

Abstract
2-(Alkoxy)propenyl bromides are readily prepared from 1,3-dibromo-2-propanol in a two-step sequence involving hydroxyl protection and sodium hydride-induced dehydrobromination. Indium-mediated allylation of aldehydes, ketones, and sulfonimines with 2-(alkoxy)propenyl bromides furnishes the corresponding homoallylic alcohols and sulfonamines in good yields. The products can be easily transformed into β-hydroxy ketones and esters, as well as substituted dihydropyrans and protected β-amino acids. Chiral 2-(alkoxy)propenyl halides, derived from (−)-menthol and D-glucal, furnish diastereomerically enriched products.
The indium-mediated allylation of aldehydes and ketones is a powerful and stereoselective method for carbon-carbon bond formation useful in organic synthesis.1 The reaction takes place successfully in a variety of solvents including water, with allylindium (III) sesquihalide intermediates likely for processes occurring in organic solvents2 and allylindium (I) species likely for processes occurring in aqueous media.3 The allylation reaction has been applied to the synthesis of a variety of complex products, and tandem or sequential indium-mediated carbon-carbon bond-forming reactions have been shown to be especially useful in this regard.4
A variety of 2-substituted allyl halides have been employed with great utility in aldehyde and ketone allylation reactions. For example, it has been shown that 2-(carboxyalkyl)-substituted allyl halides are useful reagents for the indium-mediated synthesis of α-methylene-γ-butyrolactones.5 Furthermore, 2-(trimethylsilylmethyl)allyl halides have been employed in the one-pot preparation of 2,6-disubstituted tetrahydropyrans6 and oxa-bridged 7- and 8 membered carbocycles7 via indium-mediated allylation of mono- and dicarbonyl compounds, respectively, in aqueous media. In this letter we wish to report our studies on the preparation and indium-mediated reactions of 2-(alkoxy)allyl halides.
Horning has shown that 3-bromo-2-(tetrahydropyranyloxy)propene, prepared by sodium hydride-induced elimination of hydrogen bromide from 1,3-dibromo-2-(tetrahydropyran-2-y1oxy)propane, is a useful “masked” acetonyl bromide equivalent, reacting with a variety of carbon nucleophiles to generate products containing new carbon-carbon bonds (Scheme 1).8 We were interested not in the electrophilic properties9 of this reagent but rather in its potential to form an allyl nucleophile upon exposure to indium metal.11 To test this hypothesis, we decided to prepare MEM derivative 3-bromo-2-((2-methoxyethoxy)methoxy)propene (3) and investigate its reactivity with aldehydes and ketones in the presence of indium metal.
Scheme 1.

(a) Horning’s preparation of 1 and its utility as an electrophile. (b) Preparation of MEM derivative 3.
Exposure of 1,3-dibromo-2-propanol to MEM-Cl10 (1.5 equiv), i-Pr2NEt (2 equiv) and TBAI (10 mol%) in CH2Cl2 for 36 hours gave rise to 1,3-dibromo-2-((2-methoxyethoxy)methoxy)propane 2 in 60% yield. Addition of a DMF solution of 2 to NaH (2.2 equiv) in DMF at 0°C, followed by warming to room temperature, furnished bromide 3 in 72% yield. This substance was not purified but used directly in allylation reactions. It was subsequently found that trace amounts of water in the DMF solvent employed for the preparation of 3 from 2 gave rise to allylic alcohol 4, which proved difficult to separate from allylation products 5 (vide infra). The formation of this byproduct could be completely avoided by storing the DMF used for the dehydrobromination reaction over activated 4Å molecular sieves for several days prior to use. It is worthy of note that 1,3-dibromo propanol derivatives bearing non-acetal-type protecting groups (e.g., methyl or menthyl ether) undergo dehydrobromination with NaH in only very poor yields (<10%) at room temperature.
The reaction of 3 with representative aldehydes and ketones in the presence of indium metal is presented in Table 1. The optimal solvent for the reaction was DMF; reactions performed in THF were slower, and as previously shown by Saicic,11 mixtures of THF and water produced β-hydroxyketone 6 (Scheme 2) as the main product, presumably formed via hydrolysis of the enol ether functionality of 5 due to the rising acidity6 of the reaction medium as the reaction progresses. Tetra-n-butylammonium iodide or sodium iodide (1 equiv) was found to be an important additive necessary for increasing the reactivity of 3 toward aldehydes and ketones, leading to decreased reaction times (>24h→5h). The optimal conditions for the reaction involved combining the aldehyde or ketone (1 equiv) with 3 (2 equiv), indium metal (1.5 equiv), and TBAI or NaI (1 equiv) in DMF (2M) at room temperature for 4–6 h. Allylation of glyceraldehyde acetonide (entry 6) led to the expected homoallylic alcohol as a 5:1 mixture of trans:cis diastereomers.4
Table 1.
Scope of indium-mediated allylation of aldehydes and ketones with 3.a
 | ||||
|---|---|---|---|---|
| entry | R1 | R2 | 5 | % yield 5 |
| 1 | 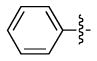 |
H | a | 87 |
| 2 | H | b | 91 | |
| 3 | 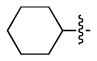 |
H | c | 68 |
| 4 | H | d | 61 | |
| 5 | H | e | 65 | |
| 6 | 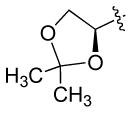 |
H | f | 78b |
| 7 | CH3 | CH3 | g | 92 |
| 8 | CH3 | 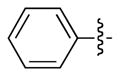 |
h | 89 |
Reaction Conditions: 3 (2 equiv) aldehyde or ketone (1 equiv), TBAI (1 equiv) and indium metal (1.5 equiv) combined in DMF (2M) and stirred at room temperature for 4–6 h.
Anti:syn diastereomer ratio=5:1
Scheme 2.
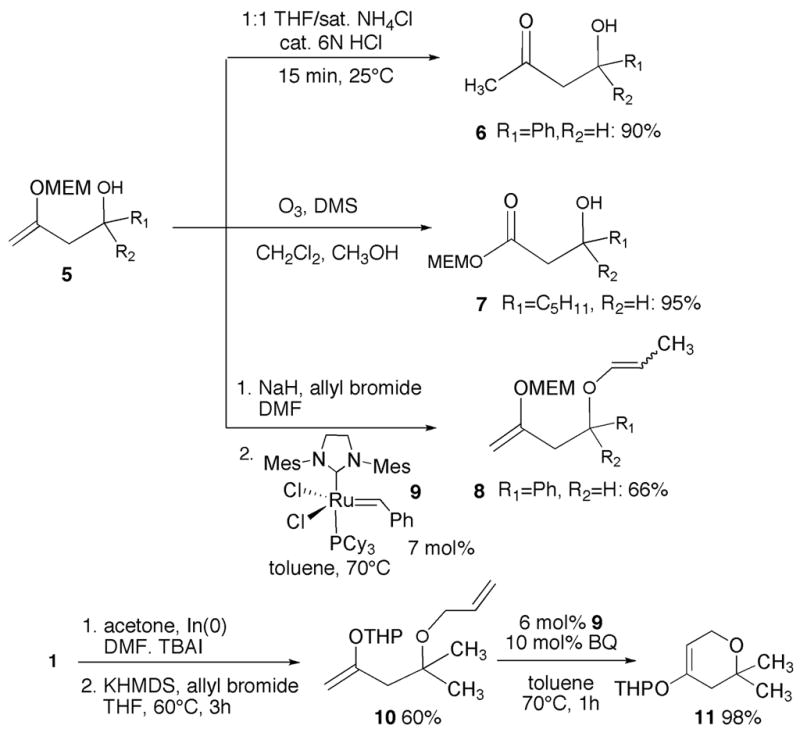
Useful transformations of alcohol 5 and THP ether 10.
Enol ethers 5 may be readily transformed into β-hydroxy ketones and esters. Stirring compound 5a in a 1:1 THF:saturated NH4Cl solution containing a drop of 6N HCl for 15 minutes results in conversion to ketone 6 in 90% isolated yield; bubbling ozone through a solution of 5b in 1:1 CH2Cl2:CH3OH at −78°C for 30 minutes, followed by exposure to DMS and warming to room temperature, gives rise to MEM ester 7 in 95% yield (Scheme 2). Compound 5a could be easily transformed into the corresponding allyl ether by treatment with NaH/allyl bromide in DMF; however, attempted ring-closing metathesis with Grubb’s second-generation catalyst 912 led only to the isomerized acyclic products 8. Repetition of this reaction in the presence of the isomerization inhibitor benzoquinone (10 mol%) gave no reaction.13 Since many examples of enol ether metathesis exist in the literature,14 we surmised that the methoxyethoxymethyl ether moiety may be inhibiting ring-closing metathesis by coordination to ruthenium. Thus, we prepared allyl ether 10 from Horning’s THP ether (1) and subjected it to ring closing metathesis. A 98% yield of dihydropyran 11 was obtained after stirring 10 for 1 hour at 70°C in toluene in the presence of 6 mol% 9 and 10 mol% benzoquinone (BQ).
Toluenesulfonyl imine 1215 also underwent reaction with 1 (3 equiv) in the presence of indium metal (3 equiv) to produce homoallylic sulfonamide 13 as a 1:1 mixture of diastereomers in 90% yield (Scheme 3). Treatment of 13 with OsO4/KIO4 under Jin’s conditions16 cleanly gave rise to sulfonyl-protected β-amino acid 14 in 84% yield.
Scheme 3.
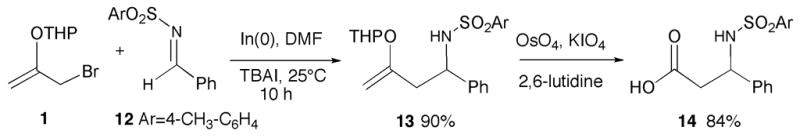
Indium-mediated allylation of sulfonimine 12.
To explore the extension of this method beyond the use of an acetone enolate equivalent, we prepared MEM ether 16 from the known cis, cis-2,6-dibromocyclohexanol (15, Scheme 4).17 Treatment of 16 with NaH in DMF, followed by exposure to benzaldehyde and indium metal in DMF overnight, led to the production of enol ether 17 in 45% yield as a 6:1 mixture of syn/anti diastereomers;18 diene 18,19 formed competitively in both the dehydrobromination and allylation reactions, was also isolated in significant quantities.
Scheme 4.

Reactions of the allylindium reagent derived from dibromide 16.
Finally, we envisioned that chiral 3-bromo-2-alkoxypropenyl halides could be easily prepared by the protocols outlined above and participate in diastereoselective indium-mediated carbonyl allylation reactions. Menthyloxymethyl ether 20 was thus synthesized from 1,3-dibromo-2-propanol and symmetrical acetal 1920 (derived from (−)-menthol) under TMSOTf catalysis (Scheme 5). Dehydrobromination under standard conditions, followed by indium-mediated allylation of benzaldehyde, furnished homoallylic alcohol 21 in 80% yield. To assess the stereoselectivity of the reaction, it was necessary to hydrolyze 21 to β-hydroxyl ketone 6 and prepare the corresponding Mosher ester21 derivative. Analysis of the 1H NMR spectrum of ester 22 revealed a disappointing 1.4:1 diastereomer ratio.
Scheme 5.
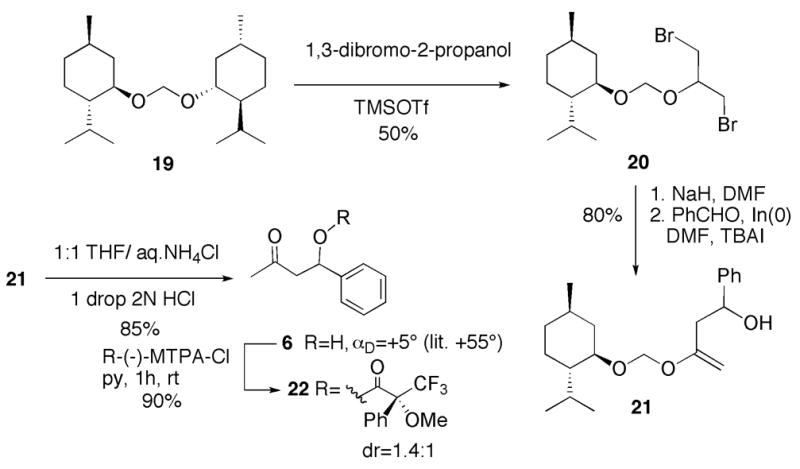
Attempted use of the menthyloxymethyl ether as a chiral auxiliary in indium - mediate alllylation.
Given that tetrahydropyranyl ether 1 undergoes smooth indium-mediated carbon-carbon bond-forming reactions, we believed that the use of a carbohydrate auxiliary might give rise to enhanced diastereoselection in the allylation process. Thus, 3,4,6-tri-O-methyl-D-glucal22 was treated with 1,3-dibromo-2-propanol (1.5 equiv) and triphenylphosphine hydrobromide (5 mol%)23 in CH2Cl2 at room temperature for 16 hours to afford O-glycoside 23 in 80% yield as a 10:1 mixture of α/β diastereomers which could be easily separated by flash chromatography (Scheme 6). Dehydrobromination of the α-stereoisomer was carried out in DMF with NaH, and crude enol ether 24 (2 equivalents) was reacted with benzaldehyde (1 equiv) and indium metal (1.5 equiv) in DMF overnight. The corresponding homoallylic alcohol 26 was obtained in 90% yield as 3:1 mixture of diastereomers. Repetition of the reaction in THF or dioxane furnished less than 10% of 26; reaction in 4:1 THF/DMF provided 26 as a 3:1 mixture of diastereomers in 65% yield after 48 hours. Treatment of intermediate allyl bromide 24 with NaI in acetone, isolation of iodide 25, and stirring of crude 25 in THF with indium metal prior to the addition of benzaldehyde gave 26 in 55% yield, but again as a 3:1 mixture of diastereomers.
Scheme 6.

Synthesis and indium-mediated allylation reactions of chiral allyl bromide 24.
Aqueous acidic hydrolysis of the enol ether function of 26 gave β-hydroxy ketone 6, whose sign of optical rotation indicated that the predominant enantiomer possessed the S-configuration.24 The preferential formation of this stereoisomer may be rationalized based on a transition state assembly in which the pyran ether oxygen and the C.6 oxygen atom of the carbohydrate simultaneously coordinate the allylic indium species, as shown in scheme 6.
In conclusion, we have demonstrated the synthetic utility of the indium reagents derived from 2-alkoxy-substituted allyl bromides. Studies directed toward improving the diastereoselectivity of this process, as well as explorations of the utility of this method in natural products synthesis, are currently under way and will be reported in due course.
General procedure for the synthesis of homoallylic alcohols 5a-h from 2
A solution of 2 (603 mg, 2 mmol) in DMF (0.6 mL) was added rapidly to a 0°C solution of NaH (400 mg, 10 mmol, 60% dispersion in mineral oil, washed 2× with dry pentane) in DMF (4 mL) and the resulting solution was allowed to warm to room temperature and stir for 30 minutes. The mixture was diluted with ether (20 mL) and quenched with saturated NaHCO3 solution (10 mL). The layers were separated and the organic layer was washed with an additional portion of saturated NaHCO3 solution (10 mL). The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo to provide 3 as a light yellow oil.
To a solution of crude 3 in DMF (0.5 mL) was added the appropriate aldehyde or ketone (1.0 mmol), indium metal (171 mg, 1.5 mmol), TBAI (369 mg, 1.0 mmol) and 4Å molecular sieves (200 mg). The solution was allowed to stir overnight with protection from light. The reaction mixture was then diluted with 1:1 ether:ethyl acetate (10 mL) and saturated NaHCO3 solution (10 mL) and the phases were separated. The aqueous phase was extracted again with ether (10 mL) and the combined organics were dried over anhydrous sodium sulfate. Filtration and concentration in vacuo furnished a crude yellow oil. Purification by flash chromatography provided homoallylic alcohols 5a–h.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (SC2 GM081064-01) and the Henry Dreyfus Teacher-Scholar Award for their generous support of our research program.
Footnotes
Supplementary data associated with this article can be found, in the online version, at:
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.For reviews, see: Li CJ. Chem Rev. 2005;105:3095. doi: 10.1021/cr030009u.Li CJ, Chan TH. Organic Reactions in Aqueous Media. Wiley; New York: 1997. Li CJ, Chan TH. In: Organic Synthesis in Water. Grieco PA, editor. Thomson Science; Glasgow, Scotland: 1998. Li CJ, Chan TH. Tetrahedron Lett. 1991;32:7017.Chan TH, Yang Y. J Am Chem Soc. 1999;121:3228.
- 2.Araki S, Ito H, Butsugan Y. J Org Chem. 1988;53:1831. [Google Scholar]
- 3.Chan TH, Yang Y. J Am Chem Soc. 1999;121:3228. [Google Scholar]
- 4.Moral JA, Moon SJ, Rodriguez-Torres S, Minehan TG. Org Lett. 2009;11:3734. doi: 10.1021/ol901353f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choudhury PK, Foubelo F, Yus M. Tetrahedron Lett. 1998;39:3581. [Google Scholar]
- 6.Pham M, Allatabakhsh A, Minehan TG. J Org Chem. 2008;73:741. doi: 10.1021/jo7016857. [DOI] [PubMed] [Google Scholar]
- 7.Allatabakhsh A, Pham M, Minehan TG. Heterocycles. 2007;72:115. [Google Scholar]
- 8.(a) Horning DE, Kavadias G, Muchowski JM. Can J Chem. 1970;48:975. [Google Scholar]; (b) Jacobson RM, Ratha RA, McDonald JH. J Org Chem. 1977;42:2545. [Google Scholar]
- 9.(a) Janicki SZ, Fairgrieve JM, Petillo PA. J Org Chem. 1998;63:3694. [Google Scholar]; (b) Gu XP, Nishida N, Ikeda I, Okahara M. J Org Chem. 1987;52:3192. [Google Scholar]; (c) Gu XP, Kirito Y, Ikeda I, Okahara M. J Org Chem. 1990;55:3390. [Google Scholar]
- 10.Corey EJ, Gras JL, Ulrich P. Tetrahedron Lett. 1976;17:809. [Google Scholar]
- 11.Maslak V, Tokic-Vujosevic Z, Ferjancic Z, Saicic RN. Tetrahedron Lett. 2009;50:6709. [Google Scholar]
- 12.Scholl M, Ding S, Lee CW, Grubbs RH. Org Lett. 1999;1:953. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 13.Hong SH, Sanders DP, Lee CW, Grubbs RH. J Am Chem Soc. 2005;127:17160. doi: 10.1021/ja052939w. [DOI] [PubMed] [Google Scholar]
- 14.(a) Roberts SW, Rainier JD. Org Lett. 2007;9:2227. doi: 10.1021/ol0707970. [DOI] [PubMed] [Google Scholar]; (b) van Otterlo WAL, Ngidi EL, Coyanis EM, de Koning CB. Tetrahedron Lett. 2003;44:311. [Google Scholar]; (c) Fuwa H, Sasaki M. Org Lett. 2008;10:2549. doi: 10.1021/ol800815t. [DOI] [PubMed] [Google Scholar]; (d) Sturino CF, Wong JCY. Tetrahedron Lett. 1998;39:9623. [Google Scholar]; (e) Okada A, Ohshima T, Shibasaki M. Tetrahedron Lett. 2001;42:8023. [Google Scholar]
- 15.Fan R, Pu D, Wen F, Ye Y, Wang X. J Org Chem. 2008;73:3623. doi: 10.1021/jo800009t. [DOI] [PubMed] [Google Scholar]
- 16.Yu W, Mei Y, Kang Y, Hua Z, Jin Z. Org Lett. 2004;6:3217. doi: 10.1021/ol0400342. [DOI] [PubMed] [Google Scholar]
- 17.Wolinsky J, Thorstenson JH, Killinger TA. J Org Chem. 1978;43:875. [Google Scholar]
- 18.The ratio and identity of the syn/anti diastereomers was determined by hydrolysis of 17 (1:1 THF:saturated NH4Cl, 3 drops 6N HCl) to the corresponding β-hydroxy ketone and comparison of the NMR spectrum with the data for the individual syn and anti diastereomers reported by Xiao et al.: Chen J-R, Lu H-H, Li X-Y, Cheng L, Wan J, Xiao W-J. Org Lett. 2005;7:4543. doi: 10.1021/ol0520323.
- 19.Diene 18 was observed as a minor byproduct (10–20%) in the NaH-induced dehydrobromination of 16.
- 20.Shatzmiller S, Dolithzky B-Z, Bahar E. Liebigs Ann Chem. 1991:375. [Google Scholar]
- 21.Dale JA, Dull DL, Mosher HS. J Org Chem. 1969;39:2543. [Google Scholar]
- 22.3,4,6-Tri-O-methyl-D-glucal was prepared from commerically available 3,4,6-tri-O-acetyl-D-glucal by acetate hydrolysis (cat. KCN, MeOH) followed by methylation (NaH, MeI, DMF).
- 23.Bolitt V, Mioskowski C. J Org Chem. 1990;55:5812. [Google Scholar]
- 24.Schneider F. Synthesis. 1986;7:582. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.