placed in a 50 ml conical tube with 2 ml of a 1:50 PBS dilution of affinity-purified sheep anti-MuSK antibody. The tube was incubated overnight at 4°C on a rolling bed. The supernatant was used for immunofluorescent staining at a final dilution of 1:50. Indirect immunofluorescent staining and quantitative microscopy using a Zeiss LSM 510 Meta confocal microscope were described previously (Cole et al. 2008). While both the human and sheep antibodies bound to the 62 kD extracellular domain of MuSK, immunofluorescent staining of endplates with our affinity-purified sheep anti-MuSK antibodies was not inhibited when muscle sections were preincubated with high concentrations of MuSK2 IgG (Supplementary data S3).
Figure 6. MuSK autoantibodies induce tyrosine phosphorylation of MuSK.
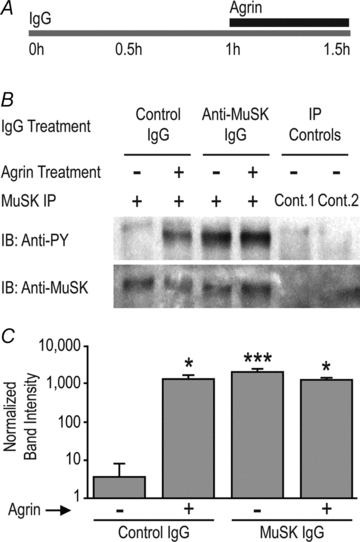
A, C2 myotubes were exposed to human IgG (2 mg ml−1) for 1 h followed by neural agrin (1 nm) for a further 30 min in the continued presence of IgG. B, sample immunoblot showing increased anti-phosphotyrosine staining (anti-PY) of the MuSK band (lower panel) in myotubes exposed to neural agrin and/or MuSK2 IgG. Controls involved substitution of normal human IgG for the immunoprecipitating antibody (Cont.1), or omission of the precipitating antibody (Cont.2). C, quantification of MuSK phosphorylation. MuSK2 IgG increased tyrosine phosphorylation of the MuSK band and precluded any further effect of neural agrin. Bars represent the mean +s.e.m. of n= 3 culture experiments. Asterisks represent bars that were significantly different from the far left control bar.
Quantification of endplate staining intensities and AChR–AChR FRET
The relative intensity of postsynaptic membrane fluorescence was determined in transverse optical sections as previously described (Gervásio et al. 2007). ImageJ 1.31v software (National Institutes of Health; Bethesda, MD, USA; http://rsb.info.nih.gov/ij) was used to measure the areas and relative fluorescence intensities. Fluorescence resonance energy transfer (FRET) between tightly packed postsynaptic AChRs was assessed by the photobleaching of acceptor method (Gervásio et al. 2007; Brockhausen et al. 2008). AChRs were labelled with a mixture of α-bungarotoxin conjugates serving as the donor and acceptor fluorophors (Alexafluor 555 and Alexafluor 647, respectively). To test linkage of postsynaptic AChRs to the cytoskeleton, unfixed cryosections were incubated with a solution of saponin (1 mg ml−1), Triton X-100 (10%), sodium m-periodate (21 mg ml−1) and l-lysine hydrochloride, (180 mg ml−1) in PBS for 10 min at room temperature prior to staining with fluorescent α-bungarotoxin as above.
Myotube cultures and associated assays
Mouse C2 muscle cells were passaged and AChR clustering experiments performed as previously described (Ngo et al. 2004; Brockhausen et al. 2008). We used a recombinant C-terminal fragment of rat agrin containing the 3, 4 and 8 amino acid inserts at the X, Y and Z mRNA splicing sites, respectively (Rupp et al. 1992; R&D Systems, Minneapolis, MN, USA). For immunoprecipitation experiments 6 cm plates of myotubes were washed twice with PBS–2 mm Na3VO4. Cells were lysed with 0.5 ml of lysis buffer (50 mm Tris pH 7.5, 150 mm NaCl, 10 mm NaF, 10 mm Na4P2O4, 1 mm Na3VO4, 1% NP-40, containing 0.01% Sigma protease inhibitor cocktail no. 1). Human anti-MuSK antibody affinity purified from MuSK2 IgG was added to the lysate supernatant overnight at 4°C. Protein-G Sepharose beads (75 μl per tube; Amersham; Pittsburgh, PA, USA) were then added for 5 h. After washing three times with lysis buffer and once with wash buffer (50 mm Tris, pH 8), proteins were eluted with 5× SDS sample buffer containing 100 mm DTT. MuSK protein was resolved by SDS-PAGE, transferred to a PVDF membrane (Amersham) and probed with anti-phosphotyrosine antibody 4G10 (1:1000; Chemicon, Temecula, CA, USA). Blots were later stripped and re-probed with affinity-purified sheep anti-MuSK (1:125). Bands were visualized with anti-mouse-HRP (1:2000; Santa Cruz Biotech, Santa Cruz, CA, USA) or anti-sheep IgG-HRP (1:2000; Sigma, St Louis, MO, USA) as appropriate, followed by ECL-plus chemiluminescence (Amersham). Controls included omission of anti-MuSK antibody from the immunoprecipitation step and substitution with control human IgG (10 mg ml−1).
To assess tyrosine phosphorylation of the AChR β-subunit, cell surface AChRs were labelled for 2 h at 4°C with 200 nm biotin–α-bungarotoxin (Invitrogen). After washing and lysis as above, AChRs were selectively precipitated with streptavidin beads (75 μl per tube; Thermo Scientific, Rockford, IL, USA) overnight at 4°C. As a control for the specificity of AChR immunoprecipitation, biotin–α-bungarotoxin was omitted. Immunoblots were probed for phosphotyrosine followed by stripping and re-probing for the AChR β-subunit with mab124. ImageJ 1.31v software was used for measurement of integrated pixel intensity values of bands (Brockhausen et al. 2008).
Time-lapse imaging of MuSK–EGFP internalization
C2 myoblasts were grown on 35 mm glass-bottom dishes (Matek, Homer, MA, USA) and transfected for 24 h with MuSK fused to enhanced green fluorescent protein (MuSK–EGFP; Cole et al. 2008). Myoblasts were imaged on a Zeiss LSM 510 Meta confocal microscope using a C-Apochromat 40× 1.2 NA water-immersion objective (Fig. 8 and Supplementary Movie) in the following solution (in mm): 140 NaCl, 5 KCl, 2.5 CaCl2, 1 MgCl2, 10 glucose. Cells were repeatedly imaged at 90 s intervals over an 80 min period at ambient temperature (∼22°C). A 4-dimensional laser scanning confocal microscopy analysis was performed (Z-stack over time) with optical sections collected at 1 μm intervals. ImageJ software was used to compare total and intracellular MuSK–EGFP fluorescence intensity and the area of the cells. Maximum intensity Z-projections were used to compare total cellular MuSK–EGFP level within a cell over time. To assess the degree of MuSK–EGFP internalization, single optical slices through the core of the cell were analysed.
Figure 8. MuSK autoantibodies cause internalization of MuSK.
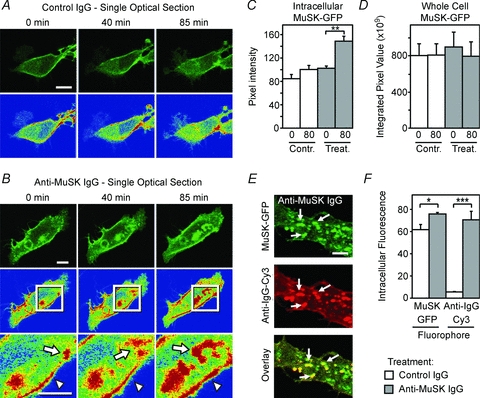
A, time-lapse imaging of MuSK–EGFP in a mouse C2 myoblast. Optical sections are shown through the core of a single myoblast 0, 40 and 85 min after adding control IgG (2 mg ml−1) to the culture medium. Pseudo-colour images (lower panels) revealed little net change in the subcellular distribution of MuSK over 85 min (red represents the most intense staining). B, time-lapse imaging of MuSK–EGFP in a single myoblast 0, 40 and 85 min after adding MuSK2 IgG to the culture medium. MuSK–EGFP accumulated in large intracellular compartments via a series of endocytic events (arrows in inset). C, increase in the average intensity of MuSK–EGFP fluorescence deep in the cytoplasm following the addition of MuSK2 IgG at time zero. D, maximum-projection images revealed no significant change in total cellular MuSK–EGFP over 80 min with either MuSK2 or control IgG. E, double labelling reveals human IgG (anti-IgG-Cy3) co-internalized with MuSK–EGFP (arrows). F, increased intracellular fluorescence intensity reflecting net internalization of both MuSK–EGFP and human IgG (anti-IgG-Cy3) in MuSK2 IgG-treated cells, compared with cells exposed to control human IgG. Data in panels C and D represent n= 3 cells imaged from 3 separate cultures. Data in panel F represent n= 3 independent culture experiments. Scale bar is 10 μm for panels A and B and 5 μm for panel E.
Analysis of mRNAs
RNA samples were prepared from muscle using an RNeasy Mini Kit (Qiagen, Germany) according to manufacturer's instructions. Reverse transcription and real-time PCR for rapsyn were performed as previously described (Brockhausen et al. 2008). AChR α1-subunit complementary DNA fragments (74 bp) were amplified using 100 nm AChR TaqMan probe that straddled the exon 3/4 boundary (5′-6FAMCGTCTGAAACAGCAATGGGTMGBNFQ-3′, Applied Biosystems), 1× TaqMan Universal PCR MasterMix, and 600 nm of both forward (5′-AGTAAATCAGATTGTGACAACCA-3′) and reverse (3′-CTGGATTCCATTTCAAGTTGTA-5′) primers. MuSK fragments (88 bp) were amplified using the TaqMan Gene Expression Assay for MuSK. The MuSK TaqMan probe straddled the exon 4/5 boundary.
Statistical analysis
For pair-wise comparison we used a paired Student's t test (Fig. 1J and K). Multiple treatment groups were compared by one-way ANOVA (Figs 1D–G, I, 2, 4A, B and D) or two-way ANOVA (Figs 5F, 6C, 7C and 8C, D and F), with Bonferroni's multiple comparison post-test. Data for AChR cluster areas in Fig. 5G was skewed, so the non-parametric Mann–Whitney test was used. Significance is indicated throughout as follows: *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 1. MuSK IgG reduced postsynaptic AChR packing density.
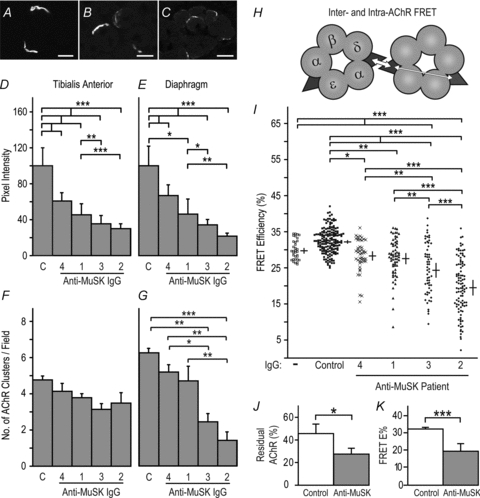
Mice were injected with human IgG (45 mg day−1, i.p.) for 14 days before killing. A, transverse optical section showing bright postsynaptic AChR plaques at NMJs in the TA muscle of a mouse injected with control human IgG. B and C, reduced postsynaptic AChR staining in mice injected with MuSK3 or MuSK2 IgGs, respectively. D and E, intensity of postsynaptic AChR staining at synapses in the TA and diaphragm muscles, respectively. The source of the IgG (patients 1–4 or control, C) is indicated beneath each bar. F and G, counts of the number of AChR plaques (NMJs) per photomicrograph field in the TA and diaphragm muscles, respectively. H, fluorescently labelled α-bungarotoxin molecules bound to AChRs function as donor and acceptor fluorophors (filled triangles) for FRET. Inter-AChR FRET (thick arrow) is much more efficient than intra-AChR FRET (thin arrow; ∼30% cf. ∼5%) because FRET efficiency declines sharply with distance (Gervásio et al. 2007). I, efficiency of postsynaptic AChR-AChR FRET. Symbols show the range of FRET efficiencies for individual synapses in TA muscles. Crossed bars show the mean endplate FRET efficiencies for the 3 mice in each treatment group (±s.e.m.). J, percentage of postsynaptic fluorescent α-bungarotoxin staining intensity remaining after extracting cryosections with non-ionic detergent. Mice injected with MuSK2 IgG for 14 days showed a significant reduction in the fraction of AChR that was resistant to detergent extraction (residual AChR), compared to control mice. K, FRET data for MuSK2 IgG-injected mice re-plotted from panel I for comparison. Data in panels D–G and I–K represent mean +s.e.m. for n= 3 mice. Scale bars represent 30 μm for panels A and B and 20 μm in panel C.
Figure 2. Expression of mRNAs encoding MuSK, AChR α-subunit and rapsyn.
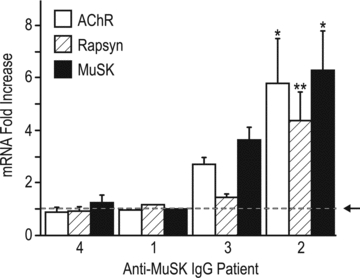
After 14 days of injections with human IgG (45 mg day−1), mice were killed and levels of mRNAs were compared by real-time PCR using Taqman probes. The horizontal dashed line (arrow) represents the average level for mice injected with control human IgG. No change was detected in mice injected with MuSK1 and MuSK4 IgGs. Mice injected with MuSK2 IgG showed significant increases in all three mRNAs. Data show the mean +s.e.m. for n= 3 mice for MuSK2, 3 and 4. For this experiment the remaining IgG from patient 1 was sufficient to inject only one mouse. Asterisks indicate significance relative to the control value.
Figure 4. Loss of postsynaptic MuSK precedes impaired AChR packing.
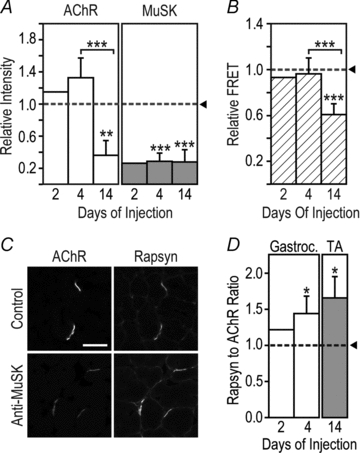
Mice were injected with MuSK2 (45 mg i.p. day−1) for 2, 4 or 14 days. A, intensity of postsynaptic staining for MuSK and AChR. Data were normalized to the mean value for mice injected with control human IgG (horizontal dotted line). B, efficiency of postsynaptic AChR–AChR FRET. C, double staining of endplates for rapsyn and AChR. Mice were injected for 14 days with MuSK2 IgG (anti-MuSK) or control IgG. Scale bar is 30 μm. D, the postsynaptic rapsyn-to-AChR fluorescence ratio was elevated in mice injected with MuSK2 IgG compared to control mice (horizontal dotted line). Data for gastrocnemius (Gastroc.) and TA muscles are shown. Data represent the mean +s.e.m. for n= 3 mice injected for either 4 days or 14 days, and the mean for n= 2 mice injected for 2 days.
Figure 5. MuSK autoantibodies cause disassembly of pre-existing AChR clusters in culture.
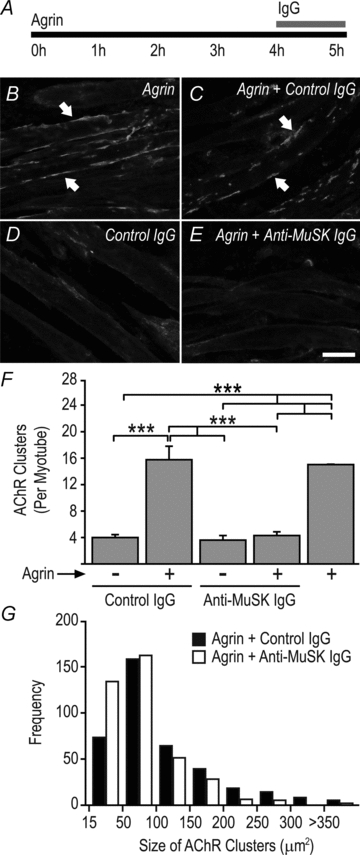
A, mouse C2 myotubes were exposed to 1 nm agrin for 4 h to form large AChR clusters. Human IgG (4.5 mg ml−1 final) was then added to the culture for 1 h in the continued presence of agrin. B, agrin-treated myotubes (no IgG) displaying large AChR clusters (arrows). C, agrin treatment followed by control human IgG. D, myotubes treated with control IgG alone (no agrin). E, agrin treatment followed by exposure to MuSK2 IgG for 1 h. Scale bar is 25 μm. F, counts of the number of AChR clusters per myotube segment. All AChR clusters larger than 15 μm2 were counted. Bars show mean +s.e.m. for n= 3 culture experiments. G, frequency distributions representing the size of AChR clusters from myotubes treated with agrin followed by either MuSK2 IgG or control IgG for 1 h.
Figure 7. MuSK autoantibodies increased tyrosine phosphorylation of the AChR β-subunit.
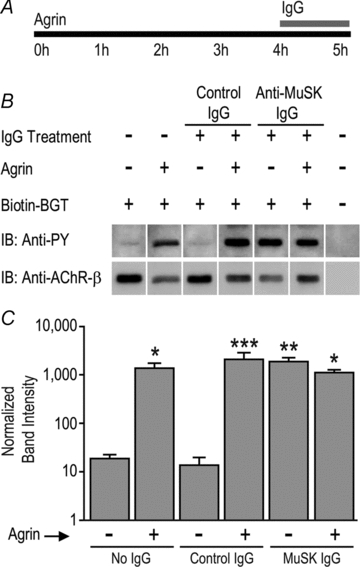
A, myotubes were exposed to neural agrin (1 nm) for 4 h followed by human IgG (4.5 mg ml−1) for a further 1 h in the continued presence of agrin. B, sample immunoblot showing bands labelled with anti-phosphotyrosine (anti-PY; upper panel) and re-probed with anti-AChR-β-subunit (mab124; lower panel). The last lane is a control for non-specific precipitation (biotin–α-bungarotoxin was omitted). C, quantification of AChR β-subunit phosphorylation. Bars represent the mean +s.e.m. of n= 3 culture experiments. Asterisks indicate bars that were significantly different from the far left control bar.
Results
MuSK autoantibody-positive IgG reduces postsynaptic AChR packing
Mice received daily injections of IgG from one of four myasthenia gravis patients (MuSK1–MuSK4) or control human IgG (45 mg day−1i.p.). After 14 days the intensity of postsynaptic AChR staining was significantly reduced in mice injected with IgG from each of the MuSK patients (Fig. 1A–E). MuSK autoantibody titres were highest for IgG from MuSK2 and MuSK3 (Supplementary data S1). Mice injected with these IgG preparations also displayed the most pronounced reductions in postsynaptic AChR staining intensities (Fig. 1B–E) and developed muscle weakness and weight loss (Cole et al. 2008). Average postsynaptic AChR staining intensities were reduced in both the diaphragm and tibialis anterior (TA) muscles (Fig. 1D and E). However, AChR staining of some endplates in the diaphragm was particularly impaired, with pixel intensities just above the threshold for measurement (Supplementary data S4). Moreover, blind counts revealed fewer AChR plaques per microscope field in the diaphragm muscles of mice injected with MuSK2 and MuSK3, compared to control mice (Fig. 1G), suggesting outright loss of some NMJs. Counts revealed no significant change in the number of AChR plaques per field in the TA muscle (Fig. 1F).
The reduced postsynaptic AChR staining might have resulted from impairment in either the biosynthesis, clustering or metabolic stability of the postsynaptic AChRs (Salpeter & Harris, 1983). To address the first possibility we examined the levels of mRNAs encoding the AChR α-subunit, MuSK and rapsyn in the TA muscle. Mice injected for 14 days with MuSK1 or MuSK4 IgG showed no change in the level of these mRNAs when compared to mice injected with control IgG. Mice injected with MuSK2 or MuSK3 IgG showed increases (rather than decreases) in AChR α-subunit, MuSK and rapsyn mRNAs (Fig. 2). The latter increases may have been secondary to muscle inactivity (see Discussion). In summary, we found no evidence of a reduction of mRNAs for MuSK, AChR-α or rapsyn in the TA muscle of mice injected with MuSK autoantibody-positive IgG despite serious impairment of postsynaptic AChR staining.
To assess the tightness of packing of postsynaptic AChR we employed a confocal fluorescence resonance energy transfer (FRET) assay. AChR–AChR FRET detects non-radiative energy transfer between fluorescent α-bungarotoxin molecules bound to AChRs within the postsynaptic membrane (Gervásio et al. 2007; Brockhausen et al. 2008). High efficiency inter-AChR FRET occurs only when AChRs are assembled at membrane densities of about 104μm−2 (Fig. 1H; Gervásio et al. 2007). Thus, FRET reveals the tight AChR packing that occurs at the lips of the postjunctional membrane infoldings, subjacent to sites of presynaptic ACh release (Salpeter & Harris, 1983). The scatter-plot in Fig. 1I shows the efficacy of AChR–AChR FRET at individual NMJs in the TA muscle. Cross-bars show the means for the three mice in each treatment group (±s.e.m.). As expected, untreated mice showed high AChR–AChR FRET efficiency (∼30%), as did mice injected with control human IgG for 14 days (Fig. 1I). The mean postsynaptic FRET efficiency was reduced in mice injected for 14 days with IgG from MuSK1–MuSK4 (Fig. 1I). MuSK2 and MuSK3 IgG produced the greatest reductions in postsynaptic AChR packing. The increased spread of scatter points (cf. controls) suggest that, at the time of killing, some NMJs were more severely affected by MuSK autoantibody injections than other NMJs in the same muscle (Fig. 1I).
To see if some postsynaptic AChRs might have become detached from the AChR scaffold, we subjected cryosections to detergent extraction. Detergent treatment extracted more AChR from the postsynaptic membrane of mice injected with MuSK2 IgG than from control endplates (Fig. 1J). Resistance of AChRs to detergent extraction was reduced roughly in proportion to the reduction in AChR–AChR FRET (compare Fig. 1J and K). AChRs that detach from the AChR scaffolds but remain within the postsynaptic membrane cause a proportional reduction in FRET efficiency (Gervásio et al. 2007; Brockhausen et al. 2008). Thus, the reduction in FRET efficiency might be explained simply by impaired retention of AChRs within the postsynaptic AChR scaffold.
Loss of MuSK precedes AChR lattice disassembly
Sheep anti-MuSK antibodies were used to study the spatial distribution of MuSK. Mice injected for 14 days with MuSK2 and MuSK3 IgGs showed marked reductions in the intensity of postsynaptic MuSK immunofluorescence compared to control mice (Fig. 3A; P < 0.01). Indeed, when results for all the mice were plotted, reduced levels of postsynaptic MuSK staining intensity correlated directly with reductions in AChR staining intensity and AChR–AChR FRET (Fig. 3B). The greater the autoantibody-induced loss of MuSK, the greater was the reduction in postsynaptic AChR.
Figure 3. MuSK autoantibodies reduced postsynaptic MuSK staining.
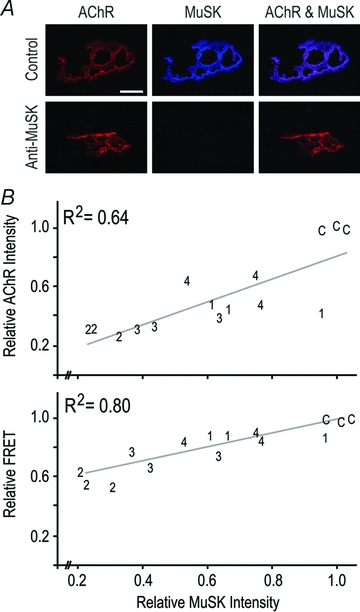
Mice were injected with human IgG as above for 14 days. A, extended focus images show double labelling of endplates for AChR (Alexa488–α-bungarotoxin) and MuSK (sheep anti-MuSK). In the case of mice injected with MuSK2 IgG the brightness and contrast was increased for reproduction. Scale bar is 10 μm. B, scatter plots depict postsynaptic MuSK staining intensity versus AChR staining intensity (upper plot), and MuSK intensity versus the efficiency of AChR–AChR FRET (lower plot). Each number on the plot is a data point (the average value for endplates from a single mouse). Numbers refer to the patient from whom the IgG was derived (C, control IgG). The R2 value shown in each panel is the square of the correlation coefficient for the least-squares line of best fit.
While IgG from all four patients produced similar trends of reduced postsynaptic MuSK, AChR and FRET, MuSK2 IgG displayed the highest autoantibody titre and produced the greatest magnitude of synaptic changes. We therefore focused on IgG from MuSK2 to study the time-course of changes in the postsynaptic membrane. Mice were injected daily with MuSK2 IgG for either 2, 4 or 14 days (45 mg day−1i.p.). TA muscles were sectioned and processed concomitantly for confocal imaging. At each time-point examined, postsynaptic MuSK immunofluorescence was reduced to about one quarter of control levels (Fig. 4A filled bars, dotted line represents the control mean). In contrast, postsynaptic AChR staining intensity and AChR–AChR FRET were reduced only later in the injection series (Fig. 4A open bars, Fig. 4B). Thus, in mice injected daily with MuSK2 IgG, MuSK was lost from the postsynaptic membrane before any impairment in AChR packing (Fig. 4A and B).
To assess the effect of MuSK2 IgG upon rapsyn (an effector of MuSK), we measured the intensity of postsynaptic anti-rapsyn immunofluorescence relative to AChR staining (Fig. 4C). The rapsyn-to-AChR fluorescence ratio was elevated in mice injected with MuSK2 IgG compared to mice injected with control IgG (Fig. 4D), suggesting possible activation of the MuSK–rapsyn system by the autoantibodies.
MuSK autoantibodies caused disassembly of AChR clusters in culture
When added to cultured C2 myotubes, MuSK2 IgG (4.5 mg ml−1) caused disassembly of pre-existing (agrin-induced) AChR clusters within 1 h (Fig. 5A–F). Control human IgG caused no change in the number of AChR clusters. Those AChR clusters that survived MuSK2 IgG treatment were significantly smaller than for control cultures (Fig. 5G, P < 0.001).
MuSK autoantibodies caused activation and internalization of MuSK
Myotubes were treated with neural agrin and/or human IgG to examine the effect upon activation of MuSK (Fig. 6A). In the presence of control human IgG, neural agrin caused the expected increase in phosphotyrosine staining of the MuSK band (Fig. 6B). However, MuSK2 IgG also caused tyrosine phosphorylation of the MuSK (Fig. 6B and C). Tyrosine phosphorylation of the AChR β-subunit was also strongly induced by both neural agrin (as expected) and MuSK2 IgG (Fig. 7B and C). Myotubes treated with a combination of neural agrin and MuSK2 IgG produced no greater tyrosine phosphorylation of either the MuSK or AChR β-subunit bands, suggesting that MuSK2 IgG was sufficient to maximally activate the MuSK–rapsyn pathway.
It has recently been reported that activation of MuSK involves MuSK internalization from the plasma membrane (Lu et al. 2007; Zhu et al. 2008). We therefore tested the effect of MuSK2 IgG upon the subcellular distribution of MuSK. For these studies we used C2 myoblasts (rather than myotubes) to simplify the collection and interpretation of time-lapse images. Myoblasts transfected with MuSK–EGFP were live-imaged at 90 s intervals over 80 min. Human IgG was added at time zero. MuSK–EGFP was initially spread across the peripheral membrane and was also concentrated in some intracellular compartments (consistent with ongoing biosynthesis and trafficking). Addition of control human IgG produced no marked change in the subcellular distribution of MuSK–EGFP (Fig. 8A). In contrast, cells exposed to MuSK2 IgG revealed an increase in MuSK–EGFP deep in the cytoplasm over the 80 min treatment (Fig. 8B and C). Internalization appeared to involve large endocytic engulfment events (Fig. 8B arrows; Supplementary movie). Double labelling of fixed cultures confirmed that MuSK2 IgG was specifically co-internalized with MuSK–EGFP (Fig. 8E and F). The percentage increase in intracellular MuSK–EGFP was smaller than for IgG because of the considerable intracellular MuSK–EGFP prior to addition of the antibodies (Fig. 8B–F). The increase in intracellular MuSK–EGFP could not be explained by any change in total MuSK–EGFP in the cells (Fig. 8D). These results suggest that human MuSK autoantibodies can cause internalization of MuSK from the plasma membrane, providing a potential explanation for the loss of postsynaptic MuSK in MuSK IgG-injected mice.
Discussion
Recent work in animal models confirms the potential of MuSK autoantibodies to cause myasthenia gravis, but the mechanism of antibody action has remained uncertain (Jha et al. 2006; Shigemoto et al. 2006; Cole et al. 2008; ter Beek et al. 2009). The in vivo and in vitro experiments described here suggest the following process. Binding of autoantibodies caused activation of MuSK, presumably by dimerization (Hopf & Hoch, 1998). Depletion of MuSK from the postsynaptic membrane occurred early in the injection series, possibly due to internalization of activated MuSK. This loss of MuSK was followed, after a delay of several days, by impaired postsynaptic AChR packing. The loss of postsynaptic AChRs might be explained by disassembly of the postsynaptic AChR scaffold and leaching of AChRs. In short, the results suggest the patient MuSK autoantibodies cause depletion of postsynaptic MuSK leading to disassembly of the AChR scaffold.
Postsynaptic AChR plaques were reduced in size and/or AChR staining intensity when animals were actively immunized with MuSK (Jha et al. 2006; Shigemoto et al. 2006) or received injections of IgG from MuSK autoantibody-positive patients but the in vivo mechanism was not determined (Cole et al. 2008). When added to cultured myotubes, rabbit anti-MuSK antibodies inhibited the ability of agrin and other agents to assemble AChR clusters (Shigemoto et al. 2006). Plasma and IgG from MuSK MG patients also inhibited formation of AChR clusters (Hoch et al. 2001; Farrugia et al. 2007; Cole et al. 2008) and caused disassembly of pre-existing clusters (current study). In these culture experiments, at least, the possibility of complement-mediated or cellular attack on the AChR cluster could be excluded. Thus, the autoantibodies appear to act by interfering with the expression or function of MuSK. Interestingly, the diaphragm muscle was more severely affected than the TA muscle in mice actively immunized with MuSK (Xu et al. 2006) and in mice injected with MuSK patient IgG (current work). A similar predilection for respiratory muscle involvement is reported for anti-MuSK-positive MG patients (Evoli et al. 2003). Counts revealed fewer AChR plaques from the diaphragm muscle of the most severely affected mice (those injected with MuSK2 and MuSK3). This raises the possibility that outright loss of NMJs from certain muscles might contribute to the particular severity of the MuSK autoantibody form of MG.
A pertinent question is whether MuSK autoantibodies impair NMJs by inhibiting MuSK-dependent transcription of postsynaptic genes. Cell culture studies suggest that MuSK signalling can influence the rate of transcription of mRNAs encoding MuSK and certain AChR subunits at the synapse (Lacazette et al. 2003; Strochlic et al. 2004). The influence of such pathways upon gene expression at the adult NMJ remains uncertain. Studies in which plasma from MuSK patients was added to muscle cell cultures differed. Some reported reductions in the level of mRNAs encoding AChR subunits while others found no change (Boneva et al. 2006; Farrugia et al. 2007). Mice injected with MuSK1 and MuSK4 IgGs did not become weak and their TA muscles showed no change in mRNAs encoding AChR α-subunit, rapsyn or MuSK. Mice injected with MuSK2 became weak and showed significant elevation of these same mRNAs. Muscle electrical activity strongly represses the expression of AChR-α, MuSK and rapsyn mRNAs in functioning muscle (Witzemann et al. 1991; Bowen et al. 1998) (data not shown for rapsyn). Thus, elevation of synaptic mRNAs can most easily be explained by lack of muscle activity in the mice that became weak. We have not studied the expression of all the genes that could potentially influence postsynaptic differentiation. We have studied mRNA in whole muscle rather than micro-dissecting the postsynaptic portion of each fibre. Notwithstanding these limitations, our approach permitted precise comparison of mRNA levels for MuSK, AChR-α and rapsyn, and provided no indication that reduced AChR staining in the postsynaptic membrane might be due to impaired transcription of these key synaptic genes.
A question remains as to why it has been difficult to demonstrate muscle weakness in mice following repeated injections of IgG from MuSK autoantibody-positive patients. At healthy NMJs the endplate current exceeds that needed to reach threshold potential by 2- to 5-fold (Wood & Slater, 2001). This ‘safety factor’ masks neuromuscular impairment until such impairment becomes extreme. Early experiments involving passive transfer of plasma or IgG from patients seronegative for anti-AChR (prior to the discovery of MuSK) produced sub-clinical impairment of neuromuscular transmission (Burges et al. 1994). More recently, ter Beek and colleagues, injected plasma from MuSK autoantibody-positive MG patients into the flexor digitorum brevis muscle of mice (40 μl plasma day−1 for 7–14 days; ter Beek et al. 2009). Again, the effect of the antibodies was sub-clinical. Nerve-evoked contraction force was used to test for evidence of impaired neuromuscular transmission. Significant impairment could be demonstrated only under conditions where the quantal content of transmission was artificially compromised ([Ca2+]o≤ 0.5 mm). In comparison to the studies mentioned above, we injected greater amounts of total IgG systemically (45 mg IgG per day i.p. for 14 days) and studied muscles that are more proximal and are clinically more affected in MuSK autoantibody patients. Following the classical study of Toyka et al. (1977) we also injected cyclophosphamide 24 h after the first IgG injection to prevent an active immune response to the human IgG. Such a response could otherwise have quelled the injected autoantibodies. The autoantibody titre should also be considered. We have now studied the effects of IgG from a total of five MuSK MG patients. Three of the five produced muscle weakness, weight loss and a decremental compound muscle action potential during repetitive stimulation of the nerve (Cole et al. 2008; and data not shown). These were the IgG preparations with the highest autoantibody titres. High titres were associated with larger reductions in postsynaptic AChR packing and development of muscle weakness. From a broader perspective, the relatively short-term injections series that we, and others, have used in mice may not adequately replicate longer-term chronic effects of MuSK autoantibodies on NMJs of MG patients.
Our results are consistent with the idea that MuSK MG patient autoantibodies activate the MuSK–rapsyn system. MuSK2 IgG caused strong tyrosine phosphorylation of MuSK and the AChR β-subunit, consistent with the effects of MuSK antibodies raised in animals (Hopf & Hoch, 1998; Shigemoto et al. 2006). Agrin-induced activation of MuSK in myotubes involves internalization of MuSK (Lu et al. 2007; Zhu et al. 2008), so internalization of MuSK in response to anti-MuSK IgG is consistent with MuSK activation. However, myoblasts lack key components of the MuSK complex that are needed for the (physiological) activation of MuSK by agrin (Linnoila et al. 2008; Inoue et al. 2009). For this reason we are not sure whether MuSK internalization produced by agrin and anti-MuSK represent the same kind of trafficking response. The results in vivo also seem consistent with MuSK activation. The postsynaptic rapsyn-to-AChR immunofluorescence ratio was elevated in mice injected with MuSK2 IgG. Similar elevation of the rapsyn-to-AChR ratio occurred at NMJs when healthy muscles were exposed to exogenous neural agrin (Brockhausen et al. 2008). By contrast, ter Beek et al. (2009) found more subtle effects of MuSK patient plasma following intramuscular injections. They found no effect upon healthy NMJs. However, their MuSK patient plasma modestly inhibited the growth of postsynaptic AChR plaques that were regenerating after i.m. injections of a myotoxin. Reduced rapsyn staining suggested that the autoantibodies may have partially inhibited MuSK–rapsyn-dependent growth of regenerating AChR plaques. The distribution of MuSK at NMJs was not reported. Why the differing results? Patient autoantibodies appear to vary somewhat in their binding sites within the extracellular domain of MuSK (McConville et al. 2004). While our results strongly suggest activation of MuSK by our patient autoantibodies, it is quite conceivable that the autoantibodies of some other patients might interact differently with the MuSK–LRP4 complex, inhibiting MuSK activation.
Recent studies have begun to reveal negative-feedback pathways through which MuSK activation can foster disassembly of existing AChR clusters (Qian et al. 2008). It is possible that by activating MuSK our MuSK autoantibodies might have aberrantly driven some such disassembly pathway, leading to the loss of postsynaptic AChRs. While we cannot exclude this possibility, knockdown of MuSK gene expression was sufficient to break up postsynaptic AChR plaques (Kong et al. 2004; Hesser et al. 2006). The simplest explanation remains that the autoantibody-induced displacement of MuSK from the postsynaptic membrane caused the break-up of postsynaptic AChRs. Two observations support the latter idea: (1) the loss of MuSK preceded the reduction in AChR packing and (2) at the end of the injection series the autoantibody-induced reductions in postsynaptic MuSK correlated with the residual levels of postsynaptic AChR staining and AChR–AChR FRET.
The impaired postsynaptic AChR packing was not the result of any deficiency in rapsyn, suggesting that in addition to the MuSK–rapsyn pathway MuSK serves another, vital function. The loss of postsynaptic MuSK preceded the first evidence of impaired AChR packing by several days. Thus, it seems unlikely that MuSK per se physically holds together the postsynaptic AChRs at the adult NMJ. However, the postsynaptic membrane comprises a meshwork of specialized transmembrane, sub-membrane, cytoskeletal and basement membrane proteins that may serve multiple structural and signalling functions (Banks et al. 2003; Strochlic et al. 2005). Notwithstanding activation of the MuSK–rapsyn system, prolonged displacement of MuSK may have de-stabilized this extended scaffold (and/or its docking sites for rapsyn and AChR). Autoantibodies now provide probes to perturb the postsynaptic scaffold complex. They should help us identify key functional interactions among the structural and signalling components that sustain the postsynaptic membrane scaffold.
Acknowledgments
We thank Sr Beth Newman and the staff of the apheresis unit and molecular medicine laboratory at Concord Hospital for collection of plasma fractions, patients from around Australia who contributed their plasma to this research, and Drs Steve Assinder and Louise Cole for valuable technical advice. This work was supported by grants from MDA (4172), NHMRC Australia (570930), the Australian Myasthenic Association in NSW and the Brain Foundation of Australia.
Glossary
Abbreviations
- AChR
acetylcholine receptor
- Alexa
Alexafluor
- EGFP
enhanced green fluorescent protein
- FRET
fluorescence resonance energy transfer
- Gastroc.
gastrocnemius
- MuSK
muscle-specific kinase
- MuSK1
MuSK autoantibody-positive MG patient number 1
- MG
myasthenia gravis
- NMJ
neuromuscular junction
- TA
tibialis anterior
Author contributions
W.D.P. and S.W.R. jointly designed the study. R.N.C. was responsible for the passive IgG transfer experiments, N.G. for the cell culture experiments and O.L.G. for the live cell imaging. S.T.N. developed and supervised the MuSK immunoprecipitation analyses. S.W.R. was responsible for all clinical aspects of the study and for human IgG purification. All authors contributed to writing and/or revision of the manuscript and approved the final draft. The experiments were conducted at the University of Sydney.
Supplemental material
Supplementary data S1
Supplementary data S2
Supplementary data S3
Supplementary data S4
Supplementary Movie
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Antolik C, Catino DH, Resneck WG, Bloch RJ. The tetratricopeptide repeat domains of rapsyn bind directly to cytoplasmic sequences of the muscle-specific kinase. Neuroscience. 2007;145:56–65. doi: 10.1016/j.neuroscience.2006.03.035. [DOI] [PubMed] [Google Scholar]
- Banks GB, Fuhrer C, Adams ME, Froehner SC. The postsynaptic submembrane machinery at the neuromuscular junction: requirement for rapsyn and the utrophin/dystrophin-associated complex. J Neurocytol. 2003;32:709–726. doi: 10.1023/B:NEUR.0000020619.24681.2b. [DOI] [PubMed] [Google Scholar]
- Bezakova G, Rabben I, Sefland I, Fumagalli G, Lomo T. Neural agrin controls acetylcholine receptor stability in skeletal muscle fibers. Proc Natl Acad Sci U S A. 2001;98:9924–9929. doi: 10.1073/pnas.171539698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boneva N, Frenkian-Cuvelier M, Bidault J, Brenner T, Berrih-Aknin S. Major pathogenic effects of anti-MuSK antibodies in myasthenia gravis. J Neuroimmunol. 2006;177:119–131. doi: 10.1016/j.jneuroim.2006.05.017. [DOI] [PubMed] [Google Scholar]
- Borges L, Yechikov S, Lee YI, Rudell JB, Friese MB, Burden SJ, Ferns M. Identification of a motif in the acetylcholine receptor β-subunit whose phosphorylation regulates rapsyn association and postsynaptic receptor localization. J Neurosci. 2008;28:11468–11476. doi: 10.1523/JNEUROSCI.2508-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen DC, Park JS, Bodine S, Stark JL, Valenzuela DM, Stitt TN, Yancopoulos GD, Lindsay RM, Glass DJ, DiStefano PS. Localisation and regulation of MuSK at the neuromuscular junction. Dev Biol. 1998;199:309–319. doi: 10.1006/dbio.1998.8936. [DOI] [PubMed] [Google Scholar]
- Brockhausen J, Cole RN, Gervásio OL, Ngo ST, Noakes PG, Phillips WD. Neural agrin increases postsynaptic ACh receptor packing by elevating rapsyn protein at the mouse neuromuscular synapse. Dev Neurobiol. 2008;68:1153–1169. doi: 10.1002/dneu.20654. [DOI] [PubMed] [Google Scholar]
- Burges J, Vincent A, Molenaar PC, Newsom-Davis J, Peers C, Wray D. Passive transfer of seronegative myasthenia gravis to mice. Muscle Nerve. 1994;17:1393–1400. doi: 10.1002/mus.880171208. [DOI] [PubMed] [Google Scholar]
- Cole RN, Reddel SW, Gervásio OL, Phillips WD. Anti-MuSK patient antibodies disrupt the mouse neuromuscular junction. Ann Neurol. 2008;63:782–789. doi: 10.1002/ana.21371. [DOI] [PubMed] [Google Scholar]
- Conti-Fine BM, Milani M, Kaminski HJ. Myasthenia gravis: past, present, and future. J Clin Invest. 2006;116:2843–2854. doi: 10.1172/JCI29894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evoli A, Tonali PA, Padua L, Monaco ML, Scuderi F, Batocchi AP, Marino M, Bartoccioni E. Clinical correlates with anti-MuSK antibodies in generalized seronegative myasthenia gravis. Brain. 2003;126:2304–2311. doi: 10.1093/brain/awg223. [DOI] [PubMed] [Google Scholar]
- Farrugia ME, Bonifatia DM, Clovera L, Cossinsa J, Beesona D, Vincent A. Effect of sera from AChR-antibody negative myasthenia gravis patients on AChR and MuSK in cell cultures. J Neuroimmunol. 2007;185:136–144. doi: 10.1016/j.jneuroim.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Fuhrer C, Gautam M, Sugiyama JE, Hall ZW. Roles of rapsyn and agrin in interaction of postsynaptic proteins with acetylcholine receptors. J Neurosci. 1999;19:6405–6416. doi: 10.1523/JNEUROSCI.19-15-06405.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervásio OL, Armson PF, Phillips WD. Developmental increase in the amount of rapsyn per acetylcholine receptor promotes postsynaptic receptor packing and stability. Dev Biol. 2007;305:262–275. doi: 10.1016/j.ydbio.2007.02.008. [DOI] [PubMed] [Google Scholar]
- Gervásio OL, Phillips WD. Increased ratio of rapsyn to ACh receptor stabilizes postsynaptic receptors at the mouse neuromuscular synapse. J Physiol. 2005;562:673–685. doi: 10.1113/jphysiol.2004.077685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass DJ, Bowen DC, Stitt TN, Radiejewski C, Brunno J, Ryan TE, Gies DR, Shah S, Mattsson K, Burden SJ, DiStefano PS, Valenzuela DM, DeChiara TM, Yancopoulos GD. Agrin acts via a MuSK receptor complex. Cell. 1996;85:513–523. doi: 10.1016/s0092-8674(00)81252-0. [DOI] [PubMed] [Google Scholar]
- Hesser BA, Henschel O, Witzemann V. Synapse disassembly and formation of new synapses in postnatal muscle upon conditional inactivation of MuSK. Mol Cell Neurosci. 2006;31:470–480. doi: 10.1016/j.mcn.2005.10.020. [DOI] [PubMed] [Google Scholar]
- Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, Vincent A. Autoantibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med. 2001;7:365–368. doi: 10.1038/85520. [DOI] [PubMed] [Google Scholar]
- Hopf C, Hoch W. Dimerization of the muscle-specific kinase induces tyrosine phosphorylation of acetylcholine receptors and their aggregation on the surface of myotubes. J Biol Chem. 1998;273:6467–6473. doi: 10.1074/jbc.273.11.6467. [DOI] [PubMed] [Google Scholar]
- Inoue A, Setoguchi K, Matsubara Y, Okada K, Sato N, Iwakura Y, Higuchi O, Yamanashi Y. Dok-7 activates the muscle receptor kinase MuSK and shapes synapse formation. Sci Signal. 2009;2:ra7. doi: 10.1126/scisignal.2000113. [DOI] [PubMed] [Google Scholar]
- Jha S, Xu K, Maruta T, Oshima M, Mosier DR, Atassi MZ, Hoch W. Myasthenia gravis induced in mice by immunization with the recombinant extracellular domain of rat muscle-specific kinase (MuSK) J Neuroimmunol. 2006;175:107–117. doi: 10.1016/j.jneuroim.2006.03.016. [DOI] [PubMed] [Google Scholar]
- Kim N, Stiegler AL, Cameron TO, Hallock PT, Gomez AM, Huang JH, Hubbard SR, Dustin ML, Burden SJ. Lrp4 is a receptor for agrin and forms a complex with MuSK. Cell. 2008;135:334–342. doi: 10.1016/j.cell.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong XC, Barzaghi P, Ruegg MA. Inhibition of synapse assembly in mammalian muscle in vivo by RNA interference. EMBO Rep. 2004;5:183–188. doi: 10.1038/sj.embor.7400065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacazette E, Le Calvez S, Gajendran N, Brenner HR. A novel pathway for MuSK to induce key genes in neuromuscular synapse formation. J Cell Biol. 2003;161:727–736. doi: 10.1083/jcb.200210156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land BR, Salpeter EE, Salpeter MM. Acetylcholine receptor site density affects the rising phase of miniature endplate currents. Proc Natl Acad Sci U S A. 1980;77:3736–3740. doi: 10.1073/pnas.77.6.3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnoila J, Wang Y, Yao Y, Wang ZZ. A mammalian homologue of Drosophila tumorous imaginal discs, TID1, mediates agrin signaling at the neuromuscular junction. Neuron. 2008;60:625–641. doi: 10.1016/j.neuron.2008.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Je H-S, Young P, Gross J, Lu B, Feng G. Regulation of synaptic growth and maturation by a synapse-associated E3 ubiquitin ligase at the neuromuscular junction. J Cell Biol. 2007;177:1077–1089. doi: 10.1083/jcb.200610060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo ZG, Wang Q, Zhou JZ, Wang J, Luo Z, Liu M, He X, Wynshaw-Boris A, Xiong WC, Lu B, Mei L. Regulation of AChR clustering by dishevelled interacting with MuSK and PAK1. Neuron. 2002;35:489–505. doi: 10.1016/s0896-6273(02)00783-3. [DOI] [PubMed] [Google Scholar]
- McConville J, Farrugia ME, Beeson D, Kishore U, Metcalf R, Newsom-Davis J, Vincent A. Detection and characterization of MuSK antibodies in seronegative myasthenia gravis. Ann Neurol. 2004;55:580–584. doi: 10.1002/ana.20061. [DOI] [PubMed] [Google Scholar]
- Madhavan R, Peng HB. Molecular regulation of postsynaptic differentiation at the neuromuscular junction. IUBMB Life. 2005;57:719–730. doi: 10.1080/15216540500338739. [DOI] [PubMed] [Google Scholar]
- Moransard M, Borges LS, Willmann R, Marangi PA, Brenner HR, Ferns MJ, Fuhrer C. Agrin regulates rapsyn interaction with surface AChRs which underlies cytoskeletal anchoring and clustering. J Biol Chem. 2003;278:7350–7359. doi: 10.1074/jbc.M210865200. [DOI] [PubMed] [Google Scholar]
- Ngo ST, Balke C, Phillips WD, Noakes PG. Neuregulin potentiates agrin-induced acetylcholine receptor clustering in myotubes. Neuroreport. 2004;15:2501–2505. doi: 10.1097/00001756-200411150-00014. [DOI] [PubMed] [Google Scholar]
- Peng HB, Froehner SC. Association of the postsynaptic 43K protein with newly formed acetylcholine receptor clusters in cultured muscle cells. J Cell Biol. 1985;100:1698–1705. doi: 10.1083/jcb.100.5.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian YK, Chan AWS, Madhavan R, Peng HB. The function of Shp2 tyrosine phosphatase in the dispersal of acetylcholine receptor clusters. BMC Neurosci. 2008;9:70. doi: 10.1186/1471-2202-9-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupp F, Ozçelik T, Linial M, Peterson K, Francke U, Scheller R. Structure and chromosomal localization of the mammalian agrin gene. J Neurosci. 1992;12:3535–3544. doi: 10.1523/JNEUROSCI.12-09-03535.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salpeter MM, Harris R. Distribution and turnover rate of acetylcholine receptors throughout the junction folds at a vertebrate neuromuscular junction. J Cell Biol. 1983;96:1781–1785. doi: 10.1083/jcb.96.6.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci. 2001;2:791–805. doi: 10.1038/35097557. [DOI] [PubMed] [Google Scholar]
- Shigemoto K, Kubo S, Maruyama N, Hato N, Yamada H, Jie C, Kobayashi N, Mominoki K, Abe Y, Ueda N, Matsuda S. Induction of myasthenia by immunization against muscle-specific kinase. J Clin Invest. 2006;116:1016–1024. doi: 10.1172/JCI21545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacy S, Gelb BE, Koop BA, Windle JJ, Wall KA, Krolick KA, Infante AJ, Kraig E. Split tolerance in a novel transgenic model of autoimmune myasthenia gravis. J Immunol. 2002;169:6570–6579. doi: 10.4049/jimmunol.169.11.6570. [DOI] [PubMed] [Google Scholar]
- Strochlic L, Cartaud A, Cartaud J. The synaptic muscle-specific kinase (MuSK) complex: new partners, new functions. Bioessays. 2005;27:1129–1135. doi: 10.1002/bies.20305. [DOI] [PubMed] [Google Scholar]
- Strochlic L, Cartaud A, Mejat A, Grailhe R, Schaeffer L, Changeux JP, Cartaud J. 14-3-3γ associates with muscle specific kinase and regulates synaptic gene transcription at vertebrate neuromuscular synapse. Proc Natl Acad Sci U S A. 2004;101:18189–18194. doi: 10.1073/pnas.0406905102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Beek PW, Martínez-Martínez P, Losen M, de Baets MH, Wintzen AR, Verschuuren JJ, Niks EH, Duinen SGV, Vincent A, Molenaar PC. The effect of plasma from muscle-specific tyrosine kinase myasthenia gravis patients on regenerating endplates. Am J Path. 2009;175:1536–1544. doi: 10.2353/ajpath.2009.090040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyka KV, Drachman DB, Griffin DE, Pestronk A, Winkelstein JA, Fishbeck KH, Kao I. Myasthenia gravis. Study of humoral immune mechanisms by passive transfer to mice. N Engl J Med. 1977;296:125–131. doi: 10.1056/NEJM197701202960301. [DOI] [PubMed] [Google Scholar]
- Weston C, Yee B, Hod E, Prives JM. Agrin-induced acetylcholine receptor clustering is mediated by the small guanosine triphosphatases Rac and Cdc42. J Cell Biol. 2000;150:205–212. doi: 10.1083/jcb.150.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witzemann V, Brenner HR, Sakmann B. Neural factors regulate AChR subunit mRNAs at rat neuromuscular junctions. J Cell Biol. 1991;114:125–141. doi: 10.1083/jcb.114.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood SJ, Slater CR. Safety factor at the neuromuscular junction. Prog Neurobiol. 2001;64:393–429. doi: 10.1016/s0301-0082(00)00055-1. [DOI] [PubMed] [Google Scholar]
- Xu K, Jha S, Hoch W, Dryer SE. Delayed synapsing muscle are more severely affected in an experimental model of MuSK-induced myasthenia gravis. Neuroscience. 2006;143:655–659. doi: 10.1016/j.neuroscience.2006.09.022. [DOI] [PubMed] [Google Scholar]
- Zhang B, Luo S, Wang Q, Suzuki T, Xiong WC, Mei L. LRP4 serves as a coreceptor of agrin. Neuron. 2008;60:285–297. doi: 10.1016/j.neuron.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Glass DJ, Yancopoulos GD, Sanes JR. Distinct domains of MuSK mediate its abilities to induce and to associate with postsynaptic specializations. J Cell Biol. 1999;146:1133–1146. doi: 10.1083/jcb.146.5.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D, Yang Z, Luo Z, Luo S, Xiong WC, Mei L. Muscle-specific receptor tyrosine kinase endocytosis in acetylcholine receptor clustering in response to agrin. J Neurosci. 2008;28:1688–1696. doi: 10.1523/JNEUROSCI.4130-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.