

Abstract
Hydrophobic bile salts are thought to contribute to the disruption of gallbladder smooth muscle (GBSM) function that occurs in gallstone disease, but their mechanism of action is unknown. The current study was undertaken to determine how hydrophobic bile salts interact with GBSM, and how they reduce GBSM activity. The effect of hydrophobic bile salts on the activity of GBSM was measured by intracellular recording and calcium imaging using wholemount preparations from guinea pig and mouse gallbladder. RT-PCR and immunohistochemistry were used to evaluate expression of the G protein-coupled bile acid receptor, GPBAR1. Application of tauro-chenodeoxycholate (CDC, 50–100 μm) to in situ GBSM rapidly reduced spontaneous Ca2+ flashes and action potentials, and caused a membrane hyperpolarization. Immunoreactivity and transcript for GPBAR1 were detected in gallbladder muscularis. The GPBAR1 agonist, tauro-lithocholic acid (LCA, 10 μm) mimicked the effect of CDC on GBSM. The actions of LCA were blocked by the protein kinase A (PKA) inhibitor, KT5720 (0.5–1.0 μm) and the KATP channel blocker, glibenclamide (10 μm). Furthermore, LCA failed to disrupt GBSM activity in Gpbar1−/− mice. The findings of this study indicate that hydrophobic bile salts activate GPBAR1 on GBSM, and this leads to activation of the cyclic AMP–PKA pathway, and ultimately the opening of KATP channels, thus hyperpolarizing the membrane and decreasing GBSM activity. This inhibitory effect of hydrophobic bile salt activation of GPBAR1 could be a contributing factor in the manifestation of gallstone disease.
Introduction
Decreased gallbladder contractility is a hallmark of both calculous and acalculous gallbladder disease and is associated with inflammation and gallstone formation. Diminished gallbladder emptying in response to a meal and/or cholecystokinin (CCK) infusion is observed in the vast majority of patients with cholesterol gallstones (Jazrawi et al. 1995; Shoda et al. 1995). These individuals also have increased fasting residual gallbladder volumes, as well as a decreased ejection fraction after a meal. Moreover, the interrelationship between bile stasis, inflammation and stone formation is still poorly understood.
Decreased contractility in gallstone disease may be caused by elevation of two main bile components, cholesterol and hydrophobic bile salts. Gallbladder smooth muscle (GBSM) strips from patients with cholesterol gallstones have impaired contractile responses to a variety of stimuli in vitro (Behar et al. 1989). When ground squirrels are fed a lithogenic (stone forming) diet rich in cholesterol and cholate, they exhibit a progressive increase in biliary cholesterol that is associated with a decrease in gallbladder contractility, cholesterol stone formation and cholecystitis (Xu & Shaffer, 1993). Gallbladders of C57L mice fed a lithogenic diet also have altered histology, are larger and have greater residual fasting volume (Wang et al. 1997; van Erpecum et al. 2006). Their bile shows cholesterol supersaturation, accumulation of mucin gel, and the formation of cholesterol crystals and gallstones. Cellular mechanisms responsible for the impaired GBSM activity are still unclear.
GBSM activity is characterized by rhythmic spontaneous action potentials that cause synchronized increases in intracellular [Ca2+] called Ca2+ flashes (Zhang et al. 1993; Balemba et al. 2006b). While voltage-dependent Ca2+ channels (VDCCs) are mainly responsible for the fast depolarization component of the action potential, the repolarization involves various members of the K+ channel family (Zhang et al. 1993). Coupling between interstitial cells of Cajal (ICC), known as the pacemaker cells of the GI tract, and GBSM is in part responsible for the synchronicity and rhythmicity of the GBSM activity (Lavoie et al. 2007). This spontaneous activity in GBSM, which underlies the maintenance of tone and gallbladder contractions, is disrupted by activation of the cAMP–PKA pathway (Zhang et al. 1994b). PKA-mediated suppression of gallbladder activity occurs primarily via the activation of ATP-sensitive K+ (KATP) channels (Zhang et al. 1994a).
In addition to cholesterol, bile also contains both hydrophobic and hydrophilic bile salts referred to as ‘bad’ and ‘good’ bile salts, respectively, because of their relative effects on gallbladder function. Hydrophobic bile salts attenuate gallbladder contractility, an effect that has been directly related to the level of bile salt hydrophobicity (Xu et al. 1997). Using cannulated gallbladders in vitro, Rutishauser (1978) showed that hydrophobic bile salts inhibit gallbladder motor activity. Subsequently, Shaffer and colleagues demonstrated that hydrophobic bile salts cause a concentration-dependent inhibition of CCK-induced gallbladder muscle strip contractions (Xu et al. 1997).
The cellular mechanisms by which hydrophobic bile salts disrupt GBSM function have not been resolved, but in other tissues, hydrophobic bile salts relax smooth muscle (Bomzon & Ljubuncic, 2001; Dopico et al. 2002). Furthermore, hydrophobic bile salts appear to activate different K+ channels in various cell types. For example, in murine small intestinal ICCs, they inhibit pacemaker currents by activating KATP channels (Jun et al. 2005), whereas in rabbit mesenteric artery myocytes, they increase large conductance, Ca2+-activated K+ (BK) channel function (Dopico et al. 2002).
Specific bile acid receptors have been identified and they have been divided into two classes. The first class includes the nuclear bile acid receptors, farnesoid X receptor and pregnane X receptor, which control transcriptional level of enzymes involved in synthesis of bile acid. More recently a plasma membrane bile acid receptor called the G protein-coupled bile acid receptor (GPBAR1; also termed TGR5, M-Bar, or GPR131) has been characterized (Kawamata et al. 2003). It is a member of the rhodopsin-like family of G protein-coupled receptors that is ubiquitously expressed, but is most highly expressed in the gallbladder (Vassileva et al. 2006; Watanabe et al. 2006). GPBAR1 activation may be involved in gallstones disease because Gpbar1−/− mice do not form gallstones when fed a lithogenic diet (Vassileva et al. 2006). GPBAR1 activation by bile salts stimulates the Gs–cAMP–PKA pathway (Kawamata et al. 2003; Watanabe et al. 2006). Increased cAMP levels by β-receptor agonists, vasoactive intestinal peptide (VIP) and calcitonin gene-related peptide (CGRP) induce gallbladder relaxation. Activation of the cAMP–PKA pathway mediates the inhibition of gallbladder contractility by activating KATP channels, which, in turn, causes a membrane potential hyperpolarization of GBSM. We have previously shown in detail how CGRP, a smooth muscle relaxant, activates the opening of KATP channels via the Gs–cAMP–PKA pathway (Zhang et al. 1994a; Morales et al. 2004). The aim of this study was to test the hypothesis that hydrophobic bile salts disrupt GBSM function by binding to the Gs-coupled receptor GPBAR1 and activating a cAMP-mediated opening of KATP channels.
Methods
Animals
In this study, adult (female and male, 250–350 g) guinea pigs and C57BL/6J mice (male 7–15 weeks) were used. The Gpbar1−/− mice were generated directly into C57BL/6J genetic background by using C57BL/6J-derived ES cells and mating the chimeric males to C57BL/6J females (Vassileva et al. 2006, 2010).
The animals were anaesthesized with isoflurane and exsanguinated using protocols that were approved by the Institutional Animal Care and Use Committee of the University of Vermont. The abdominal cavity was opened and the gallbladder removed. Tissue was collected into ice-cold modified Krebs solution composed of (in mm): 121 NaCl, 5.9 KCl, 2.5 CaCl2, 1.2 MgCl2, 25 NaHCO3, 1.2 NaHPO4 and 8 glucose (pH 7.4).
Gallbladders were opened from fundus to the cystic duct, stretched and pinned mucosa side up on a Sylgard dish filled with ice-cold Krebs solution. In guinea pig preparations, the mucosal layer was removed with sharp forceps to enhance penetration of Fluo-4AM and visualization of the GBSM, whereas full-thickness wholemounts were used for mice, which are thinner and removal of the mucosa was not necessary.
Imaging and analysis of Ca2+ events
Each segment of tissue was stretched and pinned between two pieces of Sylgard. Tissue was equilibrated in HEPES buffer containing (in mm): 110 NaCl, 5.4 KCl, 1.8 CaCl2, 1.0 MgCl2, 10 HEPES and 5 glucose (pH 7.4) and then loaded for 1 h at room temperature in a HEPES buffer containing 10 μm fluo-4AM (Invitrogen) and 2.5 μg-ml−1 pluronic acid (Invitrogen). These preparations were washed in HEPES for at least 30 min at room temperature to allow deesterification.
The preparations were then placed in a Ca2+ imaging chamber, and superfused with aerated physiological saline solution (PSS) containing (in mm): 119 NaCl, 7.5 KCl, 1.6 CaCl2, 1.2 MgCl2, 23.8 NaHCO3, 1.2 NaH2PO4, 0.023 EDTA and 11 glucose (pH 7.3) at 35–37°C. After a 15–20 min equilibration, Ca2+ events were analysed using a Nikon TMD inverted microscope with a 60× water immersion lens attached to a Noran Oz laser confocal system. Movie files were acquired over periods of 20–30 s (30 frames per second). In some experiments, Ca2+ events were visualized using an Andor iXonEM+ 897 back-illuminated EMCCD camera attached to an inverted fluorescent Olympus IX70 microscope equipped with a 40× objective. Movies were analysed using SparkAN, a custom software written at the University of Vermont (A. D. Bonev).
Intracellular recording
Wholemount sections of guinea pig gallbladder muscularis propria were stretched in a Sylgard covered recording chamber serosa side up on a Nikon TMD inverted microscope equipped with Hoffman modulation contrast optics (Modulation Optics, Greenvale, NY, USA) to visualize smooth muscle bundles. Tissue was continuously superfused with aerated (95% O2–5% CO2) Krebs solution at 35–37°C. Electrical activity was recorded using glass microelectrodes filled with 2 m KCl (resistance range of 70–150 MΩ) and a negative-capacity compensation amplifier (Axoclamp 2A, Molecular Devices, Sunnyvale, CA, USA). Membrane potential traces were recorded and analysed using PowerLab/4SP and Chart5 software packages (ADInstruments, Colorado Springs, CO, USA).
Detection of Gpbar1 mRNA in the gallbladder by RT-PCR
Total RNA was isolated from mouse and guinea pig gallbladder using RNAeasy Midi kit (Qiagen Inc., Valencia, CA, USA). For each reaction 2 μg of total RNA was combined with oligo(dT), dNTP, RNasin, and M-MLV RT enzyme and buffer (Promega, Madison, WI, USA). The PCR reaction was performed using the Hot Start IT taq with the following primer sets: mouse Gpbar1: forward (5′-TGGAAGTTTATGGCCTCCTG-3′) and reverse (5′-CCAACACAGCAAGAAGAGCA-3′) primers; guinea pig Gpbar1: forward (5′-TGGAAGTTTATGGCCTCCTG-3′) and reverse (5′-CCAACACAGCAAGAAGAGCA-3′) primers; mouse Muc5ac: forward (5′-GTCTGGCAGAAACAGTGGAGATT-3′) and reverse (5′-TCGTGGCTTCTCACAGAACTTG-3′) primers. The 208 bp mouse and 246 bp guinea pig Gpbar1 products, and the 78 bp mouse Muc5ac were detected on an ethidium bromide-stained agarose gel and sequenced to confirm identity.
Immunohistofluorescence to detect GPBAR1 localization in gallbladder tissue
Mouse and guinea pig gallbladders were fixed in 4% paraformaldehyde (0.1 m phosphate buffered saline (PBS), pH 7.4, overnight, 4°C). Gallbladders were cleared with dimethyl sulfoxide (DMSO), washed with PBS, and incubated in 30% sucrose PBS (overnight, 4°C). They were embedded in Optimal Cutting Temperature compound (Tissue-Tek, Sakura Finetek, Torrance, CA, USA), sectioned (10 μm), and thaw-mounted onto slides. Sections were incubated in PBS containing 4–10% normal serum and 0.1–0.5% Triton X-100 (1 h, room temperature). Sections were incubated with GPBAR1 antibody (Poole et al. 2010), smooth muscle actin (Sigma-Aldrich, St Louis, MO, USA, 1:10,000) and GPBAR1 antibody preadsorbed with immunizing peptide (50 μm, overnight, 4°C). Slides were washed and incubated with secondary antibody conjugated to Cy3, Rhodamine Red X or FITC (Jackson Immunoresearch Laboratories Inc., West Grove, PA, USA, 1:200–500, for 1–3 h at room temperature). Mouse sections were mounted using ProlongGold (Invitrogen, Calsbad, CA, USA) and observed using a Zeiss LSM510 META confocal system. Guinea pig preparations were mounted using Citi-Fluor (Electron Microscopy Sciences Co., Ft Washington, PA, USA) and observed using an inverted Olympus IX-70 fluorescence microscope equipped with a MagnaFire CCD camera (Optronics, Goleta, CA, USA).
Statistical analyses
Statistical analyses were done using Graph Pad Prism (GraphPad Software Inc., San Diego, CA, USA). Data are expressed as means ±s.e.m., and n refers to the number of preparation from different animals used. P values ≤ 0.05 were considered statistically significant (Student's t test for two samples or one-way ANOVA and Dunnett's multiple comparison test for more than two samples).
Chemicals
Unless otherwise specified drugs used in this study including tauro-chenodeoxycholate, tauro-lithocholate, glibenclamide, KT-5720, pinacidil, and forskolin were from Sigma-Aldrich (St Louis, MO, USA).
Results
The hydrophobic bile salt tauro-chenodeoxycholate disrupts GBSM function
We have previously shown that GBSM activity is characterized by rhythmic firing of spontaneous action potentials that cause Ca2+ flashes in all of the GBSM cells of a given bundle, which in turn stimulate tissue contraction (Zhang et al. 1993; Balemba et al. 2006b). We therefore used Ca2+ imaging and intracellular recording techniques to evaluate the effect of the hydrophobic bile salts on the basal rhythmic activity in the guinea pig GBSM bundles.
In guinea pig, Ca2+ flashes in GBSM cells were synchronized within a given bundle (Fig. 1A). The basal frequency of Ca2+ flashes recorded in the current study was 0.22 ± 0.02 Hz (n= 19), which is similar to that previously reported (Balemba et al. 2006b). Addition of the hydrophobic bile salt tauro-chenodeoxycholate (CDC, 50–100 μm) to the bathing solution caused a rapid reduction of spontaneous Ca2+ activity in GBSM, with elimination of flashes as soon as 2.5 min after the application of CDC to the bath.
Figure 1. The hydrophobic bile salt tauro-chenodeoxycholate (CDC) disrupts spontaneous activity in guinea pig GBSM.
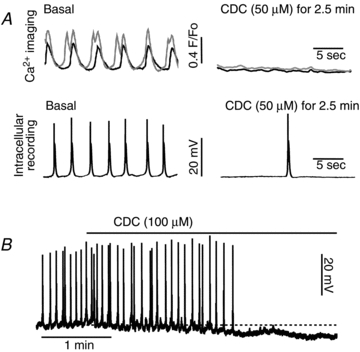
A, the hydrophobic bile salt CDC (50–100 μm) rapidly reduces the rhythmic discharge of Ca2+ flashes (top traces; two muscle cells in the same bundle are represented) and eliminates action potentials (bottom traces; recorded in a separate preparation). B, the intracellular recording demonstrates a membrane potential hyperpolarization that was induced by CDC (the dashed line indicates the resting membrane potential). Resting potentials were: A, −47.4 mV; B, −40.7 mV.
When recording from GBSM with intracellular microelectrodes, the addition of CDC (100 μm) resulted in a loss of action potentials within 2.5 min of administration (Fig. 1B). The disruption of action potential activity was associated with a membrane hyperpolarization of 8.2 ± 2.5 mV (basal, −44.8 ± 4.2 mV; CDC, −53.0 ± 6.1 mV; P < 0.05, paired t test; n= 4) (Fig. 1C). Washout reversed the effect of CDC at 50 and 100 μm within 5 min.
GPBAR1 is expressed by GBSM
The rapid action of CDC on GBSM supports the concept that hydrophobic bile salts disrupt GBSM function via activation of a membrane receptor. Recent studies have shown that the membrane bile acid receptor GPBAR1 is highly expressed in the gallbladder, but it was thought to be primarily expressed by epithelial cells on the basis of in situ hybridization studies (Vassileva et al. 2006). Using mouse and guinea pig Gpbar1 sequences from the GenBank and Ensembl databases, respectively, we designed species-specific primer sets that allowed us to amplify Gpbar1 cDNA fragments. Gpbar1 mRNA transcripts were expressed in the full thickness of the gallbladder as well as in the mucosal and muscularis layer of the tissue (Fig. 2A and B). In mouse, we used primers for Muc5ac (Wang et al. 2004), a gene expressed in gallbladder epithelial cells, to verify that the muscularis sample did not contain epithelial cells. Muc5ac mRNA transcripts were expressed in the full-thickness of the gallbladder as well as in the mucosal layer, but were absent from the muscularis layer. Since the Gpbar1 sequence is located in a single exon, appropriated controls (DNase-treated and no RT samples) were amplified, and no band was amplified (data not shown).
Figure 2. The bile salt receptor GPBAR1 is expressed in mouse and guinea pig GBSM.
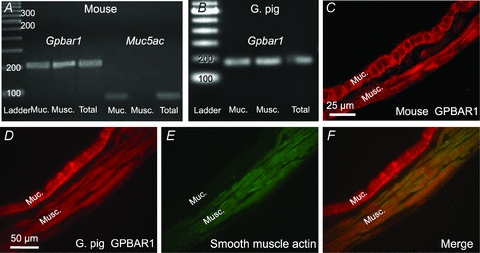
RT-PCR results demonstrating that the Gpbar1 transcript is detectable in RNA extracted from mouse (A) and guinea pig (B) gallbladder muscularis, as well as RNA from the mucosa, suggesting that this receptor is present in GBSM. The predicted product size was 208 bp for the mouse and 246 bp for the guinea pig. In the mouse, the Muc5ac transcript (78 bp) was only detected in RNA from the mucosa indicating that the muscularis propria sample did not contain epithelial cells. Cross-sections of full-thickness mouse (C) and guinea pig (D) gallbladder show that GPBAR1 is expressed in mucosa and muscularis layer. GBSM cells that express smooth muscle actin (E) also express GPBAR1 (F) as shown using double immunostaining of guinea pig gallbladder. Muc., mucosal layer; musc., muscularis layer.
An antibody against the human GPBAR1 was used for immunohistochemistry to confirm the presence of GPBAR1 in the mouse and guinea pig gallbladders and determine its cellular distribution. In cryostat cross-sections, intense immunoreactivity for GPBAR1 was detected in the epithelial layer, which corresponds to the previous in situ hybridization studies, as well as the muscularis propria (Fig. 2C and D). The presence of GPBAR1 in GBSM was confirmed by double label immunohistochemistry for smooth muscle actin (Fig. 2D–F). Collectively, these findings demonstrated the presence of GPBAR1 transcript and immunoreactivity in the muscularis propria of the gallbladder, indicating that GBSM cells express GPBAR1.
The GPBAR1 agonist lithocholic acid disrupts GBSM function
Lithocholic acid (LCA) has a high affinity for, and acts as an agonist of, GPBAR1 (Iguchi et al. 2009). We therefore tested whether LCA disrupts GBSM Ca2+ transients and action potentials in a manner similar to that observed in response to CDC.
Addition of LCA (10 μm) to the perfusion solution caused a significant reduction in the frequency of GBSM Ca2+ flashes (Fig. 3A). LCA decreased the frequency of Ca2+ flashes from 0.22 ± 0.04 Hz to 0.05 ± 0.01 Hz within 5 min (n= 9; P < 0.001). The decrease in GBSM Ca2+ flash frequency persisted for at least 20 min (0.05 ± 0.01 Hz; n= 7; P < 0.001 at 20 min).
Figure 3. The GPBAR1 agonist lithocholic acid (LCA) decreases GBSM activity.
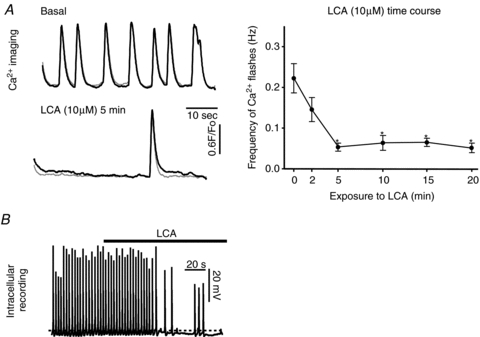
A, application of LCA (10 μm) to the bath rapidly reduced the spontaneous Ca2+ flashes in the GBSM (A). (Traces represent two separate cells in the same muscle bundle.) The effect of LCA was maintained during prolonged incubations (right panel). B, intracellular recording from guinea pig GBSM demonstrates that soon after application of LCA (10 μm), the membrane potential is hyperpolarized (the dashed lined denotes the resting membrane potential), and spontaneous action potential activity is dramatically reduced. *Significantly different from basal (P < 0.001). Resting potential: −46.6 mV. n values for data in the graph are 7–9.
Intracellular recordings demonstrated that bath application of LCA (10 μm) elicits a similar effect on GBSM to that of CDC. LCA caused a rapid reduction of action potential frequency that was associated with a membrane potential hyperpolarization of 5.22 ± 1.4 mV (Fig. 3B) (basal, −49.6 ± 2.1 mV; LCA −54.9 ± 1.9 mV; P < 0.05, paired t test; n= 4). The hyperpolarization elicited by LCA was comparable to that caused by CDC (P= 0.34; unpaired t test). The response to 10 μm LCA partially recovered following washout.
GPBAR1 activates the cAMP–PKA pathway in GBSM
GPBAR1 is known to act via stimulation of the cAMP–PKA pathway (Kawamata et al. 2003). Therefore, we tested the hypothesis that blocking this pathway would protect GBSM from the disruptive effects of hydrophobic bile salts.
The selective PKA inhibitor KT5720 (0.5–1.0 μm) was added to the bathing solution at least 30 min prior to LCA application. KT5720 (0.5–1 μm) alone did not have any detectable effect on the basal Ca2+ flash activity in GBSM (PSS, 0.20 ± 0.05 Hz, n= 4; KT5720 after 30 min: 0.26 ± 0.05 Hz; n= 6; P > 0.05). In presence of KT5720, LCA (10 μm) failed to disrupt Ca2+ flash activity in GBSM (Fig. 4A) (LCA plus KT5720, 0.20 ± 0.05 Hz; n= 6; P > 0.05 vs. KT5720). Frequencies remained unchanged even after prolonged exposure to both compounds (0.20 ± 0.07 Hz after 15 min; n= 5; P > 0.05). The inhibition of Ca2+ flashes in response to CDC was also attenuated by KT5720 (n= 2; data not shown).
Figure 4. Inhibition of protein kinase A (PKA) or KATP channels protects guinea pig muscularis against adverse effects of LCA.
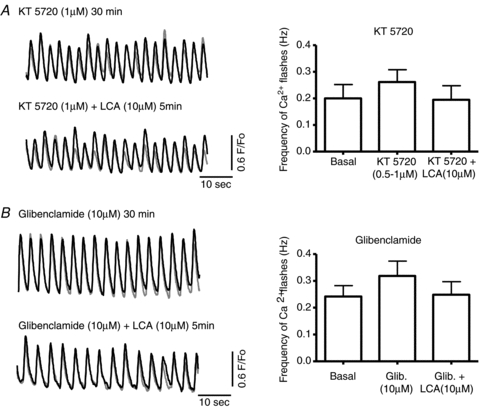
A, the addition to the bath of the PKA inhibitor KT5720 (0.5–1 μm) alone did not affect Ca2+ flashes in guinea pig GBSM (P > 0.05; n= 6). However, in the presence of KT5720, LCA (10 μm) failed to inhibit GBSM function (P > 0.05; n= 6). B, the KATP channel blocker glibenclamide (10 μm) blocked the effect of LCA (10 μm) on the frequency of Ca2+ flashes (P > 0.05; n= 6).
Hydrophobic bile salt disruption of GBSM is inhibited by the KATP blocker glibenclamide
We have previously demonstrated that stimulation of the cAMP–PKA pathway leads to an activation of a KATP current in GBSM (Zhang et al. 1994a). Furthermore, calcitonin gene-related peptide (CGRP), which stimulates receptors that act via the cAMP–PKA pathway, activates the KATP current in GBSM and suppresses action potentials with an accompanying hyperpolarization of the membrane potential (Zhang et al. 1994b). Therefore, if hydrophobic bile salts act on GPBAR1, which in turn activates the cAMP–PKA pathway, it would be expected to inhibit GBSM function by opening KATP channels. We tested this hypothesis by applying LCA in the presence of the KATP channel blocker glibenclamide.
In the presence of glibenclamide (10 μm), LCA (10 μm) failed to alter the frequency of Ca2+ flashes (Fig. 4B) (glibenclamide: 0.32 ± 0.06 Hz; LCA plus glibenclamide: 0.25 ± 0.05 Hz; n= 6; P > 0.05). The flash frequency did not decrease during prolonged exposure to both compounds (0.28 ± 0.05 Hz after 20 min; n= 6; P > 0.05). The inhibition of Ca2+ flashes in response to CDC was also attenuated by glibenclamide (n= 3; data not shown).
LCA failed to disrupt GBSM function in Gpbar1−/− mice
To investigate whether LCA disrupts GBSM function via activation of GPBAR1, we evaluated its effects on GBSM in preparations from mice that lack GPBAR1. These Gpbar1−/− mice have been characterized previously, and interestingly, they are less prone to developing gallstones when fed a lithogenic diet (Vassileva et al. 2006, 2010).
In control C57BL/6J mice, the basal rate of GBSM Ca2+ flashes was very similar to that observed in guinea pig tissue. In these preparations, 10 μm LCA caused a rapid and persistent disruption of Ca2+ flash activity (Fig. 5A) that was similar to the effect of LCA on Ca2+ flashes in guinea pig GBSM. In these experiments involving mouse gallbladder whole mount preparations with intact mucosa, Ca2+ waves were more predominant than in the guinea pig muscularis preparation. This was not unexpected because we had previously reported that Ca2+ waves are more common in full-thickness preparations (Balemba et al. 2006a).
Figure 5. In Gpbar1 knockout mice, the rhythmic activity of GBSM is not affected by LCA.
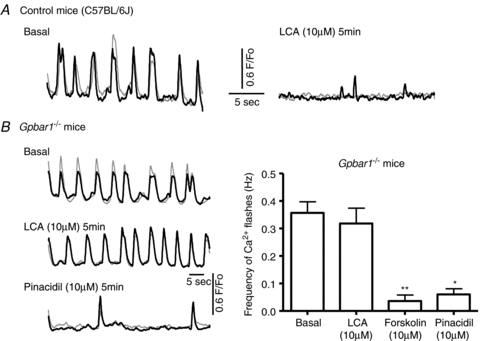
A, basal activity of GBSM in C57BL/6J mice is comparable to the basal activity of guinea pig GBSM. In control mice, LCA (10 μm) rapidly reduced the frequency of Ca2+ flashes (n= 6). B, in Gpbar1−/− mice, LCA (10 μm) did not alter the activity of Ca2+ flashes (P > 0.05; n= 11). However GBSM activity was quickly disrupted by forskolin, an activator of the PKA pathway (**P < 0.001, n= 5), and pinacidil (10 μm), a KATP channel opener (*P < 0.01; n= 4). These results support the hypothesis that LCA activates KATP channels.
In Gpbar1−/− mice basal activity of the GBSM was comparable to that observed in C57BL/6J mice and the guinea pig. Unlike the responses to LCA in wild-type mice described above, addition of LCA (10 μm) to preparations from Gpbar1−/− mice failed to alter spontaneous Ca2+ flash activity (Fig. 5; basal: 0.36 ± 0.04 Hz; LCA after 5 min: 0.32 ± 0.06 Hz; n= 10; P > 0.05). To verify that necessary components of the cAMP–PKA–KATP pathway were present and functional in tissue from the Gpbar1−/− mice, we tested the effects of the PKA activator forskolin (10 μm) and the KATP channel opener pinacidil (10 μm) on the basal activity of GBSM in the Gpbar1−/− mice. In preparations from Gpbar1−/− mice, both forskolin (10 μm) and pinacidil (10 μm) rapidly decreased the frequency of Ca2+ flashes (Fig. 5B) (forskolin 5 min: 0.04 ± 0.02 Hz, n= 5, P < 0.001; pinacidil 5 min: 0.06 ± 0.02 Hz, n= 4, P < 0.01).
Discussion
The present study was undertaken to investigate the effects of hydrophobic bile acids on spontaneous activity in gallbladder smooth muscle cells, and to elucidate the mechanisms by which these compounds decrease GBSM contractility. The findings of this study support the concept that hydrophobic bile acids disrupt GBSM function by activating a G protein-coupled receptor that leads to membrane hyperpolarization via the activation of a KATP channel (Fig. 6). Evidence reported here in support of this include the following: (1) the membrane-bound bile acid receptor GPBAR1 is expressed by smooth muscle cells in the mouse and guinea pig gallbladder; (2) stimulation of GPBAR1 with CDC or LCA inhibited GBSM activity; (3) in the presence PKA or KATP channel inhibitors, LCA failed to alter GBSM function; and (4) gallstone-resistant mice that do not express the GPBAR1 protein were not responsive to LCA.
Figure 6. Proposed pathway for the action of hydrophobic salts on GBSM activity.
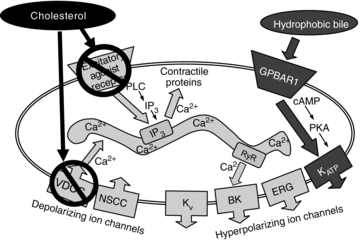
Based on the results of this study, we proposed that activation of GPBAR1 on the plasma membrane of the GBSM by hydrophobic bile salts leads to a rapid activation of the cAMP–PKA pathway causing a hyperpolarization of the muscle cells due to the opening of the ATP-sensitive KATP channels and a subsequent decrease in GBSM activity. In Gpbar1−/− mice, which do not express GPBAR1, GBSM activity was not affected by the hydrophobic bile salt LCA. Our data also support that cholesterol and bile salts are affecting GBSM activity via two distinct pathways. Previous investigations have demonstrated that cholesterol disrupts GBSM function by inhibiting voltage-dependent Ca2+ channel (VDCC) activity, and by preventing activation of excitatory agonist receptors on GBSM.
Bile acids inhibit GBSM activity via G protein-coupled GPBAR1 receptors
Bile acids are now recognized as signalling molecules that can exert effects non-genomically by activating the G protein-coupled receptor, GPBAR1/TGR5, and genomically by activating the farnesoid X receptor, a member of the nuclear hormone receptor superfamily (Nguyen & Bouscarel, 2008).
GPBAR1 is widely distributed, but it is expressed at particularly high concentrations in the gallbladder, where it was previously thought to be present primarily in epithelial cells (Vassileva et al. 2006; Keitel et al. 2009). The current study provides new evidence that GPBAR1 mRNA and protein are present in GBSM in addition to epithelial cells in mouse and guinea pig gallbladders. Tauro-chenodeoxycholate and LCA, a relatively selective GPBAR1 agonist present in the human bile, hyperpolarized GBSM cells and eliminated spontaneous action potential generation. The disruptive effect of LCA on GBSM activity occurred rapidly and in the absence of the epithelial cells, supporting a direct activation of G protein-coupled receptors on the smooth muscle cells. Interestingly, LCA also mediated its action on GBSM in full-thickness preparations, with the epithelial layer exposed directly to the bathing solution, supporting the concept that ‘intraluminal’ bile could gain access to GBSM by penetrating the epithelial layer. In the full-thickness preparations, the basal Ca2+ flash activity and the time course of LCA action were similar to those observed in muscularis preparations.
It is possible that GPBAR1 plays a role in the development of gallstone disease. Bile acids have been shown to decrease GBSM contractility in response to agonists or nerve stimulation, suggesting that they may contribute to the alterations in motor activity that are observed in gallstone disease. Also, Gpbar1−/− mice do not develop gallstones when fed a lithogenic diet that leads to cholesterol crystal and gallstone formation as well as cholecystitis in wild-type animals. Interestingly, the Gpbar1−/− mice develop normally and do exhibit altered levels of cholesterol, bile acids or any other bile constituents (Vassileva et al. 2006). Therefore, the protective effect of the gene deletion may involve the lack of a target for the bile acids in GBSM rather than, or in addition to, an effect on bile synthesis. Consistent with this concept, gallstone disease in humans is associated with elevated gallbladder Gpbar1 mRNA levels (Keitel et al. 2009).
KATP channels and smooth muscle relaxation
In human gallbladder, KATP channels can mediate GBSM relaxation (Bird et al. 2002). Interestingly the action of the smooth muscle relaxant CGRP, which has been well characterized in gallbladder and arterial smooth muscles, is known to activate the ATP-sensitive KATP via PKA (Quayle et al. 1994; Zhang et al. 1994a). Activation of the cAMP–PKA pathway by other signalling molecules such as β-adrenergic receptor agonists and VIP also leads to muscle relaxation. The signal transduction mechanisms underlying the activation of the KATP channels by GPBAR1 are similar to that described for CGRP. The GPBAR1 agonist, LCA, like CGRP, hyperpolarized smooth muscle cells by activation of the cAMP–PKA pathway, which leads to the opening of KATP channels. Other channels may also be activated by the cAMP–PKA pathway in the GBSM. However, when applied alone, the KATP channel inhibitor glibenclamide prevented the LCA-induced decrease of GBSM activity, supporting a critical role of KATP channels. Physiologically, the hyperpolarization mediated by KATP channel activation would greatly diminish the ability of VDCCs to open by driving the membrane potential to a level below their activation threshold. This is critical because the activity of these channels is fundamental for action potentials and associated Ca2+ entry to occur (Balemba et al. 2006b), and for the maintenance of intracellular Ca2+ stores (Morales et al. 2005).
Cholesterol vs. bile acid in gallstone disease
Taken together these findings also indicate that while both cholesterol and hydrophobic bile acids have harmful effects on gallbladder contractility, their mechanisms of action are different (Fig. 6). The mechanism by which cholesterol interacts with GBSM is fairly well understood. Cholesterol accumulates in the plasma membrane of GBSM, predominantly in the caveolar regions, where it acts by decreasing membrane fluidity and by attenuating the function of CCK receptors, which are abundant in these regions (Jennings et al. 1999; Xiao et al. 2000, 2007). CGRP-induced hyperpolarizations are reduced by cholesterol enrichment, supporting changes in receptor–ligand binding and/or second messenger interactions. Cholesterol reduces the action potential firing in intact tissue by disrupting Ca2+-mediated events via inhibition of Ca2+ channel function. In isolated cells, Ca2+ currents are reduced but the activity of the KATP and voltage-activated K+ channels are not affected by cholesterol (Jennings et al. 1999). In contrast, hydrophobic bile salts inhibit GBSM contractility via a hyperpolarization caused by activation of KATP channels.
Bile acid receptors and metabolic disease
In addition to their role on G protein-coupled receptor GPBAR1, bile acids can also signal through the activation of specific nuclear receptors (farnesoid X receptor, pregnane X receptor, and vitamin D receptor) and intracellular kinase (c-jun N-terminal kinase 1/2, AKT, and ERK 1/2) in the liver and gastrointestinal tract (Hylemon et al. 2009). Their role includes the regulation of bile acid, lipid and glucose metabolism, energy homeostasis, and insulin sensitivity.
In addition to their involvement in gallstone disease, bile acids and their receptors have been associated with a number of metabolic disorders including obesity and diabetes (Houten et al. 2006; Thomas et al. 2008; Lefebvre et al. 2009; Vassileva et al. 2010). For example, GPBAR1 activation induces the release of intestinal glucagon-like peptide, which in obese mice can improve liver and pancreatic functions as well as glucose tolerance (Thomas et al. 2009). Interestingly, although type II diabetic patients are likely candidates for gallbladder disorders, genetic variation on the Gpbar1 gene is not associated with the increased risk of the metabolic disorder (Mussig et al. 2009).
The development of specific bile acid receptor ligands offers new avenues to prevent and treat lipid and glucose-related metabolic diseases such as obesity and type II diabetes. However, in light of the findings reported here, demonstrating a disruptive effect of GPBAR1 activation on GBSM function, it is quite possible that the development of gallstone disease could be an adverse effect of these compounds.
Our results suggest that blocking GBSM KATP channels or increasing GBSM excitability could prevent gallbladder stasis and gallstone formation to hydrophobic bile salts. Sulfonylurea drugs inhibit KATP channels in smooth muscle and pancreatic β cells, and therefore would not be a viable approach. However, KATP channels in smooth muscle and pancreatic β cells have different structures: SUR2B/Kir6.2 and SUR1/Kir6.1, respectively. Therefore, identification of smooth muscle-specific KATP channel antagonists may represent a possible approach. Conversely, gain-of-function polymorphisms of the SUR2B/Kir6.1 may increase the risk of gallbladder dysfunction. Nonetheless, our results provide a mechanistic basis of how hydrophobic bile salts influence gallbladder function, and thereby suggest therapeutic opportunities for intervention.
Acknowledgments
The authors would like to thank E. M. Brooks, Dr A. B. Bartoo and Dr D. Poole, for their assistance, and to Dr A. D. Bonev for developing the software used to analyse Ca2+ events. This work was funded by National Institutes of Health (NIH) Grants NS-26995/DK-080480 and DK-62267 to G.M.M., and R37DK053832 and R01DK065947 to M.T.N. The Center of Biomedical Research Excellence imaging-physiology core facility is funded by NIH Grant NCRR P20 RR16435. This work was also supported by a grant from the Department of Veterans Affairs, Biomedical Laboratory Research and Development to C.U.C. The authors have no conflicts of interest to disclose.
Glossary
Abbreviations
- CCK
cholecystokinin
- CDC
tauro-chenodeoxycholic acid
- CGRP
calcitonin gene-related peptide
- GBSM
gallbladder smooth muscle
- GPBAR1
G protein-coupled bile acid receptor
- ICC
interstitial cells of Cajal
- KATP
ATP-sensitive potassium channel
- LCA
tauro-lithocholic acid
- PKA
protein kinase A
- VDCC
voltage-dependent calcium channel
Author contributions
Conception and design of the experiments: B.L., G.V., C.U.C., M.T.N. and G.M.M.; collection of data: B.L., O.B.B., C.G., C.A.W.; analysis and interpretation of data: B.L., O.B.B., C.G., C.A.W., C.U.C., G.M.M.; drafting of the manuscript: B.L., G.M.M.; critical revision of the manuscript for important intellectual content: B.L., O.B.B., G.V., C.U.C., M.T.N., G.M.M.; obtained funding: C.U.C., G.M.M. All authors approved the final version of the manuscript.
Author's present address
O. B. Balemba: Department of Biological Sciences/WWAMI, University of Idaho, Moscow, ID, USA.
References
- Balemba OB, Heppner TJ, Bonev AD, Nelson MT, Mawe GM. Calcium waves in intact guinea pig gallbladder smooth muscle cells. Am J Physiol Gastrointest Liver Physiol. 2006a;291:G717–727. doi: 10.1152/ajpgi.00035.2006. [DOI] [PubMed] [Google Scholar]
- Balemba OB, Salter MJ, Heppner TJ, Bonev AD, Nelson MT, Mawe GM. Spontaneous electrical rhythmicity and the role of the sarcoplasmic reticulum in the excitability of guinea pig gallbladder smooth muscle cells. Am J Physiol Gastrointest Liver Physiol. 2006b;290:G655–664. doi: 10.1152/ajpgi.00310.2005. [DOI] [PubMed] [Google Scholar]
- Behar J, Lee KY, Thompson WR, Biancani P. Gallbladder contraction in patients with pigment and cholesterol stones. Gastroenterology. 1989;97:1479–1484. doi: 10.1016/0016-5085(89)90392-2. [DOI] [PubMed] [Google Scholar]
- Bird NC, Ahmed R, Chess-Williams R, Johnson AG. Active relaxation of human gallbladder muscle is mediated by ATP-sensitive potassium channels. Digestion. 2002;65:220–226. doi: 10.1159/000063815. [DOI] [PubMed] [Google Scholar]
- Bomzon A, Ljubuncic P. Ursodeoxycholic acid and in vitro vasoactivity of hydrophobic bile acids. Dig Dis Sci. 2001;46:2017–2024. doi: 10.1023/a:1010663904820. [DOI] [PubMed] [Google Scholar]
- Dopico AM, Walsh JV, Jr, Singer JJ. Natural bile acids and synthetic analogues modulate large conductance Ca2+-activated K+ (BKCa) channel activity in smooth muscle cells. J Gen Physiol. 2002;119:251–273. doi: 10.1085/jgp.20028537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houten SM, Watanabe M, Auwerx J. Endocrine functions of bile acids. EMBO J. 2006;25:1419–1425. doi: 10.1038/sj.emboj.7601049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hylemon PB, Zhou H, Pandak WM, Ren S, Gil G, Dent P. Bile acids as regulatory molecules. J Lipid Res. 2009;50:1509–1520. doi: 10.1194/jlr.R900007-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iguchi Y, Yamaguchi M, Sato H, Kihira K, Nishimaki-Mogami T, Une M. Bile alcohols function as the ligands of membrane-type bile acid-activated G protein-coupled receptor. J Lipid Res. 2009;51:1432–1441. doi: 10.1194/jlr.M004051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazrawi RP, Pazzi P, Petroni ML, Prandini N, Paul C, Adam JA, Gullini S, Northfield TC. Postprandial gallbladder motor function: refilling and turnover of bile in health and in cholelithiasis. Gastroenterology. 1995;109:582–591. doi: 10.1016/0016-5085(95)90348-8. [DOI] [PubMed] [Google Scholar]
- Jennings LJ, Xu QW, Firth TA, Nelson MT, Mawe GM. Cholesterol inhibits spontaneous action potentials and calcium currents in guinea pig gallbladder smooth muscle. Am J Physiol Gastrointest Liver Physiol. 1999;277:G1017–1026. doi: 10.1152/ajpgi.1999.277.5.G1017. [DOI] [PubMed] [Google Scholar]
- Jun JY, Choi S, Chang IY, Yoon CK, Jeong HG, Kong ID, So I, Kim KW, You HJ. Deoxycholic acid inhibits pacemaker currents by activating ATP-dependent K+ channels through prostaglandin E2 in interstitial cells of Cajal from the murine small intestine. Br J Pharmacol. 2005;144:242–251. doi: 10.1038/sj.bjp.0706074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, Fukusumi S, Habata Y, Itoh T, Shintani Y, Hinuma S, Fujisawa Y, Fujino M. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278:9435–9440. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- Keitel V, Cupisti K, Ullmer C, Knoefel WT, Kubitz R, Haussinger D. The membrane-bound bile acid receptor TGR5 is localized in the epithelium of human gallbladders. Hepatology. 2009;50:861–870. doi: 10.1002/hep.23032. [DOI] [PubMed] [Google Scholar]
- Lavoie B, Balemba OB, Nelson MT, Ward SM, Mawe GM. Morphological and physiological evidence for interstitial cell of Cajal-like cells in the guinea pig gallbladder. J Physiol. 2007;579:487–501. doi: 10.1113/jphysiol.2006.122861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. 2009;89:147–191. doi: 10.1152/physrev.00010.2008. [DOI] [PubMed] [Google Scholar]
- Morales S, Camello PJ, Mawe GM, Pozo MJ. Cyclic AMP-mediated inhibition of gallbladder contractility: role of K+ channel activation and Ca2+ signaling. Br J Pharmacol. 2004;143:994–1005. doi: 10.1038/sj.bjp.0706006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales S, Camello PJ, Mawe GM, Pozo MJ. Characterization of intracellular Ca2+ stores in gallbladder smooth muscle. Am J Physiol Gastrointest Liver Physiol. 2005;288:G507–513. doi: 10.1152/ajpgi.00385.2004. [DOI] [PubMed] [Google Scholar]
- Mussig K, Staiger H, Machicao F, Machann J, Schick F, Schafer SA, Claussen CD, Holst JJ, Gallwitz B, Stefan N, Fritsche A, Haring HU. Preliminary report: genetic variation within the GPBAR1 gene is not associated with metabolic traits in white subjects at an increased risk for type 2 diabetes mellitus. Metabolism. 2009;58:1809–1811. doi: 10.1016/j.metabol.2009.06.012. [DOI] [PubMed] [Google Scholar]
- Nguyen A, Bouscarel B. Bile acids and signal transduction: role in glucose homeostasis. Cell Signal. 2008;20:2180–2197. doi: 10.1016/j.cellsig.2008.06.014. [DOI] [PubMed] [Google Scholar]
- Poole DP, Godfrey C, Cattaruzza F, Cottrell GS, Kirkland JG, Pelayo JC, Bunnett NW, Corvera CU. Expression and function of the bile acid receptor GpBAR1 (TGR5) in the murine enteric nervous system. Neurogastroenterol Motil. 2010;22:814–e228. doi: 10.1111/j.1365-2982.2010.01487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quayle JM, Bonev AD, Brayden JE, Nelson MT. Calcitonin gene-related peptide activated ATP-sensitive K+ currents in rabbit arterial smooth muscle via protein kinase A. J Physiol. 1994;475:9–13. doi: 10.1113/jphysiol.1994.sp020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutishauser SC. Effects of bile salts on the motor activity of the guinea-pig gall-bladder in vitro. Q J Exp Physiol Cogn Med Sci. 1978;63:265–276. doi: 10.1113/expphysiol.1978.sp002440. [DOI] [PubMed] [Google Scholar]
- Shoda J, He BF, Tanaka N, Matsuzaki Y, Osuga T, Yamamori S, Miyazaki H, Sjovall J. Increase of deoxycholate in supersaturated bile of patients with cholesterol gallstone disease and its correlation with de novo syntheses of cholesterol and bile acids in liver, gallbladder emptying, and small intestinal transit. Hepatology. 1995;21:1291–1302. [PubMed] [Google Scholar]
- Thomas C, Auwerx J, Schoonjans K. Bile acids and the membrane bile acid receptor TGR5-connecting nutrition and metabolism. Thyroid. 2008;18:167–174. doi: 10.1089/thy.2007.0255. [DOI] [PubMed] [Google Scholar]
- Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, Macchiarulo A, Yamamoto H, Mataki C, Pruzanski M, Pellicciari R, Auwerx J, Schoonjans K. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10:167–177. doi: 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Erpecum KJ, Wang DQ, Moschetta A, Ferri D, Svelto M, Portincasa P, Hendrickx JJ, Schipper M, Calamita G. Gallbladder histopathology during murine gallstone formation: relation to motility and concentrating function. J Lipid Res. 2006;47:32–41. doi: 10.1194/jlr.M500180-JLR200. [DOI] [PubMed] [Google Scholar]
- Vassileva G, Golovko A, Markowitz L, Abbondanzo SJ, Zeng M, Yang S, Hoos L, Tetzloff G, Levitan D, Murgolo NJ, Keane K, Davis HR, Jr, Hedrick J, Gustafson EL. Targeted deletion of Gpbar1 protects mice from cholesterol gallstone formation. Biochem J. 2006;398:423–430. doi: 10.1042/BJ20060537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassileva G, Hu W, Hoos L, Tetzloff G, Yang S, Liu L, Kang L, Davis H, Hedrick J, Lan H, Kowalski T, Gustafson E. Gender-dependent effect of Gpbar1 genetic deletion on the metabolic profiles of diet-induced obese mice. J Endocrinol. 2010;205:225–232. doi: 10.1677/JOE-10-0009. [DOI] [PubMed] [Google Scholar]
- Wang DQ, Paigen B, Carey MC. Phenotypic characterization of Lith genes that determine susceptibility to cholesterol cholelithiasis in inbred mice: physical-chemistry of gallbladder bile. J Lipid Res. 1997;38:1395–1411. [PubMed] [Google Scholar]
- Wang HH, Afdhal NH, Gendler SJ, Wang DQ. Targeted disruption of the murine mucin gene 1 decreases susceptibility to cholesterol gallstone formation. J Lipid Res. 2004;45:438–447. doi: 10.1194/jlr.M300468-JLR200. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, Messaddeq N, Harney JW, Ezaki O, Kodama T, Schoonjans K, Bianco AC, Auwerx J. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439:484–489. doi: 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- Xiao Z, Schmitz F, Pricolo VE, Biancani P, Behar J. Role of caveolae in the pathogenesis of cholesterol-induced gallbladder muscle hypomotility. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1641–1649. doi: 10.1152/ajpgi.00495.2006. [DOI] [PubMed] [Google Scholar]
- Xiao ZL, Chen Q, Amaral J, Biancani P, Behar J. Defect of receptor-G protein coupling in human gallbladder with cholesterol stones. Am J Physiol Gastrointest Liver Physiol. 2000;278:G251–258. doi: 10.1152/ajpgi.2000.278.2.G251. [DOI] [PubMed] [Google Scholar]
- Xu QW, Freedman SM, Shaffer EA. Inhibitory effect of bile salts on gallbladder smooth muscle contractility in the guinea pig in vitro. Gastroenterology. 1997;112:1699–1706. doi: 10.1016/s0016-5085(97)70053-2. [DOI] [PubMed] [Google Scholar]
- Xu QW, Shaffer EA. Cisapride improves gallbladder contractility and bile lipid composition in an animal model of gallstone disease. Gastroenterology. 1993;105:1184–1191. doi: 10.1016/0016-5085(93)90966-g. [DOI] [PubMed] [Google Scholar]
- Zhang L, Bonev AD, Mawe GM, Nelson MT. Protein kinase A mediates activation of ATP-sensitive K+ currents by CGRP in gallbladder smooth muscle. Am J Physiol Gastrointest Liver Physiol. 1994a;267:G494–499. doi: 10.1152/ajpgi.1994.267.3.G494. [DOI] [PubMed] [Google Scholar]
- Zhang L, Bonev AD, Nelson MT, Mawe GM. Ionic basis of the action potential of guinea pig gallbladder smooth muscle cells. Am J Physiol Cell Physiol. 1993;265:C1552–1561. doi: 10.1152/ajpcell.1993.265.6.C1552. [DOI] [PubMed] [Google Scholar]
- Zhang L, Bonev AD, Nelson MT, Mawe GM. Activation of ATP-sensitive potassium currents in guinea-pig gall-bladder smooth muscle by the neuropeptide CGRP. J Physiol. 1994b;478:483–491. doi: 10.1113/jphysiol.1994.sp020267. [DOI] [PMC free article] [PubMed] [Google Scholar]