

Abstract
KCNQ gene expression was previously shown in various rodent blood vessels, where the products of KCNQ4 and KCNQ5, Kv7.4 and Kv7.5 potassium channel subunits, respectively, have an influence on vascular reactivity. The aim of this study was to determine if small cerebral resistance arteries of the rat express KCNQ genes and whether Kv7 channels participate in the regulation of myogenic control of diameter. Quantitative reverse transcription polymerase chain reaction (QPCR) was undertaken using RNA isolated from rat middle cerebral arteries (RMCAs) and immunocytochemistry was performed using Kv7 subunit-specific antibodies and freshly isolated RMCA myocytes. KCNQ4 message was more abundant than KCNQ5=KCNQ1, but KCNQ2 and KCNQ3 message levels were negligible. Kv7.1, Kv7.4 and Kv7.5 immunoreactivity was present at the sarcolemma of freshly isolated RMCA myocytes. Linopirdine (1 μm) partially depressed, whereas the Kv7 activator S-1 (3 and/or 20 μm) enhanced whole-cell Kv7.4 (in HEK 293 cells), as well as native RMCA myocyte Kv current amplitude. The effects of S-1 were voltage-dependent, with progressive loss of stimulation at potentials of >−15 mV. At the concentrations employed linopirdine and S-1 did not alter currents due to recombinant Kv1.2/Kv1.5 or Kv2.1/Kv9.3 channels (in HEK 293 cells) that are also expressed by RMCA myocytes. In contrast, another widely used Kv7 blocker, XE991 (10 μm), significantly attenuated native Kv current and also reduced Kv1.2/Kv1.5 and Kv2.1/Kv9.3 currents. Pressurized arterial myography was performed using RMCAs exposed to intravascular pressures of 10–100 mmHg. Linopirdine (1 μm) enhanced the myogenic response at ≥20 mmHg, whereas the activation of Kv7 channels with S-1 (20 μm) inhibited myogenic constriction at >20 mmHg and reversed the increased myogenic response produced by suppression of Kv2-containing channels with 30 nm stromatoxin (ScTx1). These data reveal a novel contribution of KCNQ gene products to the regulation of myogenic control of cerebral arterial diameter and suggest that Kv7 channel activating drugs may be appropriate candidates for the development of an effective therapy to ameliorate cerebral vasospasm.
Introduction
Cerebral blood flow regulation is dependent on the integration of multiple physiological factors that affect force generation by cerebral vascular smooth muscle cells (VSMCs) and, thereby, cerebral arterial diameter (Davis & Hill, 1999). These factors include the intrinsic response of VSMCs to intravascular pressure, substances released from cell types (e.g. endothelium, nerve varicosities and astrocytes) or present within the bloodstream, and electrical coupling with the endothelium (Davis & Hill, 1999). The mechanical stress of intravascular pressure on the vessel wall leads to a state of partial constriction of VSMCs that is referred to as the myogenic response. This ability of resistance arteries to react to elevated pressure with constriction, and to pressure reduction with dilatation, can be traced to cellular mechanisms inherent to VSMCs (Davis & Hill, 1999). Considerable progress has been made in determining the mechanisms underlying the myogenic response, but our understanding remains incomplete.
Force generation in myogenic constriction is dependent in part on the level of membrane potential of VSMCs, as voltage-gated Ca2+ channels (VGCCs) are a major source of Ca2+ influx to support contraction (Knot & Nelson, 1998; Davis & Hill, 1999; Hill et al. 2001, 2006). The prevailing view holds that the myogenic response results from: (1) pressure-induced depolarization of membrane potential, (2) voltage-dependent activation of VGCCs, (3) a rise in cytosolic Ca2+ level ([Ca2+]i), (4) Ca2+-dependent activation of myosin light chain kinase, (5) phosphorylation of 20 kDa myosin light chain subunits, (6) initiation of cross-bridge cycling, (7) Ca2+ sensitization of the myofilaments owing to Rho kinase-mediated phosphorylation of myosin light chain phosphatase, and (8) increased force generation (Knot & Nelson, 1998; Davis & Hill, 1999; Johnson et al. 2009). Regenerative, feed-forward activation of VGCCs in response to myogenic depolarization could cause excessive Ca2+ influx and an inappropriate level of constriction, vasospasm and/or vasomotion. Substantial evidence has been obtained to suggest that myogenic depolarization is precisely controlled by a negative-feedback mechanism involving the activation of VSMC K+ channels. For example, previous studies support the view that large conductance Ca2+ activated (BKCa) channels, as well as voltage-gated Kv1 and Kv2 pore-forming subunit-containing channels contribute to the control of myogenic depolarization and the regulation of resistance arterial diameter at transmural pressures of greater than ∼40 mmHg (Nelson et al. 1995; Knot & Nelson, 1995; Nelson & Quayle, 1997; Knot et al. 1998; Albarwani et al. 2003; Amberg & Santana, 2006; Chen et al. 2006; Yang et al. 2009; Zhong et al. 2010). However, it is likely that additional types of K+ channels are also involved in this critical physiological mechanism; expression of message and/or protein for members of other families of K+ channels (Kv and non-Kv) by VSMCs has also been reported (see Jackson, 2005; Cole et al. 2005; Greenwood & Ohya, 2009). For example, VSMCs in different vessels are reported to express Kv3, Kv4, Kv7 and voltage-independent, inwardly rectifying channels (e.g. Kir2; Zaritsky et al. 2000), ATP-sensitive K+ channels (Kir6.1; see Teramoto, 2006) and twin-pore K+ channels (Bryan et al. 2006; but see Namiranian et al. 2010). These channels may be directly activated in response to myogenic depolarization or they may be sensitive to extrinsic factors that modulate the myogenic response. We currently lack an understanding of how this diverse complement of K+ channels contribute to the regulation of the myogenic response and this deficiency impairs the development of novel strategies for the treatment of conditions with abnormal control of arterial diameter.
Given the crucial role that VSMC K+ channel activity plays in controlling cerebral arterial diameter in health, and possibly disease, it is essential that we have a comprehensive knowledge of all of the varieties of K+ channels that are involved. Recently, VSMCs of several blood vessels have been shown to possess a novel variety of K+ channel owing to expression of KCNQ genes (Ohya et al. 2003; Brueggemann et al. 2007; Yeung et al. 2007, Mackie et al. 2008; Joshi et al. 2009). The Kv7.1, Kv7.4 and Kv7.5 pore-forming subunits encoded by these genes assemble to form homotetrameric and/or heterotetrameric voltage-gated K+ channels that exhibit minimal inactivation and a relatively negative activation threshold, and are known to be key regulators of the cardiac action potential and neuronal resting membrane potential (Jentsch, 2000; Robbins, 2001; Greenwood & Ohya, 2009). Blockade of Kv7 channels causes contraction of conduit and resistance vessels and/or increase the efficacy of vasoconstrictor agonists (e.g. Yeung & Greenwood, 2005; Joshi et al. 2006, Brueggemann et al. 2007, Yeung et al. 2007, Mackie et al. 2008). In contrast, drugs such as flupirtine and retigabine that stimulate Kv7 channel activity hyperpolarize VSMCs and relax pre-contracted aorta, pulmonary and mesenteric conduit arteries (Yeung et al. 2007, 2008; Mackie et al. 2008; Joshi et al. 2009). In addition, the cyclo-oxygenase inhibitors meclofenamic acid and celecoxib also activate Kv7 channels and relax blood vessels (Yeung et al. 2007; Brueggemann et al. 2009).
With these findings in mind, we postulated that Kv7 subunit-containing K+ channels may play a role in regulating cerebral blood flow by contributing to the control of myogenic depolarization. Here, we use a combination of approaches including molecular, immunocytochemical, patch clamp electrophysiology and pressurized arterial myography to assess KCNQ gene expression and Kv7 channel function in rat middle cerebral arteries (RMCAs). Our findings indicate that Kv7 subunits are expressed by RMCA myocytes and contribute to the regulation of the cerebral myogenic response. Moreover, we show that the stimulation of Kv7 channel activity can reverse vasoconstriction and enhanced myogenic reactivity of RMCAs induced by pharmacological inhibition of non-Kv7 K+ channels. Thus, Kv7 channels represent a novel potential therapeutic target for treatment of cerebral vasospasm.
Methods
Ethical approval
Male Sprague–Dawley rats (250–275 g; Charles River, Montréal, Quebec, Canada, Charles River, UK) were killed by halothane inhalation and exsanguination according to the standards of the Canadian Council on Animal Care and reviewed by the Animal Care Committee of the Faculty of Medicine, University of Calgary, the UK Animals (Scientific Procedures) Act 1986 and The Journal of Physiology's ethical policies and regulations as indicated in Drummond (2009). A total of 105 rats were employed in this study.
Quantitative real-time PCR
Total RNA extraction from basilar and cerebral arteries of 9- to 10-week-old male Sprague–Dawley rats and cDNA synthesis were performed as previously reported (Ohya et al. 2003). Specific primers were designed to determine relative levels of expression of KCNQ and KCNE subtypes in both arteries. cDNA samples from brain and heart were used as positive controls for primers. Quantitative, real-time PCR (QPCR) was performed with the use of Power Syber Green chemistry on an ABI 7000 sequence detector (Applied Biosystems, Foster City, CA, USA) as previously reported (Ohya et al. 2003; Yeung et al. 2007). Regression analysis of the mean values of three multiplex RT-PCRs for the log10 diluted cDNA was used to generate standard curves. Unknown quantities relative to the standard curve for a particular set of primers were calculated, yielding transcriptional quantification of KCNQ and KCNE gene products relative to the endogenous standard (β-actin). The reproducibility of the assay was tested by analysis of variance (ANOVA) comparing repeat runs of samples, and mean values generated at individual time points were compared by Student's t test. Specific primers were designed, as follows: KCNQ1 (GenBank accession number: NM_032073): 901–1034, amplicon = 134 bp; KCNQ2 (NM_133322): 1084–1184, 101 bp; KCNQ3 (AF091247): 916–1018, 103 bp: KCNQ4 (XM_233477): 1427–1559, 133 bp; KCNQ5 (XM_001071249): 2763–2882, 120 bp; KNCE1 (NM_012973): 186–291, 106 bp; KCNE2 (NM_133603): 151–251, 101 bp; KCNE3 (NM_022235): 436–567, 132 bp; KCNE4 (AF512994): 419–536, 118 bp; KCNE5 (XM_001101003): 296–416, 121 bp. Each amplified product was sequenced by the chain termination method with an ABI Prizm (model 310) (Applied Biosystems).
Immunocytochemistry
Single cells isolated from left and right middle cerebral arteries were fixed with 4% formaldehyde solution at 4°C for 4 min respectively, washed with physiological salt solution (PSS) and incubated with PSS containing 3% bovine serum albumin (BSA) and 0.3% Triton X-100. Then samples were incubated with primary antibodies in PSS containing 2% BSA and 0.3% Triton X-100 overnight at 4°C, washed with PSS and incubated for 2 h with secondary antibodies conjugated with fluorescent probes. Samples were washed with PSS, and viewed using a Zeiss LSM 510 laser scanning confocal microscope. The following primary antibodies were used: rabbit anti-Kv7.1 (Millipore, Billerica, MA, USA), dilution 1:200, rabbit anti-Kv7.4 (AbCam, UK), 1:200 and rabbit anti-Kv7.5 (Millipore), 1:200. These antibodies have been validated in previous studies (Jepps et al. 2009). The donkey anti-rabbit MFP 488 secondary antibody (dilution 1:400, from Mobitec, Göttingen, Germany) was used in all experiments. In control staining with primary antibodies was omitted.
Confocal imaging data were processed and analysed using Zeiss LSM software. An image taken approximately in the middle of the cell was selected out of the z-stack of horizontally taken images. Using such an image the average pixel fluorescence (APF) was calculated according to equation:
where i(p) is the intensity of a pixel within the confocal plane of the cell, and n(p) is the total number of pixels of the selected region. To analyse the cellular distribution of Kv channels, three different circular areas of 0.79 μm2 (diameter approx. 1 μm; referred to as region of interest 1 (ROI 1)) were randomly selected in the subplasmalemmal area of the every analysed cell (Fig. 1Ca) so that their perimeter touched the edge of the cell. Another three circular areas of 0.79 μm2 (ROI 2) were selected so that the perimeter of that circle touched the perimeter of ROI 1 and the line bisecting these circles was perpendicular to the edge of the cell, thought to be the plasma membrane. The AFP values were then compared between ROI 1 and ROI 2 and between whole cell planes and controls for each different Kv antibody labelling. Statistical evaluation and graphs were created using Origin (OriginlLab Corp., Northampton, MA, USA).
Figure 1. Expression of KCNQ genes in rat cerebral arteries.
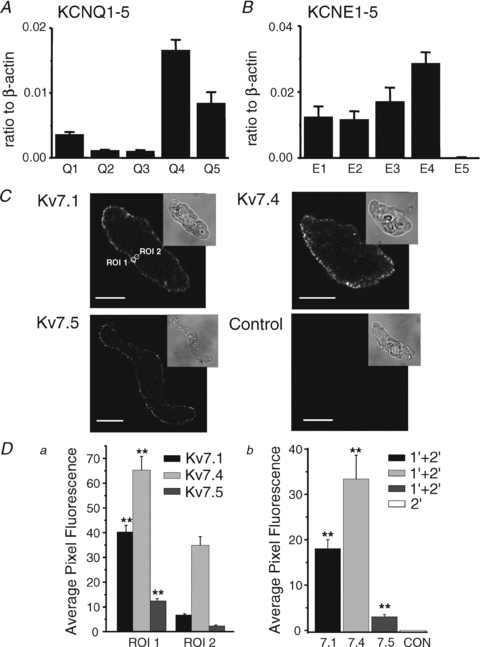
A and B, quantitative, real-time PCR detection of KCNQ (KCNQ1–5) and KCNE (KCNE1–5) subunit transcript expression relative to β-actin in mRNA extracted from RMCAs. All expression data are expressed as means ±s.e.m. (n= 4 for each). C, immunostaining of freshly isolated myocytes using anti-Kv7.1, -Kv7.4, or -Kv7.5, and a control experiment in which primary antibody was omitted. The transmitted light image for each cell is shown in upper right inset images. Circles indicate regions of interest: ROI 1 and ROI 2, which were used to analyse the localization of fluorescence (see Methods for details). The calibration bars in panel C are 10 μm. D, summarized data for localization of Kv7.1, Kv7.4 and Kv7.5 immunofluorescence in RMCA myocytes. There was significantly more fluorescence in the region of plasma membrane (ROI 1) than in deep cytoplasm (ROI 2) (Da, n= 10 myocytes for each antibody). Db shows summarized data for average pixel fluorescence in a whole-cell confocal plane compared to control myocyte that demonstrated a lack of immunofluorescence in the absence of primary antibody (n= 10 myocytes; 1′, primary antibody; 2′, secondary antibody). **Statistical significance at P < 0.01.
Intact cerebral arterial pressure myography
The brain was carefully removed and placed in ice-cold Krebs solution containing (in mm): NaCl 120, NaHCO3 25, KCl 4.8, NaH2PO4 1.2, MgSO4 1.2, glucose 11, CaCl2 2.5 (pH 7.4 when aerated with 95% air–5% CO2). Left and right RMCAs were removed from the brain, dissected free of the surrounding tissue and cut into 2 mm segments in preparation for arterial pressure myography, as previously described (Chen et al. 2006; Johnson et al. 2009). Briefly, cerebral arteries were mounted in an arteriograph chamber attached to a pressure myograph (Living Systems, Burlington, VT, USA) for measurement of outer arterial diameter with an automated edge detection system (IonOptix, Milton, MA, USA). Endothelial cells were removed from all arteries by briefly passing a stream of air through the vessel lumen and confirmed by loss of vasodilatation to 10 μm bradykinin. Arteries were allowed to warm to 37°C for 30 min in Krebs solution, then pressurized to 60 mmHg and allowed to develop active tone over 15–30 min. All arteries were subjected to two 5 min pressure steps from 20 to 80 mmHg to ensure development of stable pressure-dependent myogenic constriction. Vessels that exhibited leaks and/or did not exhibit stable constriction to pressure were discarded. After development and stabilization of myogenic constriction, pressure was reduced to 10 mmHg for 10 min prior to the start of all experiments.
RMCA myocyte isolation
Rat brains were carefully removed from the skull and placed in an ice-cold smooth muscle dissection solution (SMDS) containing (in mm): 60 NaCl, 80 sodium glutamate, 5 KCl, 2 MgCl2, 10 glucose, and 10 Hepes (pH 7.4). RMCAs were dissected and single myocytes were enzymatically isolated using a method modified from Plane et al. (2005). Briefly, arteries were equilibrated in SMDS containing bovine serum albumin (BSA; 1 mg ml−1) at 37°C for 10 min, exposed to the same solution supplemented with papain (0.5 mg ml−1; Worthington Biochemical Corp., Lakewood, NJ, USA) and dithiothrietol (1.5 mg ml−1) at 37°C for 8–10 min, washed in ice-cold SMDS (2 times), incubated in SMDS containing 100 μm Ca2+, BSA (1 mg ml−1) and collagenase (0.7 mg ml−1 type F and 0.4 mg ml−1 type H; Sigma) at 37°C for 8–10 min and then washed in ice-cold SMDS (2 times). Isolated myocytes were liberated from the digested vessels by gentle trituration with a fire-polished pipette and then kept in ice-cold SMDS containing 1 mg ml−1 BSA until use (within 12 h).
Cell culture and transfection
Human embryonic kidney 293 (HEK 293) cells were maintained, as previously described (Kerr et al. 2001; Chen et al. 2006), on 10 cm plastic culture dishes in high glucose Dulbecco's modified Eagle's medium (Gibco/Invitrogen) supplemented with 10% fetal bovine serum and 5% ampicillin–streptomycin at 37°C in 5% CO2. Transfections were performed using FuGENE 6 transfection Reagent (Roche) as per the vendor's instructions. cDNAs encoding green fluorescent protein (GFP), Kv7.4, Kv1.2, Kv1.5, Kv2.1 and Kv9.3 were subcloned into pcDNA3.1. Cells were transfected with 2 μg GFP plasmid with 2 μg Kv7.4, or 2 μg Kv1.2 and 2 μg Kv1.5, or 2 μg Kv2.1 and 6 μg Kv9.3. Transfected cells were re-plated onto 35 mm plastic culture dishes for 24–72 h after transfection prior electrophysiological recordings.
Patch clamp electrophysiology
Whole-cell currents due to native RMCA myocyte Kv current and heterologous expression of Kv7.4, Kv1.2/Kv1.5 and Kv2.1/Kv9.3 channels in HEK 293 cells were recorded and analysed as previously described (Kerr et al. 2001; Chen et al. 2006). Briefly, cells were superfused with a bath solution containing (in mm): 120 NaCl, 3 NaHCO3, 4.2 KCl, 1.2 KH2PO4, 2 MgCl2, 0.1 CaCl2, 10 glucose, and 10 Hepes (pH 7.4). The patch pipette solution contained (in mm): 110 potassium gluconate, 30 KCl, 0.5 MgCl2, 5 Hepes, 10 EGTA, 5 Na2ATP, and 1 GTP (pH 7.2). Junction potential was determined to be 15 mV for the recording conditions employed and this value was used to correct all voltage protocols. Current–voltage (I–V) relations were determined using 325 ms step pulses to between −95 and +45 mV in increments of 10 mV from a holding potential of −75 mV. In some cases, a modified protocol was employed to enhance the availability of Kv7 current in which a pre-step to −95 mV for 80 ms was applied prior to stepping to each test potential. Current amplitudes in treated and control groups were normalized to peak current at +45 mV or +25 mV under control conditions to reduce variability resulting from cell-to-cell differences in current amplitude.
Materials
All general chemicals were purchased from Sigma-Aldrich (Oakville, ON, Canada and Poole, UK) unless indicated otherwise. 4-Aminopyridine (4-AP), tetraethylammonium ion (TEA+) and linopirdine (3,3-bis(4-pyridinylmethyl)-1-phenylindolin-2-one; DUP996) were from Sigma, XE991 (10,10-bis(4-pyridinylmethyl)-9(10H)-anthracenone) was obtained from Tocris Bioscience (Ellisville, MO, USA), and stromatoxin (ScTx1) from Alomone Laboratories (Jerusalem, Israel). Retigabine (ethyl N-[2-amino-4-[(4-fluorophenyl)methylamino]phenyl]carbamate) was a gift from Dr M. Schwake (Germany) and S-1 ((S)-N-[1-(3-morpholin-4-yl-phenyl)-ethyl]-3-phenyl-acrylamide) was synthesized and supplied by NeuroSearch A/S (Denmark). Kv7.4 and Kv9.3 clones were from Thermo Scientific (Open Biosystems) (Huntsville, AL, USA).
Statistical analysis
Where applicable, values are presented as the mean ±s.e.m., with n indicating the number of tissue/cells studied for a given treatment. For the patch clamp and pressure myography experiments all cells/vessels in individual treatment groups were obtained from different cell transfections or rats. Statistical differences were determined using Student's t test or repeated-measures of ANOVA followed by Bonferroni's post hoc test. A level of P < 0.05 was considered to be statistically significant.
Results
KCNQ gene expression in RMCAs
KCNQ transcript expression in RMCAs was quantified by real-time PCR using subunit-specific primers. Of the five known KCNQ gene subtypes, KCNQ1, KCNQ4, and KCNQ5 transcripts encoding Kv7.1, Kv7.4 and Kv7.5 subunits were found to be highly expressed (Fig. 1A), but KCNQ2 and KCNQ3 expression was negligible at less than 0.002 relative to β-actin (n= 4). As positive controls, KCNQ2 and KCNQ3 expression were found to be 0.23 and 0.14 in brain extract using the same primers (n= 2), respectively. We also determined the levels of message encoding KCNE1–5 gene products whose expression products affect the biophysical and pharmacological properties of Kv7 channels (e.g. McCrossan & Abbott, 2004). KCNE1–4, but not KCNE5 were abundantly expressed in RMCAs (Fig. 1B). Identical results were obtained using mRNAs extracted from rat basilar arteries (the abundance of KCNQ1, KCNQ4, and KCNQ5 relative to β-actin were 0.0062 ± 0.0005, 0.0261 ± 0.0056 and 0.0082 ± 0.0009 (n= 4), whereas KCNQ2 and KCNQ3 were below 0.002 (n= 4) and KCNE1–4 transcripts were 0.027 ± 0.006, 0.022 ± 0.004, 0.047 ± 0.009, and 0.046 ± 0.010, respectively; n= 4, and KCNE5 was below 0.002).
Immunocytochemical analysis of KCNQ gene product expression at the protein level (i.e. Kv7.x subunits) was performed with antibodies specific for Kv7.1, Kv7.4 and Kv7.5 according to the expression profile identified by QPCR. Figure 1C shows that immunofluorescence specific to Kv7.1, Kv7.4 and Kv7.5 was localized to the sarcolemma of RMCA myocytes and not present in control experiments when primary antibodies were omitted. Mean fluorescence data for each Kv7 channel subunit are shown in Fig. 1D. Overall, these data suggest that rat cerebral arterial myocytes have a KCNQ expression profile of KCNQ4 > KCNQ5 and KCNQ1 and products of their expression show appropriate trafficking to the sarcolemmal membrane.
Identification of the presence of Kv7 currents in freshly isolated RMCA myocytes
RMCA myocytes were previously shown to express Kv1 (Albarwani et al. 2003; Chen et al. 2006) and Kv2-containing (Amberg & Santana, 2006; Zhong et al. 2010) channels exhibiting sensitivity to 4-AP (100–300 μm) or correolide (1–10 μm) and TEA+ (2–10 mm) or stromatoxin (10–300 nm; ScTx1), respectively. Based on these data, we sequentially exposed freshly dispersed RMCA myocytes to 4-AP (100 μm) and TEA+ (4 mm) prior to treatment with XE991, a blocker of all Kv7 channels with an IC50 of ∼1 μm (Wang et al. 2000; Dupuis et al. 2002; Greenwood & Ohya, 2009) to inhibit any residual Kv7 current if present. Figure 2A–C shows representative families of Kv currents, currents recorded at +25 mV for RMCA myocytes, as well as mean normalized (to peak current in control conditions at +45 mV) current–voltage (I–V) relations for RMCA myocytes exposed to control conditions and following sequential treatment with 4-AP, 4-AP and TEA+, and 4-AP, TEA+ and XE991. Sequential treatment with 4-AP and TEA+ caused a progressive inhibition of Kv current, and 10 μm XE991, as was used in previous studies on smooth muscle Kv7 channels (see Greenwood & Ohya, 2009), inhibited a small residual component of ∼20% total current amplitude. However, Fig. 2D and E shows that when XE991 was applied in the absence of 4-AP and TEA+, this drug caused a significant suppression of net Kv current and the extent of block was inconsistent with a selective inhibition of Kv7 channels alone; i.e. the amplitude of residual current was considerably less than that expected for the sum of the 4-AP-sensitive Kv1 and TEA+-sensitive Kv2 current components.
Figure 2. Suppression of RMCA myocyte Kv current by XE991.
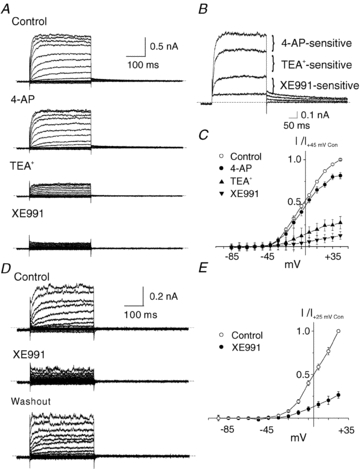
A, representative families of Kv currents of an RMCA myocyte evoked by 300 ms steps to voltages between −95 and +45 mV prior to repolarization to −55 mV (a similar protocol was used for all recordings unless indicated otherwise) in the absence (Control) and presence of 100 μm 4-AP, 4-AP plus 4 mM TEA+, and 4-AP, TEA+ and 10 μm XE991. B, representative traces from panel A for voltage steps to +25 mV showing the 4-AP-, TEA+- and XE991-sensitive components of net Kv current. C, mean ±s.e.m. normalized (to peak current at +45 mV in control condition) I–V relations for Kv current of RMCA myocytes (n= 3) sequentially treated with 4-AP, TEA+ and XE991 as in panel A. D, representative families of Kv currents from an RMCA myocyte in the absence and presence of 10 μm XE991 followed by drug washout. Voltage protocol as in panel A but from −95 to +25 mV only. E, mean ±s.e.m. normalized (to peak current at +25 mV in control condition) I–V relations for Kv current of RMCA myocytes (n= 5) in the absence (Control) and presence of 10 μm XE991. Note that XE991 caused a significant suppression of net Kv current in panels D and E that was larger than that predicted by the results of panels A–C.
To determine whether XE991 was selective for Kv7 channels at the concentration of 10 μm, this drug was applied to HEK 293 cells expressing homotetrameric Kv7.4 as a positive control, and heterotetrameric Kv1.2/Kv1.5 (Albarwani et al. 2003; Chen et al. 2006) and Kv2.1/Kv9.3 (Zhong et al. 2010) previously identified to be expressed by RMCA myocytes. Figure 3 shows representative families and mean normalized I–V relations for recombinant Kv7.4, Kv1.2/Kv1.5 and Kv2.1/Kv9.3 currents in the absence and presence of XE991. XE991 caused a marked inhibition of Kv7.4 current as expected; however, it also significantly reduced the amplitude of Kv1.2/Kv1.5 and Kv2.1/Kv9.3 currents. The block of Kv1.2/Kv1.5 channels was voltage dependent, and evident only at voltages positive to −15 mV. In contrast, the suppression of Kv2.1/Kv9.3 channels by XE991 was apparent at all potentials positive to −45 mV. These data indicate that XE991 at 10 μm has non-selective effects on the other heterotetrameric Kv channels expressed by RMCA myocytes.
Figure 3. Suppression of recombinant homomeric Kv7.4 and heteromeric Kv1.2/Kv1.5 and Kv2.1/Kv9.3 currents by XE991.
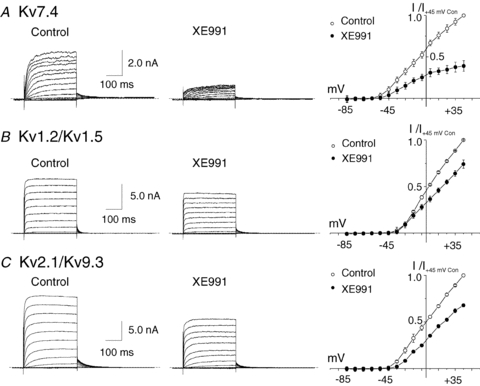
A, representative families and mean ±s.e.m. (n= 3) I–V relations for whole-cell Kv7.4 currents (HEK 293 cells) in the absence (Control) and presence of 10 μm XE991. B, representative families and mean ±s.e.m. (n= 3) normalized (to peak current at +45 mV in control condition) I–V relations for whole-cell Kv1.2/Kv1.5 currents (HEK 293 cells) in the absence (Control) and presence of 10 μm XE991 (note voltage steps were to between −95 and +25 mV only). C, representative families and mean ±s.e.m. (n= 3) normalized (to peak current at +45 mV in control condition) I–V relations for whole-cell Kv2.1/Kv9.3 currents (HEK 293 cells) in the absence (Control) and presence of 10 μm XE991. Note the suppression of Kv1.2/Kv1.5 and Kv2.1/Kv9.3 currents by XE991 at 10 μm.
Owing to the identified non-selective actions of XE991 shown in Fig. 3, we employed two additional compounds, linopirdine (Lamas et al. 1997; Dupuis et al. 2002) and S-1 (Bentzen et al. 2006), known to inhibit and activate Kv7 channels, respectively. The actions of linopirdine and S-1 were first studied using recombinant channels to identify appropriate concentrations that affected Kv7.4, but not Kv1.2/Kv1.5 or Kv2.1/Kv9.3 channels. Figures 4A and 5A show that linopirdine at 1 μm and S-1 at 3 μm significantly inhibited and increased recombinant Kv7.4 current amplitude, respectively. However, at identical concentrations, linopirdine and S-1 did not affect Kv1.2/Kv1.5 or Kv2.1/Kv9.3 channels (Figs 4B and C and 5B and C). A higher concentration of linopirdine of 10 μm caused 10–15% reduction of Kv2.1/Kv9.3 channels at +25 mV (n= 3; data not shown). Application of a higher concentration of 20 μm S-1 failed to increase either Kv1.2/Kv1.5 or Kv2.1/Kv9.3 currents, but Kv7.4 current amplitude was further augmented (n= 3 each; data not shown). Based on these findings, linopirdine was used at 1 μm and S-1 at 3 or 20 μm in subsequent experiments.
Figure 4. Suppression of recombinant homomeric Kv7.4, but not heteromeric Kv1.2/Kv1.5 and Kv2.1/Kv9.3 currents by linopirdine.
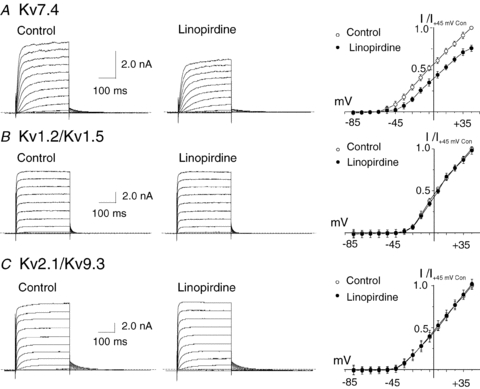
A, representative families and mean ±s.e.m. (n= 4) normalized (to peak current at +45 mV in control condition) I–V relations for whole-cell Kv7.4 currents (HEK 293 cells) in the absence (Control) and presence of 1 μm linopirdine. B, representative families and mean ±s.e.m. (n= 3) normalized (to peak current at +45 mV in control condition) I–V relations for whole-cell Kv1.2/Kv1.5 currents (HEK 293 cells) in the absence (Control) and presence of 1 μm linopirdine. C, representative families and mean ±s.e.m. (n= 3) normalized (to peak current at +45 mV in control condition) I–V relations for whole-cell Kv2.1/Kv9.3 currents (HEK 293 cells) in the absence (Control) and presence of 1 μm linopirdine. Note the lack of suppression of Kv1.2/Kv1.5 and Kv2.1/Kv9.3 currents by linopirdine at 1 μm.
Figure 5. Stimulation of recombinant homomeric Kv7.4, but not heteromeric Kv1.2/Kv1.5 and Kv2.1/Kv9.3 currents by S-1.
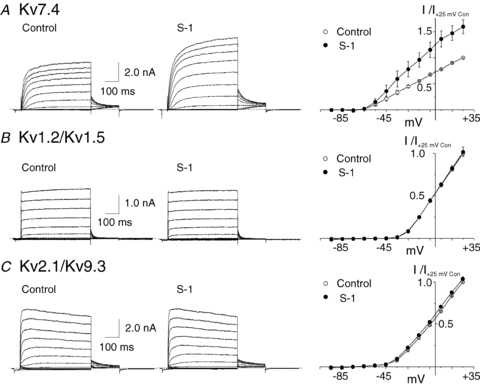
A, representative families and mean ±s.e.m. (n= 4) normalized (to peak current at +25 mV in control condition) I–V relations for whole-cell Kv7.4 currents (HEK 293 cells) in the absence (Control) and presence of 3 μm S-1. B, representative families and mean ±s.e.m. (n= 3) normalized (to peak current at +25 mV in control condition) I–V relations for whole-cell Kv1.2/Kv1.5 currents (HEK 293 cells) in the absence (Control) and presence of 3 μm S-1. C, representative families and mean ±s.e.m. (n= 3) normalized (to peak current at +25 mV in control condition) I–V relations for whole-cell Kv2.1/Kv9.3 currents (HEK 293 cells) in the absence (Control) and presence of 3 μm S-1. Note the lack of stimulation of Kv1.2/Kv1.5 and Kv2.1/Kv9.3 currents by S-1 at 3 μm.
Figures 6 and 7 show the effects of linopirdine (1 μm) and S-1 (3 μm) on native Kv current of RMCA myocytes, respectively. The amplitude of Kv current was reduced in the presence of linopirdine compared to control conditions (Fig. 6A), with the extent of block ∼20% at +5 mV (representative traces in Fig. 6B and mean normalized amplitude data in Fig. 6D). The linopirdine-sensitive component of native Kv current was time dependent, activated positive to −45 mV and showed no indication of time-dependent inactivation (Fig. 6C and E). In contrast, treatment with S-1 increased native Kv current amplitude at all potentials tested between −45 and +25 mV, as indicated in Fig. 7A–C. Importantly, S-1 increased native Kv current amplitude at −45 mV (Fig. 7B), which is within the range of membrane potential associated with the cerebral myogenic response (Knot & Nelson, 1995, 1998). Figure 7D and E show representative S-1-sensitive currents evoked by steps to between −85 and +25 mV (with the traces at −45 to +25 mV separated below for clarity) and the mean ±s.e.m.I–V relation. The increase in amplitude of the current was voltage dependent, reaching a peak at ∼−15 mV and declining with steps to increasingly positive voltages. The decline in current at positive voltages was associated with an apparent increase in time-dependent inactivation, likely to have resulted from open channel block. This is similar to the previously reported voltage-dependent, bimodal stimulation and block of murine portal vein Kv7 current by retigabine and flupirtine (Yeung et al. 2008).
Figure 6. Suppression of native RMCA myocyte Kv current by linopirdine.
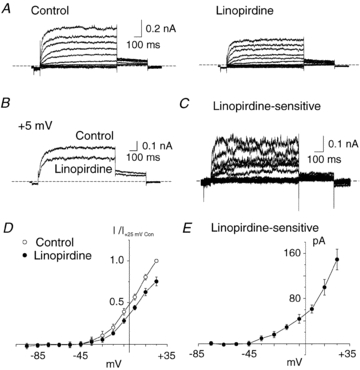
A, representative families of whole-cell RMCA myocyte Kv currents in the absence (Control) and presence of 1 μm linopirdine (note voltage clamp protocol included a 80 ms step to −95 mV prior to test steps to between −80 and +25 mV in 10 mV increments followed by repolarization to −45 mV). B, representative recordings of Kv current at +5 mV in the absence and presence of linopirdine (1 μm). C, representative family of linopirdine-sensitive Kv current obtained by digital subtraction of currents in linopirdine from those in control condition in panel A. D, mean ±s.e.m. (n= 3) normalized (to peak current at +25 mV in control condition) I–V relations for Kv current in the absence (Control) and presence linopirdine. E, mean ±s.e.m. (n= 3) I–V relation for linopirdine-sensitive Kv current determined from end-pulse difference current amplitude.
Figure 7. Stimulation of native RMCA myocyte Kv current by S-1.
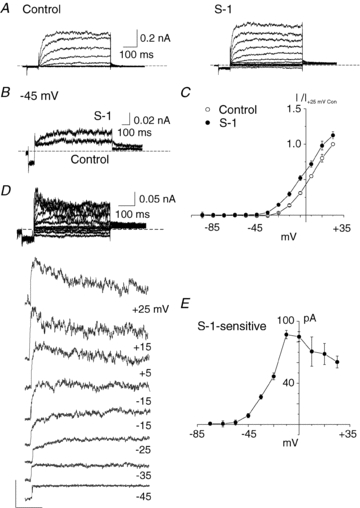
A, representative families of whole-cell RMCA myocyte Kv current in the absence (Control) and presence of 3 μm S-1 (note voltage clamp protocol included a 80 ms step to −95 mV prior to test steps). B, representative expanded traces for currents ± S-1 at −45 mV from cell in panel A. C, mean ±s.e.m. (n= 3) normalized (to peak current at +25 mV in control condition) I–V relations for Kv current in the absence (Control) and presence of S-1. D, representative S-1-sensitive Kv current obtained by digital subtraction of control from S-1 current in panel A. Expanded versions of each current recording for voltage steps to between −45 and +25 are shown below for clarity. Note the increased apparent inactivation of the S-1-sensitive current at potential positive to −5 mV. E, mean ±s.e.m. (n= 3) I–V relation of S-1-sensitive current determined from end pulse difference current amplitude. Note the decline in S-1-sensitive current positive to −15 mV, and y-axis is at −5 mV..
Kv7 channels contribute to the control of myogenic constriction of RMCA myocytes
To assess the contribution of Kv7 channels to the regulation of the cerebral arterial myogenic response, linopirdine (1 μm) and S-1 (3, 10 and 20 μm) were employed to determine whether pressure-induced constriction of RMCAs would be enhanced and inhibited, respectively, with inhibition and stimulation of Kv7 activity. The representative recordings of RMCA diameter at a constant pressure of 80 mmHg in Fig. 8B and C demonstrate the ability of linopirdine and S-1 to cause constriction and dilatation of pressurized RMCAs, respectively, but that no change in diameter occurred over the same time in untreated vessels, as shown in Fig. 8A (similar results were obtained in an additional 4 control, 4 linopirdine-treated and 2 S-1-treated vessels). The passive diameter of each treated vessel was determined by exposure to a zero Ca2+ saline solution (no added Ca2+ and 2 mm EGTA) at the end of the experiments.
Figure 8. Vasoconstriction and dilatation of RMCAs by linopirdine and S-1 at a constant pressure of 80 mmHg.
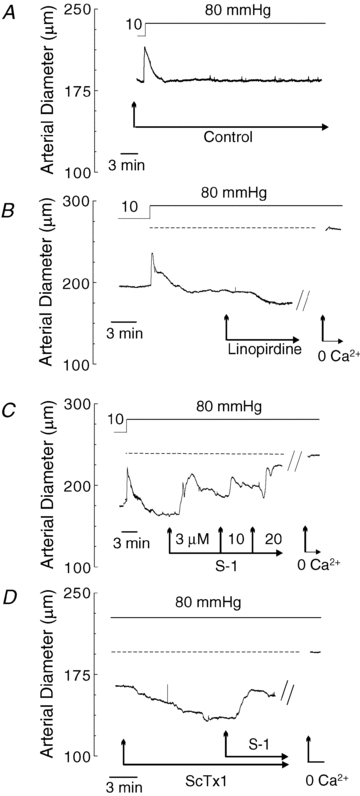
A, representative recording of RMCA diameter during a pressure step from 10 to 80 mmHg in control conditions. B, representative recording of RMCA diameter during a pressure step from 10 to 80 mmHg followed by subsequent sequential exposure to 1 μm linopirdine and zero Ca2+ solution (i.e. no added Ca2+ and 2 mm EGTA). Dashed line represents the passive diameter of the vessel in 0 Ca2+ solution at 80 mmHg. C, representative recording of RMCA diameter during a pressure step from 10 to 80 mmHg followed by subsequent sequential exposure to 3, 10 and 20 μm S-1 and zero Ca2+ solution. Dashed line represents the passive diameter of the vessel in 0 Ca2+ solution at 80 mmHg. Note concentration-dependent vasodilatation by S-1. D, representative recording of RMCA diameter at 80 mmHg during sequential exposure to 30 nm ScTx1 followed by S-1 (20 μm) and zero Ca2+ solution. Dashed line represents the passive diameter of the vessel in 0 Ca2+ solution at 80 mmHg. Note the reversal of ScTx1-induced vasoconstriction by S-1.
To assess the pressure-dependent contribution of Kv7 channels to the myogenic control of arterial diameter, RMCAs were sequentially exposed to varied intravascular pressures between 20 and 100 mmHg in 20 mmHg increments in the absence and presence of linopirdine (1 μm) or S-1 (20 μm), prior to exposure to zero Ca2+ solution to determine passive vessel diameter at each pressure. Figure 9A and B shows representative recordings of RMCA diameter and mean active constriction in control conditions and following linopirdine or S-1 treatment (i.e. active constriction is the difference between diameter ±drug and the passive diameter in absence of extracellular Ca2+ at each pressure). Linopirdine enhanced the level of active constriction of RMCAs at all pressures between 20 and 100 mmHg, whereas S-1 caused a significant reduction in active constriction at greater than 20 mmHg. We also examined a second activator, retigabine at 30 μm, and found that it also inhibited the myogenic response at >40 mmHg, although the effect was ∼50% of magnitude of the dilatation produced by 20 μm S-1 (n= 3; see online Supplemental Material). S-1 was also a more efficacious relaxant of pre-contracted RMCAs in isometric studies compared to retigabine (n= 6, see Supplemental Material).
Figure 9. Stimulation and suppression of RMCA myogenic response by linopirdine and S-1.
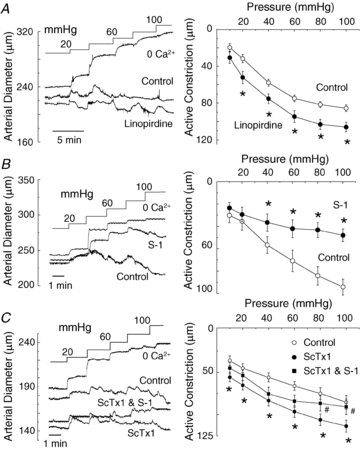
A, representative recordings of RMCA diameter during a series of pressure steps from 10 to between 20 and 100 mmHg in 20 mmHg increments in control conditions (Control), linopirdine (1 μm) and in zero Ca2+ solution, and mean ±s.e.m. (n= 5) active constriction in control and linopirdine (i.e. difference between diameter in zero Ca2+ and control or linopirdine). Note increase in active constriction in linopirdine at ≥20 mmHg. B, representative recordings of RMCA diameter during a series of pressure steps from 10 to between 20 and 100 mmHg in 20 mmHg increments in control conditions (Control), S-1 (20 μm) and in zero Ca2+ solution, and mean ±s.e.m. (n= 5) active constriction in control and S-1 (i.e. difference between diameter in zero Ca2+ and control or S-1). Note suppression of active constriction in S-1 at >20 mmHg. C, representative recordings of RMCA diameter during a series of pressure steps from 10 to between 20 and 100 mmHg in 20 mmHg increments in control conditions (Control), ScTx1 (30 nm), ScTx1 and S-1 (20 μm) and in zero Ca2+ solution, and mean ±s.e.m. (n= 3) active constriction in control, ScTx1, and ScTx1 and S-1. Note decrease in the ScTx1-enhanced active constriction in the presence of S-1 at 80 and 100 mmHg. *Significant difference at P < 0.05.
The representative recording of RMCA diameter in Fig. 8D shows that S-1 treatment also reversed the vasoconstriction caused by inhibition of Kv2-containing channels with 30 nm ScTx1 at 80 mmHg. A similar reversal of the vasoconstriction induced by ScTx1 was observed in two additional vessels treated with S-1 at 80 mmHg, and above 60 mmHg in three vessels pretreated with ScTx1 (30 nM) to cause vasoconstriction due to block of Kv2-containing channels (Fig. 9C). A final set of pressure myography experiments was performed with XE991 at a concentration of 10 μm to determine whether the potential non-selective effects of this agent on non-Kv7 channels would result in an effect on the myogenic response distinct from that observed for vessels treated with linopirdine. Figure 10 shows representative recording of diameter and average active constriction in the absence and presence of XE991. XE991 caused a pronounced vasoconstriction over the entire range of pressure studied, including at the minimal pressure of 10 mmHg, as well as a parallel increase in active constriction between 10 and 100 mmHg.
Figure 10. Stimulation of RMCA myogenic response by XE991.
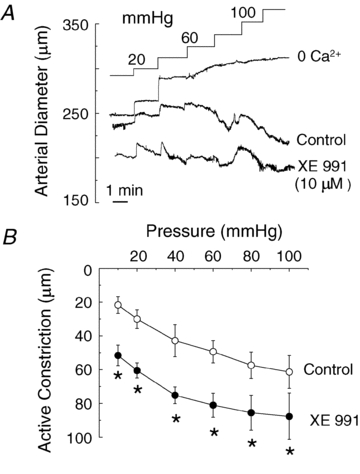
A, representative recordings of RMCA diameter during a series of pressure steps from 10 to between 20 and 100 mmHg in 20 mmHg increments in control conditions (Control), XE991 (10 μm) and in zero Ca2+ solution, and mean ±s.e.m. (n= 5) active constriction in control and XE991. Note increase in active constriction over the entire pressure range from 10 to 100 mmHg in XE991. *Significant difference at P < 0.05.
Discussion
This is the first study to identify the contribution of Kv7 channels to whole-cell Kv currents of VSMCs of small cerebral resistance arteries and to provide evidence that these channels contribute to the negative feedback regulation of the cerebral myogenic response. The view that Kv7 subunit-containing channels are expressed by RMCA myocytes and contribute to the control of the myogenic response is supported by evidence of: (1) the expression of mRNAs encoding Kv7.1, Kv7.4 and Kv7.5 pore-forming subunits, (2) Kv7.4 and Kv7.5 membrane protein expression in freshly isolated RMCA myocytes, (3) the presence of linopirdine- and S-1-sensitive components of whole-cell Kv current in RMCA myocytes, and (4) the ability of pharmacological manipulation of Kv7 channels with linopirdine and S-1 to respectively enhance and depress the myogenic response of RMCAs to changes in intravascular pressure. These novel findings have important implications for physiological control of cerebral blood flow and for the development of new pharmacological agents to treat clinical conditions of inappropriate control cerebral arterial diameter, such as vasospasm following subarachnoid haemorrhage.
The importance of vascular smooth muscle K+ channels in the physiological regulation of arterial diameter, blood pressure and organ-specific blood flow has become increasingly evident during the past 15 years. The prevailing view holds that negative-feedback activation of K+ channels plays a crucial role in controlling the myogenic response by providing hyperpolarizing membrane current to offset pressure-induced membrane depolarization owing to the activation of non-selective cation channels (Nelson et al. 1995; Knot & Nelson, 1995; Nelson & Quayle, 1997; Knot et al. 1998; Davis & Hill, 1999; Albarwani et al. 2003; Amberg & Santana, 2006; Chen et al. 2006; Yang et al. 2009; Zhong et al. 2010). This precise control of membrane potential is thought to be essential for the incremental changes in voltage-gated Ca2+ channel activation and Ca2+ influx that are required for subtle variations in arterial diameter which underlie physiological blood flow autoregulation (Nelson & Quayle, 1997; Davis & Hill, 1999). The findings of this study suggest for the first time that Kv7 channels also participate in this important physiological mechanism.
KCNQ channels have been extensively studied in neurons, cardiac myocytes and the inner ear (Jentsch, 2000), and their potential contribution to the control of membrane potential and contraction of smooth muscle has received increasing attention (reviewed in Greenwood & Ohya, 2009). The present study offers an important contribution to this knowledge by providing novel evidence of the participation of Kv7 channels in the pressure-dependent regulation of resistance arterial diameter. Previous studies have identified the expression of Kv7 subunit message and/or protein in murine portal vein (Ohya et al. 2003), as well as in myocytes of conduit and resistance vessels such as murine and/or rat thoracic aorta, pulmonary, carotid and femoral and mesenteric arteries (Joshi et al. 2006, 2009; Brueggemann et al. 2007; Yeung et al. 2007; Mackie et al. 2008). Kv7 transcripts and protein have also been identified in non-vascular, phasic tissues, such as colon (Jepps et al. 2009) and mouse and human uterine (myometrial) smooth muscle (McCallum et al. 2009, 2010). Several different Kv7 channel modulating drugs, including the inhibitors XE991 and linopirdine, and the activators S-1, retigabine and flupirtine, have been employed to obtain evidence of the contribution of Kv7 channels to membrane current and/or the regulation of contractility in these tissues. For example, these Kv7 modulators were shown to alter membrane currents of murine portal vein, and rat pulmonary and mesenteric arteries (e.g. Yeung & Greenwood, 2005; Yeung et al. 2007, 2008; Joshi et al. 2006, 2009; Mackie et al. 2008). Moreover, treatment of rat and mouse pulmonary arterial rings (Joshi et al. 2006, 2009) and conduit rat mesenteric arteries pressurized to 80 mmHg (Mackie et al. 2008) with XE991 or linopirdine induced contraction. Although in some cases the effects of the drugs were studied under conditions that minimize other Kv currents (e.g. by employing a holding potential of −4 mV; Mackie et al. 2008), or were tested for specificity by assessing their effect on non-Kv7 native K+ currents (e.g. Joshi et al. 2009), their potential non-selective effects were not considered in depth in the interpretation of the physiological responses of intact tissues.
Here, we found that XE991 at 10 μm (1) almost completely blocked Kv current of RMCA myocytes, although it is well-known that Kv1- and Kv2-containing channels contribute to the net Kv conductance of these myocytes (Albarwani et al. 2003; Chen et al. 2006; Amberg & Santana, 2006; Zhong et al. 2010), and (2) caused marked vasoconstriction at 10 mmHg, a pressure outside of the range over which linopirdine and S-1 were observed to affect the myogenic response (i.e. at >20 and 40 mmHg respectively). The increase in basal tone in the presence of XE991 at 10 mmHg was not mirrored by treatment with 1 μm linopirdine but has been observed for ScTx1 (Zhong et al. 2010), suggesting that this effect of XE991 may be the result of a non-selective block of Kv2-containing channels, as shown in this study. Although XE991 and linopirdine show selectivity for Kv7 channels, previous studies have identified inhibitory effects on homotetrameric Kv1 and Kv2 channels, as well as other Kv channels (Kv1.2, Kv4.3, EAG, ERG and ELK, Wang et al. 1998; Kv2.1, Wladyka & Kunze, 2006), and non-Kv7 neuronal transient and delayed rectifier K+ currents (e.g. Lamas et al. 1997; Schnee & Brown, 1998) at higher concentrations. For this reason, we employed concentrations of linopirdine and S-1 that were demonstrated to be without effect on the specific heteromultimeric Kv1.2/Kv1.5 and Kv2.1/Kv9.3 channels expressed by RMCA myocytes. This approach minimized the potential for non-selective effects and errors in interpretation of the mechanism of action in the physiological experiments employing intact vessels. At 1 and 3–20 μm, respectively, linopirdine and S-1 caused vasoconstriction and dilatation of RMCAs at 80 mmHg, which is within the physiological range of intravascular pressure of RMCAs (see Johnson et al. 2009). This result is consistent with the view that Kv7 channels contribute to the regulation of membrane potential in pressurized cerebral resistance arteries. We also found that linopirdine and S-1 respectively increased and decreased the myogenic response in a pressure-dependent manner at >40 mmHg. This is consistent with the expected behaviour of a K+ conductance showing voltage-dependent activation and having an increasing influence over force generation with progressively greater elevation in pressure and depolarization. Here we show that the activation range of native linopirdine- and S-1-sensitive current of RMCA myocytes at room temperature was positive to ∼−45 mV. This corresponds to the range of membrane potential associated with pressure-dependent myogenic depolarization between −65 mV at 10 mmHg and −35 mV at 120 mmHg in rat cerebral arteries (Knot & Nelson, 1998). Taken together, our findings provide evidence supporting the view that Kv7 channels contribute to the negative feedback regulation of pressure-induced myogenic constriction of cerebral arteries. A logical extension of the current study will be to determine the role of abnormal Kv7 channel (and splice variants) function in dysfunctional control of cerebral arterial membrane potential and diameter.
A limitation of the present study is that we have not determined the molecular composition of the Kv7 channels or the role of KCNE gene products in the RMCA. Variability in the biophysical properties of native M-currents of neurons are thought to result from several differences in molecular composition of the underlying Kv7 channels, including varied expression of Kv7 subunits and their splice variants (including potential dominant negative variants), heteromultimerization, and interaction with KCNE subunits (see McCrossan & Abbott, 2004; Greenwood & Ohya, 2009). The expression of Kv7.1, Kv7.4 and Kv7.5 along with KCNE1–4 transcripts suggests the possibility that both homo- and heteromultimeric channels may be present in the RMCA, as has been suggested for other vessels (see Greenwood & Ohya, 2009). Kv7.4 and Kv7.5 form functional heteromultimeric channels with properties distinct from those of their respective homomultimeric channels, but Kv7.1 only appears to interact with KNCE subunits owing to the presence of a distinct C-terminal subunit interaction domain that precludes assembly with other KCNQ gene products (Schwake et al. 2006; Bal et al. 2008). Co-expression of KCNE1–5 with Kv7.4 in Xenopus oocytes affected current amplitude (with KCNE1–4 increasing, KCNE3 decreasing and KCNE5 having little effect) and caused a leftward shift in the voltage dependence of activation (Strutz-Seebohm et al. 2006). Based on the present findings, it is impossible to offer concrete conclusions regarding the subunit composition of the RMCA Kv7-containing channels and whether KCNE gene products are present in the functional channel complex. Further experiments employing molecular approaches combined with biochemical and immunocytochemical approaches to specifically modulate different KCNQ and KCNE gene products and to detect subunit co-assembly will be required to directly address this issue, as was previously accomplished in the past in assessing the role of heteromultimeric Kv1 channels in cerebral arteries (Albarwani et al. 2003; Chen et al. 2006). Nevertheless, aspects of the present data imply the presence of heteromultimeric Kv7 channels. We base this view on the voltage-dependent stimulation of RMCA myocyte Kv current by S-1. S-1 was previously shown to preferentially activate Kv7.2–Kv7.5 channels, particularly Kv7.4 and Kv7.5, and to lack a stimulatory effect on Kv7.1 channels (Bentzen et al. 2006). The lack of effect on Kv7.1 appears to be due to the absence of a transmembrane tryptophan residue that is required for the stimulatory actions of retigabine, flutirpine and S-1 on other Kv7 channels (see Schenzer et al. 2005; Wuttke et al. 2005). Given the lack of expression of Kv7.2 and Kv7.3 in RMCAs, this result suggests that the effects of S-1 on Kv current and the myogenic response were due to modulation of Kv7.4- and/or Kv7.5-containing channels. The stimulation of homotetrameric Kv7.4 and Kv7.5 channels by S-1 progressively increases with greater depolarization (here and Bentzen et al. 2006). However, we show that the increase in RMCA myocyte Kv current amplitude reached a peak at ∼−15 mV and then progressively declined with increasing voltage in the presence of an identical concentration of S-1. A similar voltage-dependent, bimodal increase and inhibition of native Kv7 currents was previously reported for retigabine and/or flupirtine in murine portal vein myocytes (Yeung et al. 2008) and neurons (Rundfeldt, 1997; Tatulian et al. 2001). It seems likely that this difference in response to S-1 of native VSMC and recombinant homomeric Kv7 currents may be owing to heteromultimerization and possibly the presence of KCNE subunits in the native channels.
Pharmacological manipulation of Kv7 channel gating may represent an important novel option for the treatment of clinical situations of abnormal control of cerebral arterial diameter. For example, ∼66% of patients experience intense cerebral vasospasm following subarachnoid haemorrhage, but the mechanisms involved are not known with certainty and consequentially there are few effective therapies for this condition (Ferro et al. 2008; MacDonald et al. 2007). Recent data imply that reduced Kv2 channel expression and depolarization of the VSMC membrane potential may contribute to the vasospasm observed in a dog model of subarachnoid haemorrhage (Jahromi et al. 2008a,b;). Here, we show that activation of Kv7 channels with S-1 reversed the vasoconstriction of pressurized RMCAs produced by selective inhibition of Kv2-containing Kv channels with ScTx1. Moreover, this effect was pressure dependent, with the suppression of active constriction increasing between 60 and 100 mmHg. ScTx1 was employed as it causes vasoconstriction attributable solely to the inhibition of Kv2-containing channels in rat cerebral arteries (Amberg & Santana, 2006) and Zhong et al. (2010) recently reported that ScTx1 enhanced the myogenic response of RMCAs in a pressure-dependent manner. Figure 9C shows that the enhanced myogenic reactivity and increase in active vasoconstriction owing to ScTx1 treatment was reversed by S-1 activation of Kv7 channels in a pressure-dependent manner between 60 and 100 mmHg. These data indicate that the activation of Kv7 channels is sufficient to reverse the vasospasm owing to a reduction in cerebral VSMC currents due to Kv2-containing channels. At present, there is only a very limited repertoire of pharmacological agents that activate K+ channels and dilate resistance arteries/arterioles; several compounds that stimulate Ca2+-activated channels of large (BKCa) and intermediate (IKCa) conductance (e.g. NS1619 and 1-EBIO, respectively) and ATP-sensitive K+ channels (e.g. pinacidil, diazoxide, cromakalim, etc.) have been developed, but with the exception of Kv7 channel modulators, agents that stimulate Kv channel activity are lacking. Flupirtine has demonstrated effectiveness in pain management and retigabine is being considered as a possible anti-epileptic agent owing to their ability to activate Kv7.2–7.5 channels expressed in peripheral and central neurons (Main et al. 2000, Porter et al. 2007; and see Greenwood & Ohya, 2009). Whether Kv7 channel activators are also effective in the management of conditions of depressed VSMC K+ channel function, abnormal myogenic depolarization and constriction, such as are associated with cerebral vasospasm following subarachnoid haemorrhage (Ferro et al. 2008; Jahromi et al. 2008a,b; MacDonald et al. 2007), clearly warrants future examination based on our findings.
Acknowledgments
The study was supported by operating funds from the Canadian Institutes of Health Research Grant MOP-13505 to W.C.C. and from a British Heart Foundation (BHF) grant (06/057/20864 and PG/09/104) to I.A.G. X.Z.Z. was supported by salary funds from the Andrew Family Professorship in Cardiovascular Research held by W.C.C. M.I.H. is a BHF Intermediate Research Fellow (FS/06/077). We thank Professor T Bolton for use of his confocal microscope funded by The Wellcome Trust (Grants 074724 and 042293) and Emma Walsh for her help with arterial pressure myography and cell culture.
Glossary
Abbreviations
- BKCa channel
large conductance Ca2+ activated channel
- IKCa channel
intermediate conductance Ca2+ activated channel
- Kir channel
inward-rectifier K+ channel
- KV channel
voltage-gated K+ channel
- Q-PCR
quantitative reverse transcription polymerase chain reaction
- RMCA
rat middle cerebral artery
- VGCC
voltage-gated Ca2+ channel
- VSMC
vascular smooth muscle cell
- 4-AP
4-aminopyridine
- ScTx1
stromatoxin
- TEA+
tetraethylammonium ion
Author contributions
X.Z.Z. conducted all electrophysiological and pressure myograph experiments, M.I.H. conducted the immunocytochemical analysis of Kv7 channel expression in cerebral myocytes, and S.O. performed the real-time PCR analysis of Kv7 transcript expression. J.D.M. and S.P.O. were involved with the generation of preliminary data and drafting of the manuscript. X.Z.Z., W.C.C. and I.A.G. contributed to conception and design of the experiments, analysis, interpretation of the results and drafting the manuscript. All authors approved the final version of the manuscript. The experiments described in this study were conducted in the laboratories of W.C.C. in The Smooth Muscle Research Group at the University of Calgary, I.A.G. at St. George's University of London and S.O. at Nagoya City University Graduate School in Pharmaceutical Sciences. Disclosures: none.
Supplemental material
Online Data Supplement Figure 1
Online Data Supplement Figure 2
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Albarwani S, Nemetz LT, Madden JA, Tobin AA, England SK, Pratt PF, Rusch NJ. Voltage-gated K+ channels in rat small cerebral arteries: molecular identity of the functional channels. J Physiol. 2003;551:751–763. doi: 10.1113/jphysiol.2003.040014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amberg GC, Santana LF. Kv2 channels oppose myogenic constriction of rat cerebral arteries. Am J Physiol Cell Physiol. 2006;291:C348–C356. doi: 10.1152/ajpcell.00086.2006. [DOI] [PubMed] [Google Scholar]
- Bal M, Zhang J, Zaika O, Hernandez CC, Shapiro MS. Homomeric and heteromeric assembly of KCNQ (Kv7) K+ channels assayed by total internal reflection fluorescence/fluorescence resonance energy transfer and patch clamp analysis. J Biol Chem. 2008;283:30668–30676. doi: 10.1074/jbc.M805216200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentzen BH, Schmitt N, Calloe K, Dalby Brown W, Grunnet M, Olesen SP. The acrylamide (S)-1 differentially affects Kv7 (KCNQ) potassium channels. Neuropharmacology. 2006;51:1068–1077. doi: 10.1016/j.neuropharm.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Brueggemann LI, Moran CJ, Barakat JA, Yeh JZ, Cribbs LL, Byron KL. Vasopressin stimulates action potential firing by protein kinase C-dependent inhibition of KCNQ5 in A7r5 rat aortic smooth muscle cells. Am J Physiol Heart Circ Physiol. 2007;292:H1352–H1363. doi: 10.1152/ajpheart.00065.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brueggemann LI, Mackie AR, Mani BK, Cribbs LL, Byron KL. Differential effects of selective cyclooxygenase-2 inhibitors on vascular smooth muscle ion channels may account for differences in cardiovascular risk profiles. Mol Pharmacol. 2009;76:1053–1061. doi: 10.1124/mol.109.057844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan RM, Jr, You J, Phillips SC, Andresen JJ, Lloyd EE, Rogers PA, Dryer SE, Marrelli SP. Evidence for two-pore domain potassium channels in rat cerebral arteries. Am J Physiol Heart Circ Physiol. 2006;291:H770–H780. doi: 10.1152/ajpheart.01377.2005. [DOI] [PubMed] [Google Scholar]
- Chen TT, Luykenaar KD, Walsh EJ, Walsh MP, Cole WC. Key role of Kv1 channels in vasoregulation. Circ Res. 2006;99:53–60. doi: 10.1161/01.RES.0000229654.45090.57. [DOI] [PubMed] [Google Scholar]
- Cole WC, Chen TT, Clément-Chomienne O. Myogenic regulation of arterial diameter: role of potassium channels with a focus on delayed rectifier potassium current. Can J Physiol Pharmacol. 2005;83:755–765. doi: 10.1139/y05-082. [DOI] [PubMed] [Google Scholar]
- Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev. 1999;79:387–423. doi: 10.1152/physrev.1999.79.2.387. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis DS, Schrøder RL, Jespersen T, Christensen JK, Christophersen P, Jensen BS, Olesen SP. Activation of KCNQ5 channels stably expressed in HEK293 cells by BMS-204352. Eur J Pharmacol. 2002;437:129–137. doi: 10.1016/s0014-2999(02)01287-6. [DOI] [PubMed] [Google Scholar]
- Ferro JM, Canhão P, Peralta R. Update on subarachnoid haemorrhage. J Neurol. 2008;255:465–479. doi: 10.1007/s00415-008-0606-3. [DOI] [PubMed] [Google Scholar]
- Greenwood IA, Ohya S. New tricks for old dogs: KCNQ expression and function in smooth muscle. Br J Pharmacol. 2009;156:1196–1203. doi: 10.1111/j.1476-5381.2009.00131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MA, Zou H, Potocnik SJ, Meininger GA, Davis MJ. Invited review: arteriolar smooth muscle mechanotransduction: Ca2+ signaling pathways underlying myogenic reactivity. J Appl Physiol. 2001;91:973–983. doi: 10.1152/jappl.2001.91.2.973. [DOI] [PubMed] [Google Scholar]
- Hill MA, Davis MJ, Meininger GA, Potocnik SJ, Murphy TV. Arteriolar myogenic signalling mechanisms: Implications for local vascular function. Clin Hemorheol Microcirc. 2006;34:67–79. [PubMed] [Google Scholar]
- Jackson WF. Potassium channels in the peripheral microcirculation. Microcirculation. 2005;12:113–127. doi: 10.1080/10739680590896072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahromi BS, Aihara Y, Ai J, Zhang ZD, Nikitina E, Macdonald RL. Voltage-gated K+ channel dysfunction in myocytes from a dog model of subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2008a;28:797–811. doi: 10.1038/sj.jcbfm.9600577. [DOI] [PubMed] [Google Scholar]
- Jahromi BS, Aihara Y, Ai J, Zhang ZD, Weyer G, Nikitina E, Yassari R, Houamed KM, Macdonald RL. Temporal profile of potassium channel dysfunction in cerebrovascular smooth muscle after experimental subarachnoid hemorrhage. Neurosci Lett. 2008b;440:81–86. doi: 10.1016/j.neulet.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch TJ. Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neuroscience. 2000;1:21–30. doi: 10.1038/35036198. [DOI] [PubMed] [Google Scholar]
- Jepps TA, Greenwood IA, Moffatt JD, Sanders KM, Ohya S. Molecular and functional characterization of Kv7 K+ channel in murine gastrointestinal smooth muscles. Am J Physiol Gastrointest Liver Physiol. 2009;297:G107–115. doi: 10.1152/ajpgi.00057.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RP, El-Yazbi AF, Takeya K, Walsh EJ, Walsh MP, Cole WC. Ca2+ sensitization via phosphorylation of myosin phosphatase targeting subunit at threonine-855 by Rho kinase contributes to the arterial myogenic response. J Physiol. 2009;587:2537–2553. doi: 10.1113/jphysiol.2008.168252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi S, Balan P, Gurney AM. Pulmonary vasoconstrictor action of KCNQ potassium channel blockers. Respir Res. 2006;7:31. doi: 10.1186/1465-9921-7-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi S, Sedivy V, Hodyc D, Herget J, Gurney AM. KCNQ modulators reveal a key role for KCNQ potassium channels in regulating the tone of rat pulmonary artery smooth muscle. J Pharmacol Exp Ther. 2009;329:368–376. doi: 10.1124/jpet.108.147785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr PM, Clément-Chomienne O, Thorneloe KS, Chen TT, Ishii K, Sontag DP, Walsh MP, Cole WC. Heteromultimeric Kv1.2-Kv1.5 channels underlie 4-aminopyridine-sensitive delayed rectifier K+ current of rabbit vascular myocytes. Circ Res. 2001;89:1038–1044. doi: 10.1161/hh2301.100803. [DOI] [PubMed] [Google Scholar]
- Knot HJ, Nelson MT. Regulation of membrane potential and diameter by voltage-dependent K+ channels in rabbit myogenic cerebral arteries. Am J Physiol Heart Circ Physiol. 1995;269:H348–H355. doi: 10.1152/ajpheart.1995.269.1.H348. [DOI] [PubMed] [Google Scholar]
- Knot HJ, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol. 1998;508:199–209. doi: 10.1111/j.1469-7793.1998.199br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knot HJ, Standen NB, Nelson MT. Ryanodine receptors regulate arterial diameter and wall [Ca2+] in cerebral arteries of rat via Ca2+-dependent K+ channels. J Physiol. 1998;508:211–221. doi: 10.1111/j.1469-7793.1998.211br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamas JA, Selyanko AA, Brown DA. Effects of a cognition-enhancer, linopirdine (DuP 996), on M-type potassium currents (IK(M)) and some other voltage- and ligand-gated membrane currents in rat sympathetic neurons. Eur J Neurosci. 1997;9:605–616. doi: 10.1111/j.1460-9568.1997.tb01637.x. [DOI] [PubMed] [Google Scholar]
- Macdonald RL, Pluta RM, Zhang JH. Cerebral vasospasm after subarachnoid hemorrhage: the emerging revolution. Nat Clin Pract Neurol. 2007;3:256–263. doi: 10.1038/ncpneuro0490. [DOI] [PubMed] [Google Scholar]
- Mackie AR, Brueggemann LI, Henderson KK, Shiels AJ, Cribbs LL, Scrogin KE, Byron KL. Vascular KCNQ potassium channels as novel targets for the control of mesenteric artery constriction by vasopressin, based on studies in single cells, pressurized arteries, and in vivo measurements of mesenteric vascular resistance. J Pharmacol Exp Ther. 2008;325:475–483. doi: 10.1124/jpet.107.135764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Main MJ, Cryan JE, Dupere JR, Cox B, Clare JJ, Burbidge SA. Modulation of KCNQ2/3 potassium channels by the novel anticonvulsant retigabine. Mol Pharmacol. 2000;58:253–262. doi: 10.1124/mol.58.2.253. [DOI] [PubMed] [Google Scholar]
- McCallum LA, Greenwood IA, Tribe RM. Expression and function of Kv7 channels in murine myometrium throughout the oestrus cycle. Pflugers Archiv. 2009;457:1111–1120. doi: 10.1007/s00424-008-0567-5. [DOI] [PubMed] [Google Scholar]
- McCallum LA, Pierce SL, England SK, Greenwood IA, Tribe RM. The contribution of Kv7 channels to pregnant mouse and human myometrial contractility. J Cell Mol Med. 2010 doi: 10.1111/j.1582-4934.2010.01021.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrossan ZA, Abbott GW. MinK-related peptides. Neuropharmacology. 2004;47:787–821. doi: 10.1016/j.neuropharm.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633–637. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol Cell Physiol. 1997;268:C799–C822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- Namiranian K, Lloyd EE, Crossland RF, Marrelli SP, Taffet GE, Reddy AK, Hartley CJ, Bryan RM., Jr Cerebrovascular responses in mice deficient in the potassium channel, TREK-1. Am J Physiol Regul Integr Comp Physiol. 2010 doi: 10.1152/ajpregu.00057.2010. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohya S, Sergeant G, Greenwood IA, Horowitz B. Molecular variants of KCNQ channels expressed in murine portal vein myocytes: a role in delayed rectifier current. Circ Res. 2003;92:1016–1023. doi: 10.1161/01.RES.0000070880.20955.F4. [DOI] [PubMed] [Google Scholar]
- Plane F, Johnson R, Kerr P, Wiehler W, Thorneloe K, Ishii K, Chen T, Cole W. Heteromultimeric Kv1 channels contribute to myogenic control of arterial diameter. Circ Res. 2005;96:216–224. doi: 10.1161/01.RES.0000154070.06421.25. [DOI] [PubMed] [Google Scholar]
- Porter RJ, Nohria V, Rundfeldt C. Retigabine. Neurotherapeutics. 2007;4:149–154. doi: 10.1016/j.nurt.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins J. KCNQ potassium channels: physiology, pathophysiology and pharmacology. Pharmacol Ther. 2001;90:1–19. doi: 10.1016/s0163-7258(01)00116-4. [DOI] [PubMed] [Google Scholar]
- Rundfeldt C. The new anticonvulsant retigabine (D-23129) acts as an opener of K+ channels in neuronal cells. Eur J Pharmacol. 1997;336:243–249. doi: 10.1016/s0014-2999(97)01249-1. [DOI] [PubMed] [Google Scholar]
- Schnee ME, Brown BS. Selectivity of linopirdine (DuP 996), a neurotransmitter release enhancer, in blocking voltage-dependent and calcium-activated potassium currents in hippocampal neurons. J Pharmacol Exp Ther. 1998;286:709–717. [PubMed] [Google Scholar]
- Schenzer A, Friedrich T, Pusch M, Saftig P, Jentsch TJ, Grotzinger J, Schwake M. Molecular determinants of KCNQ (Kv7) K+ channel sensitivity to the anticonvulsant retigabine. J Neurosci. 2005;25:5051–5060. doi: 10.1523/JNEUROSCI.0128-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwake M, Athanasiadu D, Beimgraben C, Blanz J, Beck C, Jentsch TJ, Saftig P, Friedrich T. Structural determinants of M-type KCNQ (Kv7) K+ channel assembly. J Neurosci. 2006;26:3757–3766. doi: 10.1523/JNEUROSCI.5017-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strutz-Seebohm N, Seebohm G, Fedorenko O, Baltaev R, Engel J, Knirsch M, Lang F. Functional coassembly of KCNQ4 with KCNE-β-subunits in Xenopus oocytes. Cell Physiol Biochem. 2006;18:57–66. doi: 10.1159/000095158. [DOI] [PubMed] [Google Scholar]
- Teramoto N. Physiological roles of ATP-sensitive K+ channels in smooth muscle. J Physiol. 2006;572:617–624. doi: 10.1113/jphysiol.2006.105973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatulian L, Delmas P, Abogadie FC, Brown DA. Activation of expressed KCNQ potassium currents and native neuronal M-type potassium currents by the anti-convulsant drug retigabine. J Neurosci. 2001;21:5535–5545. doi: 10.1523/JNEUROSCI.21-15-05535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- Wang HS, Brown BS, McKinnon D, Cohen IR. Molecular basis for differential sensitivity of KCNQ and IKs channels to the cognitive enhancer XE991. Mol Pharmacol. 2000;57:1218–1223. [PubMed] [Google Scholar]
- Wladyka CL, Kunze DL. KCNQ/M-currents contribute to the resting membrane potential in rat visceral sensory neurons. J Physiol. 2006;575:175–189. doi: 10.1113/jphysiol.2006.113308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuttke TV, Seebohm G, Bail S, Maljevic S, Lerche H. The new anticonvulsant Retigabine favors voltage-dependent opening of the Kv7.2 (KCNQ2) channel by binding to its activation gate. Mol Pharmacol. 2005;67:1009–1017. doi: 10.1124/mol.104.010793. [DOI] [PubMed] [Google Scholar]
- Yang Y, Murphy TV, Ella SR, Grayson TH, Haddock R, Hwang YT, Braun AP, Peichun G, Korthuis RJ, Davis MJ, Hill MA. Heterogeneity in function of small artery smooth muscle BKCa: involvement of the β1-subunit. J Physiol. 2009;587:3025–3044. doi: 10.1113/jphysiol.2009.169920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung SYM, Greenwood IA. Electrophysiological and functional effects of the KCNQ channel blocker XE991 on murine portal vein smooth muscle cells. Br J Pharmacol. 2005;146:585–595. doi: 10.1038/sj.bjp.0706342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung SYM, Pucovský V, Moffatt JD, Saldanha L, Schwake M, Ohya S, Greenwood IA. Molecular expression and pharmacological identification of a role for Kv7 channels in murine vascular reactivity. Br J Pharmacol. 2007;151:758–770. doi: 10.1038/sj.bjp.0707284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung SYM, Schwake M, Pucovský V, Greenwood IA. Bimodal effects of the Kv7 channel activator retigabine on vascular K+ currents. Br J Pharmacol. 2008;155:62–72. doi: 10.1038/bjp.2008.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaritsky JJ, Eckman DM, Wellman GC, Nelson MT, Schwarz TL. Targeted disruption of Kir2.1 and Kir2.2 genes reveals the essential role of the inwardly rectifying K+ current in K+-mediated vasodilation. Circ Res. 2000;87:160–166. doi: 10.1161/01.res.87.2.160. [DOI] [PubMed] [Google Scholar]
- Zhong X, Walsh EJ, El-Yazbi A, Cole WC. Heteromultimeric Kv2.1/9.3 channels contribute to myogenic control of rat middle cerebral arterial (RMCA) diameter. FASEB J. 2010;24:1033.8. doi: 10.1113/jphysiol.2010.196618. (Meeting Abstracts) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.