

Abstract
The combination of artemether (ARM) and lumefantrine is currently the first-line treatment of uncomplicated falciparum malaria in mainland Tanzania. While the exposure to lumefantrine has been associated with the probability of adequate clinical and parasitological cure, increasing exposure to artemether and the active metabolite dihydroartemisinin (DHA) has been shown to decrease the parasite clearance time. The aim of this analysis was to describe the pharmacokinetics and pharmacodynamics of artemether, dihydroartemisinin, and lumefantrine in African children with uncomplicated malaria. In addition to drug concentrations and parasitemias from 50 Tanzanian children with falciparum malaria, peripheral parasite densities from 11 asymptomatic children were included in the model of the parasite dynamics. The population pharmacokinetics and pharmacodynamics of artemether, dihydroartemisinin, and lumefantrine were modeled in NONMEM. The distribution of artemether was described by a two-compartment model with a rapid absorption and elimination through metabolism to dihydroartemisinin. Dihydroartemisinin concentrations were adequately illustrated by a one-compartment model. The pharmacokinetics of artemether was time dependent, with typical oral clearance increasing from 2.6 liters/h/kg on day 1 to 10 liters/h/kg on day 3. The pharmacokinetics of lumefantrine was sufficiently described by a one-compartment model with an absorption lag time. The typical value of oral clearance was estimated to 77 ml/h/kg. The proposed semimechanistic model of parasite dynamics, while a rough approximation of the complex interplay between malaria parasite and the human host, adequately described the early effect of ARM and DHA concentrations on the parasite density in malaria patients. However, the poor precision in some parameters illustrates the need for further data to support and refine this model.
Artemisinin-based combination therapy is generally accepted as treatment of choice for acute uncomplicated Plasmodium falciparum malaria (31). A fixed-dose combination containing artemether (ARM) and lumefantrine (LUM) is currently the first-line treatment of uncomplicated falciparum malaria in mainland Tanzania. The rationale behind combining ARM with LUM is to make use of the disparate pharmacokinetic profiles of the two drugs and thereby improve treatment efficacy and delay development of drug resistance. While ARM and its primary active metabolite dihydroartemisinin (DHA) are rapidly eliminated, LUM is slowly cleared from the body.
ARM is the lipid-soluble methyl-ether derivative of DHA. It is rapidly absorbed from the gastrointestinal tract, reaching maximum concentrations (Cmax) within 2 h of administration (6, 20, 24). ARM is metabolized to DHA with a reported half-life of 0.8 to 4 h (6, 20, 24). DHA is formed through cytochrome P450-mediated demethylation (25). The metabolite reaches Cmax in plasma at approximately the same time as ARM, within 2 h of ARM administration (6, 20, 24). The reported elimination half-life of DHA ranges from 0.4 to 12.5 h (6, 24).
LUM is a fluorene derivative that was discovered at the Academy of Military Medical Sciences in China. LUM is slowly absorbed, with an estimated absorption half-life of 5.3 h. Cmax is reached in approximately 10 h (6). The exposure to LUM has been shown to increase up to 16-fold due to concomitant administration of food or a fatty drink (2, 6a). The elimination of LUM is slow, and the terminal half-life is 3 to 4 days in adult malaria patients (6).
There is a dose dependency in the efficacy of the ARM-LUM combination. The 28-day cure rate of the combination has been associated with the body-weight-normalized dose (4). While the efficacy of the six-dose regimen currently used in Tanzania is high, with PCR-corrected 28- or 42-day cure rates of 92 to 98% in pediatric patients in Africa (7, 15, 18, 19, 21), an earlier four-dose regimen resulted in reported 14-day cure rates of 86% (10a). In a study of the correlation between pharmacokinetics (PK) and pharmacodynamics (PD) of ARM plus LUM in adult patients, increasing exposure to LUM was associated with a greater chance of adequate clinical and parasitological cure but did not explain variability in parasite clearance time (PCT) (6). In contrast, increasing areas under the concentration-time curve (AUCs) of ARM and DHA both caused a decrease in PCT but did not significantly influence the cure rate.
A challenge when modeling the effect of antimalarials is to adequately describe the dynamics of the parasite population in the absence of drugs. A number of semimechanistic pharmacodynamic models based on the erythrocytic life cycle of P. falciparum have been presented (9, 9a, 23). Common features of these models are the description of the age stages of P. falciparum and the division of the parasite population into circulating and sequestered parasites. This approach allows for the description of potentially stage-specific drug action as well as renders estimates of the total, rather than visible, parasite load.
To date, models of the dynamics of untreated parasitemia are derived primarily from adult patients inoculated with P. falciparum to treat neurosyphilis (12, 22). In this analysis, we include multiple daily observations of parasitemias from asymptomatic children from the same region as the symptomatic patients to obtain parameter estimates regarding the behavior of the parasite population.
The aim of this study was to present population pharmacokinetic models describing the kinetics of ARM, DHA, and LUM in children treated for uncomplicated malaria and to model the effect of ARM (DHA) and LUM concentrations on parasite density in malaria patients. A secondary aim was to evaluate the effect of concomitant milk intake on the model-estimated PK parameters of LUM in this pediatric population.
The patient study (no. NCT00336375) is registered at www.Clinical.Trials.gov.
MATERIALS AND METHODS
Subjects and study design.
The patient study was conducted at Fukayosi Primary Health Care Centre, Bagamoyo District, Tanzania, as reported elsewhere (A. Mårtensson, A. M. Carlsson, B. Ngasala, S. Dahlström, C. Membi, M. A. Musih, S. Omary, S. M. Montgomery, L. Rombo, S. Abdullah, Z. Premji, J. P. Gil, et al., submitted for publication). Briefly, a total of 50 patients with the following characteristics were included: ages 1 to 10 years, with microscopically confirmed acute uncomplicated P. falciparum malaria with an asexual parasite density of 2,000 to 200,000/μl and fever (axillary temperature of ≥37.5°C) or a history of fever within 24 h, and whose parent/legal guardian gave their written informed consent. Exclusion criteria included a hemoglobin level of <70 g/liter, severe malnutrition, signs of severe malaria, or any other danger signs.
All patients were hospitalized. Follow-up duration was 72 h. A clinical assessment was performed at 0, 2, 4, 8, 16, 24, 36, 48, 60, and 72 h. This included a physical examination and questions related to malaria-associated symptoms/potential side effects of the study drug as well symptoms and/or signs of severe malaria.
Weight-based doses of Coartem (20 mg artemether plus 120 mg lumefantrine), manufactured by Novartis Pharma, Ltd., Switzerland, were administered at 0, 8, 24, 36, 48, and 60 h. Patients weighing 5 to 14 kg received one tablet/dose, patients weighing 15 to 24 kg received two tablets/dose, and patients weighing 25 to 34 kg received three tablets/dose. Patients were randomly allocated either to ingest each drug dose with a glass of full-fat (3.4%) cow's milk (200 ml) (n = 25) or to take the medicine with water (n = 25). Patients received oral iron supplementation at the day of discharge from the study.
Venous blood samples for drug concentration analyses and for determination of parasitemia were obtained at 0, 2, 4, 8, 16, 24, 36, 48, 60, and 72 h following treatment initiation. Whole blood (700 μl) was collected in BD Microtainer plasma tubes with lithium heparin and gel. The tubes were centrifuged according to the manufacturer's recommendations within 1 h and kept on ice for up to 24 h after sampling.
An additional capillary blood sample for the assessment of parasitemia was obtained 2 h prior to treatment initiation. Giemsa-stained thick blood films were prepared and examined at the study site by experienced microscopists. The number of parasites was calculated as the number of parasites seen against 500 leukocytes in the thick blood film, and parasite density was reported as asexual parasites/μl blood. Plasma for drug concentration analyses was stored at −20°C until transport on dry ice to Sweden and Thailand. Only patients whose parent/guardian gave their written consent were included in the study.
Parasite density data from two previously published studies in asymptomatic children in a similar coastal setting in Tanzania were included in the parasite growth model (8, 8a). In one study, capillary samples were obtained every 6 h for 5 days (n = 1), and in the other study, daily samples were obtained for 14 consecutive days (n = 10). Parasite densities were determined in Giemsa-stained thin films in 200 fields and read in random order. The included children remained asymptomatic throughout the studies.
The studies were approved by the National Institute for Medical Research, Dar es Salaam, Tanzania, and by the Regional Ethics Committee, Stockholm, Sweden.
Bioanalysis.
Concentrations of ARM and DHA in plasma were measured by high-throughput liquid chromatography-tandem mass spectrometry (LC-MS/MS) (10). In brief, plasma samples were purified by solid-phase extraction (Oasis HLB μelution plate; Waters, Milford, MA) and quantified by LC-MS/MS. Stable-isotope-labeled ARM and DHA were used as internal standards. ARM and DHA were quantified using an API 5000 triple-quadrupole mass spectrometer (Applied Biosystems/MDS SCIEX, Foster City, CA), with a TurboV ionization source (TIS) interface operated in the positive-ion mode. Quantification was performed using selected reaction monitoring (SRM) for the transitions m/z 316 to 163 and 320 to 163 for ARM and stable-isotope-labeled ARM, respectively, and 302 to 163 and 307 to 166 for DHA and stable-isotope-labeled DHA, respectively. The performance data for the assay during analysis of all samples, expressed as the coefficients of variation (CV) (relative standard deviation [RSD%]), for quality control samples were below 6% throughout the calibration range. The lower limits of quantification (LLOQ) for the assay were 4.8 nM and 5.0 nM for ARM and DHA, respectively.
LUM concentrations were determined using a solid-phase extraction (SPE) liquid chromatographic (LC) assay with UV detection as described by Annerberg et al. (1a). Briefly, plasma was precipitated with acidic acetonitrile (containing, as the internal standard, a hexyl analogue of desbutyl-lumefantrine; Novartis no. TA 213/435/16). The precipitated plasma samples were buffered and purified using a 3 M Empore C8-SD deep-well SPE 96-well plate (Presearch, Ltd., Hampshire, United Kingdom). The dried SPE eluates were reconstituted in 200 μl, and 50 μl was injected into a LaChrom Elite LC system (SB-CN column; Zorbax, Inc.) with a mobile phase containing 0.01 M sodium perchlorate and acetonitrile-phosphate buffer (pH 2; 0.1 M at 57:43 [vol/vol]). The CVs (RSD%) during the analysis were below 5% for all QC concentrations. The lower limit of quantification for the assay was 47 nM. The drug analyses were performed at the Clinical Pharmacology Laboratory in the Faculty of Tropical Medicine, Bangkok, Thailand.
PK-PD modeling.
The population PK and PD of ARM, DHA, and LUM were modeled using the nonlinear mixed effects approach as implemented in NONMEM, version VI, level 1.1 (Icon Development Solutions, MD). The first-order (FO) conditional estimation method with interaction (FOCE-I) was used throughout the PK model-building process, while the FO method was used for the PD model.
The individual post hoc parameter estimates from the PK models were introduced as fixed parameters in the PD model. The PD of ARM, DHA, and LUM were described using a semimechanistic model of parasite development, incorporating features of previously presented models such as the spleen compartment for damaged but not yet cleared parasites, separate compartments for visible and invisible parasites, and a sine function describing the oscillations in visible parasitemia (9, 22, 23).
The drug effects were modeled sequentially, beginning with ARM and DHA, as they have been shown to contribute most significantly to the immediate decline in parasitemia (30). Compounds with highly similar PK profiles need to be administered separately in order to determine the relative contribution of each substance to the effect. The parallel concentration profiles for ARM and DHA precluded the estimation of separate effect parameters for the two compounds. In vitro studies have suggested similar potencies of the two substances, making the assumption of an equal effect slope relevant (1).
Homoscedastic and heteroscedastic error models were tested to describe residual error in concentration and in parasitemias. Variability was estimated as interindividual variability (IIV) and as interoccasion variability (IOV). Possible effects of covariates milk intake, group allocation, dose number, age, weight, gender, parasitemia, fever, and concomitant administration of paracetamol (n = 23) on fixed effects of the PK and PD models were identified using the general additive method (GAM) as implemented in Xpose, version 4.0 (14).
The likelihood ratio test (LRT)—i.e., the difference in the value of the objective function (OFV)—was used to discriminate between nested models. The OFV is essentially equal to −2 × log likelihood of the data, and the difference in OFV is approximately χ2 distributed (3). Parameters producing a decrease in the OFV exceeding 3.8, indicating a better fit at the 0.05 level of significance, were retained in the full model. Backward deletion of a covariate from the full model was based on the difference in OFV being less than 6.6 (at the 0.01 level of significance) as well as failure of the covariate to explain interindividual variability.
The uncertainty in PK parameter estimates, expressed as the 95% confidence interval (95% CI), was evaluated from 1,000 nonparametric bootstrap samples. Bootstrap analyses were performed in PsN (17). The predictive performances of the final models were assessed with visual predictive checks as described by Holford (11).
RESULTS
Demographics and safety.
A total of 50 children (19 male and 31 female) were enrolled. Mean age and weight were 4 years (range, 1 to 10 years) and 14 kg (range, 8 to 30 kg), respectively. No death or serious adverse event occurred. Forty-nine children completed the study. All had adequate clinical and parasitological response at 72 h. The remaining patient was withdrawn after 48 h due to the parents' request. All data collected from this child were included in the analysis until exit of the trial. Patients allocated to receive milk with each dose completed the 200 ml on 43% of occasions. During the 72 h follow-up, two patients received antibiotics. One child was treated with cloxacillin from 48 h due to a skin/soft tissue infection related to the intravenous (i.v.) cannula. The other child received amoxicillin from 60 h after having developed high fever of unknown origin. The latter child cleared parasitemia by 36 h.
Totals of 397 ARM, 400 DHA, and 423 LUM plasma concentrations obtained from the 50 patients were included in the analysis (Fig. 1 and 2). Nine ARM, 21 DHA, and 19 LUM concentrations below the limit of detection (LOD) were excluded. Twelve ARM and 47 DHA concentrations below the LLOQ were set to LLOQ (2 and 3 nM for ARM and DHA, respectively).
FIG. 1.

Artemether (solid lines) and dihydroartemisinin (dashed lines) concentrations over time during treatment with weight-based doses of artemether and lumefantrine (Coartem) administered at 0, 8, 24, 36, 48, and 60 h. Lines are interpolated between observations.
FIG. 2.

Observed lumefantrine concentrations over time during treatment with weight-based doses of artemether and lumefantrine (Coartem) administered at 0, 8, 24, 36, 48, and 60 h. Lines are interpolated between observations.
A total of 356 peripheral parasite counts from the 50 patients (Fig. 3) and 104 from 11 asymptomatic subjects were included in the PD model. Undetectable parasitemias were excluded.
FIG. 3.

Observed parasitemia (solid lines) and body temperature (dashed lines) over time in pediatric patients during treatment with weight-based doses of artemether and lumefantrine administered at 0, 8, 24, 36, 48, and 60 h. Lines are interpolated between observations.
Pharmacokinetics of artemether, dihydroartemisinin, and lumefantrine.
There was no a priori information on the absolute bioavailability of ARM or the fraction eliminated through metabolism to DHA, and it was assumed that all ARM is eliminated via this pathway. Fixing the bioavailability of DHA to 1 also rendered the metabolite model identifiable.
The distribution of ARM was best described by a two-compartment model with first-order absorption. The parameter estimates for the ARM/DHA PK model are presented in Table 1. There was very little information on the rate of absorption in the data, and the absorption rate constant was fixed to 1/h in the final model. Altering the rate of absorption between 0.2 and 2/h did not significantly influence the remaining parameter estimates.
TABLE 1.
Population pharmacokinetic parameters for artemether and dihydroartemisinin in pediatric patients with uncomplicated falciparum malaria during a 3-day treatment with six weight-based doses of artemether and lumefantrine (Coartem)
| Parameter | Estimate (95% CI)a | IIV, % CV (95% CI)a | Parameter description |
|---|---|---|---|
| ka (per h) | 1 (fixed) | NEb | Absorption rate |
| CL/FARM (liters/h/kg) | θ1 × [1 + θ2 × (occ − 1)] | 41 (37-50) | Oral clearance |
| θ1 | 2.6 (1.5-2.6) | NE | Typical value of CL/FARM |
| θ2 | 0.57 (0.39-0.75) | NE | Fractional change in CL/FARM with dose |
| Vc/FARM (liters/kg) | 5.2 (3.5-7.1) | NE | Oral volume of distribution of the central compartment |
| QARM (liters/kg) | 1.4 (1.1-1.8) | NE | Intercompartment clearance |
| Vp/FARM (liters/kg) | 41.4 (29.0-58.1) | NE | Oral volume of distribution of the peripheral compartment |
| CL/FDHA (liters/h/kg) | 6.8 (5.8-8.0) | 47 (35-57) | Oral metabolite clearance |
| V/FDHA (liters/kg) | 3.7 (2.3-8.7) | NE | Oral metabolite volume of distribution |
| σprop ARM (%) | 61 (54-67) | Proportional residual error | |
| σadd ARM (nM) | 2 (fixed) | Additive residual error | |
| σprop DHA (%) | 82 (73-90) | Proportional residual error | |
| σadd DHA (nM) | 3 (fixed) | Additive residual error |
95% CI from 1,000 nonparametric bootstrap data sets.
NE, not estimated.
There was a trend toward a reduction in the trough concentrations of ARM and DHA, as illustrated in Fig. 4. This time dependency was described by a model including occasion (occ) as a covariate on clearance (CL)/FARM: CL/FARM = θ1 × [1 + θ2 (occ − 1)] × expη, where θ1 is the typical value of CL/FARM for the first dose and θ2 is the fractional change in CL/FARM with each occasion (occ). The inclusion of occasion as a covariate on CL/FARM resulted in ΔOFV = −167. The performance of the PK model is illustrated in Fig. 5.
FIG. 4.

Trough concentrations of artemether and dihydroartemisinin (12 h postdose) during treatment with weight-based doses of artemether and lumefantrine (Coartem) administered at 0, 8, 24, 36, 48, and 60 h. The right panel shows the dihydroartemisinin/artemether ratio over time.
FIG. 5.

Visual predictive checks of the pharmacokinetic models. The open circles are the observed concentrations. The shaded area represents the 95% prediction interval, calculated from simulated observations from 1,000 studies.
The DHA concentrations were best illustrated by a covariate-free one-compartment distribution model.
The PK of LUM was best described by a one-compartment model with a combined proportional and additive residual error. The introduction of an absorption lag time significantly improved the model (ΔOFV = −96).
The inclusion of milk intake as a covariate on the PK parameters of LUM did not explain the variability in LUM pharmacokinetics and did not result in an improvement of the model. The population pharmacokinetic parameters for LUM are presented in Table 2.
TABLE 2.
Population pharmacokinetic parameters for lumefantrine in pediatric patients with uncomplicated falciparum malaria during a 3-day treatment with six weight-based doses of artemether and lumefantrine (Coartem)
| Parameter | Estimate (95% CI)a | IIV, % CV (95% CI)a | Parameter description |
|---|---|---|---|
| Lag | 1.92 (1.86-1.96) | NEb | Absorption lag time |
| ka (per h) | 0.82 (0.45-1.61) | 156 (126-190) | Absorption rate |
| CL/F (ml/h/kg) | 77 (52-105) | NE | Oral clearance |
| V/F (liters/kg) | 8.9 (6.8-11.7) | 82 (66-102) | Oral volume of distribution |
| σprop (%) | 46 (41-52) | Proportional residual error | |
| σadd (nM) | 43 (19-66) | Additive residual error |
95% CI from 1,000 nonparametric bootstrap data sets.
NE, not estimated.
Pharmacodynamic model.
The PD model, illustrated in Fig. 6, is initiated by the introduction of a certain parasitemia (Pinit) into the compartment denoted by tiny rings, PTR, representing parasites in the earliest ring stage. The intraerythrocytic stage of the infection was assumed to have started 10 cycles prior to the first sample in asymptomatic individuals, corresponding to a time when the model infection is at steady state. The number of cycles passed since model initiation was fixed to 4 in patients. The parameter estimates are reported in Table 3 .
FIG. 6.

Pharmacodynamic model based on the blood stages of P. falciparum. Compartments within the dashed rectangle represent parasites visible in peripheral blood.
TABLE 3.
Parameter estimates for the pharmacodynamic model of the effects of artemether and dihydroartemisinin in pediatric patients with uncomplicated malaria
| Parameter | Estimate (95% CI)a | IIV, % CV (95% CI)a | Parameter description |
|---|---|---|---|
| Pinit (parasites/μl) | 1 (fixed) | 119.2 (98-161) | Initial parasitemia |
| VPT (h) | 15.5 (9.7-21.5) | NEb | Mean visible parasite transit time |
| MTT (h) | 48.5 (48.0-49.1) | NE | Mean parasite transit time |
| Patients | |||
| REPLp | 4 (3.6-4.4) | NE | Replication factor |
| Ap | 0 (fixed) | NE | Amplitude of oscillations |
| kspleen | 0.26 (fixed)c | NE | Rate of elimination of dead or injured parasites |
| Asymptomatic children | |||
| REPLa | 1 (fixed) | NE | Replication factor |
| Aa | 1.01 (0.71-1.37) | NE | Amplitude of oscillations |
| Drug effects | |||
| SARM/DHA | 0.073 (0.049-0.423) | NE | Slope of the ARM and DHA effect |
| σ (%) | 138 (125-258) | Residual error |
95% CI from 1,000 nonparametric bootstrap data sets.
NE, not estimated.
The estimate was fixed to a literature value from Gordi et al. (9).
The parasites mature through the erythrocytic stages: tiny rings (PTR), small rings (PSR), large rings (PLR), and mature trophozoites/schizonts (PMT). Parasites that are killed or injured due to drug action were assumed to remain in the blood until removed by the spleen or macrophages. These parasites are represented by the compartment denoted Pspleen. The inclusion of the spleen compartment produced a significant drop in the OFV (−61). The elimination rate from the spleen compartment, ks, was fixed to that described by Gordi et al. (9). Growth was modeled as a multiplication rate (REPL) from schizonts to tiny rings. Only the parasites in the ring stages and those injured by drugs were assumed to be visible through microscopy. The mean time to complete an intraerythrocytic cycle (MTT) was estimated to be 48.5 h, and the mean rate of transfer between visible compartments, kVPT, was modeled as 3/VPT, where VPT was estimated to 15.5 h. The mean time for transfer from the sequestered state, kIPT, was fixed to 1/(MTT − VPT).
To account for fluctuations in the parasite population over time, a sine function was applied to the input to the PTR compartment. The amplitude, A, of the sine function varies with the synchronicity in the parasite population. The sine function produced a significant drop in the OFV (−42) in asymptomatic individuals but was not supported by the sparse data in symptomatic patients. The wavelength of the sine function was fixed to the MTT.
The growth rates of the parasite population differ between patients and asymptomatic individuals. In asymptomatic children, the parasitemia was approximated by a steady state and the REPLa was fixed to 1. Patients experience a net growth of parasitemia prior to treatment, and the REPLp was estimated to 4.
The effects of ARM and DHA plasma concentrations were modeled on all developmental stages as kARM = SARM/DHA × log [ARM] and kDHA = SARM/DHA × log [DHA], where SARM/DHA is the slope of both the artemether and DHA concentration-effect correlations.
The introduction of an effect of LUM did not significantly improve the model fit, and the parameter estimates for LUM effect could not be estimated.
The rate of change of parasitemia in the different compartments was described by equations 1 to 5. The fit of the model is illustrated in a visual predictive check in Fig. 7:
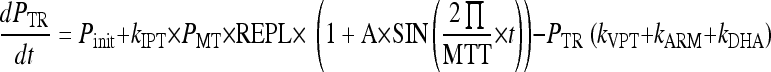 |
(1) |
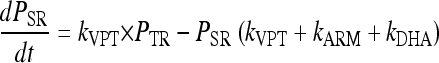 |
(2) |
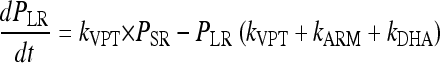 |
(3) |
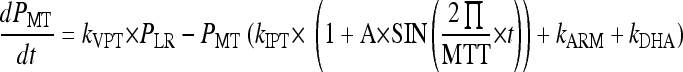 |
(4) |
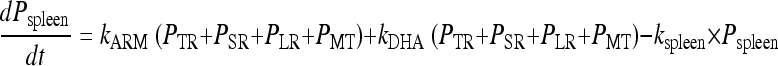 |
(5) |
FIG. 7.

Visual predictive check of the pharmacodynamic model. The open circles are the observed log-transformed parasitemias. The shaded area represents the 95% prediction interval, calculated from simulated observations from 1,000 studies, and the solid lines represent the simulated median parasitemia.
DISCUSSION
Data describing the parasitemia in pediatric patients as well as in untreated, asymptomatic children from a nearby geographic region in Tanzania were analyzed in order to describe the within-host parasite dynamics in the presence and absence of antimalarial drugs. The proposed semimechanistic model, while a rough approximation of the complex interplay between malaria parasite and the human host, adequately described the early effect of ARM and DHA concentrations on the parasite density in malaria patients (Fig. 7). To gain a better understanding of the parasite dynamics, stage-specific parasite counts should be obtained both prior to and during drug treatment.
The PK of ARM and DHA following oral administration of ARM were described by a three-compartment model, consisting of two distribution compartments for the parent compound and one for DHA. Model-estimated CL/FARM increased more than 3-fold from dose 1 to dose 6. Previous studies have indicated a time dependency in the ARM kinetics (6, 26, 27). In a study of the kinetics of ARM in Chinese adults, the Cmax following the last dose in a 2-day twice-a-day (BID) treatment was one-third of the Cmax after dose 1 (26). The decreased Cmax and/or AUC of ARM with consecutive doses have been attributed to enzyme induction (26, 27).
The introduction of IOV in the bioavailability of ARM resulted in a significant reduction of the OFV; however the data did not allow the determination of this parameter with adequate precision. A similarly variable absorption was described by Ezzet and colleagues, who found a significantly elevated bioavailability of both ARM and DHA with dose 3 compared to doses 1, 2, and 4 (6).
The pharmacokinetics of LUM during the first 3 days of treatment were adequately described by a one-compartment distribution model with first-order absorption. The predicted median LUM concentration on day 3, calculated from 1,000 realizations of the pharmacokinetic model under the study conditions, came to 9.3 μM and was similar to the median of 11.3 μM reported by Checci and colleagues from pediatric patients in Uganda (4). The typical value of CL/F was estimated to 77 ml/h/kg, which is within the range of 40 to 200 ml/h/kg reported in adults (6, 16).
Earlier pharmacokinetic models of LUM have used a two-compartment distribution model with a slow terminal elimination half-life of 3 to 4 days. The model estimated elimination half-life in our study was approximately 2 days. The trend toward underestimation of the LUM concentrations on day 3 indicates that a two-compartment model with a slower terminal phase may have provided a better description of the elimination. A two-compartment model was investigated during the model-building process but did not result in improved model performance. The shorter half-life in the present study may be an effect of the relatively short duration of sampling.
There was no significant impact of milk intake on the pharmacokinetics of LUM. A possible explanation for the discrepancy in the results compared to the previous report by Ashley and colleagues (2) is the fact that the investigated patients were unable to comply with the milk intake in almost 50% of administrations. The resulting number of doses actually administered with an adequate amount of milk may have been too small to allow the detection of a difference.
The within-host P. falciparum dynamics are complex and include stages of variable drug sensitivity as well as sequestering stages not detectable in peripheral blood (8, 8a, 28, 29). Models describing the in vivo dynamics of untreated P. falciparum infection have primarily been developed from data from adult, nonimmune, patients inoculated with malaria to treat neurosyphilis (12, 22). The proposed PD model is based on data from both symptomatic and asymptomatic children in rural settings in Tanzania. The number of compartments describing the visible parasite stages was chosen based on the feasible resolution in parasite data using microscopy: tiny rings, small rings, and immature trophozoites (8). The influence of polyclonal infections, semi-immunity, fever, and other clinical symptoms of malaria on parasite population dynamics in vivo need to be investigated in further studies. In the proposed model, we assumed that the mean transit times were equal in asymptomatic and symptomatic patients and that the rates of development through the parasite stages were the same across patients and asymptomatic carriers.
The model allows for variation in the degree of synchronicity in the infecting parasite population as studies have suggested a difference between the dynamics of parasites in symptomatic and asymptomatic patients. Parasite populations in asymptomatic individuals appear relatively synchronized, resulting in a distinct cyclical pattern in peripheral parasitemia (8, 8a, 13a). In contrast, the genotype patterns of the infecting parasite populations in the symptomatic patients in this study, which are presented elsewhere (Mårtensson et al., submitted), appear to support earlier findings of more-complex, less-synchronized dynamics, with variable clones disappearing and reappearing at different times during drug treatment (13; Mårtensson et al., submitted).
The immunological basis for the dynamic steady state of parasitemia in asymptomatic patients has not been fully elucidated. The PD model does not include an innate parasite clearance, and the steady state described in asymptomatic patients is only governed by the rate of replication at merogony. A better resolution in the data—that is, a differential count of parasites in the various developmental stages in the treated children—may have given the possibility to describe loss rates for each compartment and to increase the precision of the estimated rate of elimination of dead or injured parasites.
The REPL in symptomatic patients was estimated to be 4, which translates into a multiplication factor of 12 per life cycle. This is in accordance with previous studies that have indicated a multiplication of 8 to 20 per cycle (22, 29). The mean parasite age (since merogony) at sequestration was estimated to 15.5 h. An in vitro study has shown that cytoadherence starts at approximately 12 h and reaches 50% at 14 to 16 h (25). Similar results have also been obtained with a model describing the sequestered parasite load in adult patients with uncomplicated and severe malaria (5).
A previous study has shown the initial decline in parasitemia results mainly from the ARM/DHA component during combination treatment with lumefantrine (6). The proposed PD model supports this observation, as the addition of an effect of the LUM component did not significantly improve the model fit. The predicted median parasite clearance time (PCT) was 38 h, which compares well with the observed median PCT of 36 h. The present study was designed to describe the kinetics of ARM, DHA, and LUM during the early treatment phase in children with acute uncomplicated falciparum malaria. The short follow-up of 3 days did not allow us to model the risk of late failures and thus to accurately model the LUM effect.
In conclusion, this study describes the population PK of ARM, DHA, and LUM in pediatric patients treated for uncomplicated falciparum malaria. The time-dependent ARM kinetics described in adults was evident also in this population. Building on earlier efforts to describe the complex within-host dynamics of P. falciparum (9, 22, 23, 28), we propose a semimechanistic model of parasite dynamics, describing both visible and sequestered parasites, as well as allowing for various degrees of synchronization. The pharmacodynamic model presented adequately accounted for the effect of ARM and DHA in pediatric patients. However, the large residual error illustrates the need for further data to support and refine this model.
Acknowledgments
Financial support was received from Sida (grant SWE-2005 017). Niklas Lindegårdh and Anna Annerberg are funded by the Wellcome Trust-Mahidol University-Oxford Tropical Medicine Research Programme (077166/Z/05/Z), supported by the Wellcome Trust of Great Britain.
Footnotes
Published ahead of print on 16 August 2010.
REFERENCES
- 1.Alin, M. H., A. Bjorkman, and M. Ashton. 1990. In vitro activity of artemisinin, its derivatives, and pyronaridine against different strains of Plasmodium falciparum. Trans. R. Soc. Trop. Med. Hyg. 84:635-637. [DOI] [PubMed] [Google Scholar]
- 1a.Annerberg, A., T. Singtoroj, P. Tipmanee, N. J. White, N. P. Day, and N. Lindegardh. 2005. High throughput assay for the determination of lumefantrine in plasma. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 822:330-333. [DOI] [PubMed] [Google Scholar]
- 2.Ashley, E. A., K. Stepniewska, N. Lindegardh, A. Annerberg, A. Kham, A. Brockman, P. Singhasivanon, N. J. White, and F. Nosten. 2007. How much fat is necessary to optimize lumefantrine oral bioavailability? Trop. Med. Int. Health 12:195-200. [DOI] [PubMed] [Google Scholar]
- 3.Beal, S. L., and L. B. Sheiner. 1982. Estimating population kinetics. Crit. Rev. Biomed. Eng. 8:195-222. [PubMed] [Google Scholar]
- 4.Checchi, F., P. Piola, C. Fogg, F. Bajunirwe, S. Biraro, F. Grandesso, E. Ruzagira, J. Babigumira, I. Kigozi, J. Kiguli, J. Kyomuhendo, L. Ferradini, W. R. Taylor, and J. P. Guthmann. 2006. Supervised versus unsupervised antimalarial treatment with six-dose artemether-lumefantrine: pharmacokinetic and dosage-related findings from a clinical trial in Uganda. Malar. J. 5:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dondorp, A. M., V. Desakorn, W. Pongtavornpinyo, D. Sahassananda, K. Silamut, K. Chotivanich, P. N. Newton, P. Pitisuttithum, A. M. Smithyman, N. J. White, and N. P. Day. 2005. Estimation of the total parasite biomass in acute falciparum malaria from plasma PfHRP2. PLoS Med. 2:e204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ezzet, F., R. Mull, and J. Karbwang. 1998. Population pharmacokinetics and therapeutic response of CGP 56697 (artemether + benflumetol) in malaria patients. Br. J. Clin. Pharmacol. 46:553-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6a.Ezzet, F., M. van Vugt, F. Nosten, S. Looareesuwan, and N. J. White. 2000. Pharmacokinetics and pharmacodynamics of lumefantrine (benflumetol) in acute falciparum malaria. Antimicrob. Agents Chemother. 44:697-704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Falade, C., M. Makanga, Z. Premji, C. E. Ortmann, M. Stockmeyer, and P. I. de Palacios. 2005. Efficacy and safety of artemether-lumefantrine (Coartem) tablets (six-dose regimen) in African infants and children with acute, uncomplicated falciparum malaria. Trans. R. Soc. Trop. Med. Hyg. 99:459-467. [DOI] [PubMed] [Google Scholar]
- 8.Farnert, A., M. Lebbad, L. Faraja, and I. Rooth. 2008. Extensive dynamics of Plasmodium falciparum densities, stages and genotyping profiles. Malar. J. 7:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8a.Farnert, A., G. Snounou, I. Rooth, and A. Bjorkman. 1997. Daily dynamics of Plasmodium falciparum subpopulations in asymptomatic children in a holoendemic area. Am. J. Trop. Med. Hyg. 56:538-547. [DOI] [PubMed] [Google Scholar]
- 9.Gordi, T., R. Xie, and W. J. Jusko. 2005. Semi-mechanistic pharmacokinetic/pharmacodynamic modelling of the antimalarial effect of artemisinin. Br. J. Clin. Pharmacol. 60:594-604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9a.Gravenor, M. B., A. L. Lloyd, P. G. Kremsner, M. A. Missinou, M. English, K. Marsh, and D. Kwiatkowski. 2002. A model for estimating total parasite load in falciparum malaria patients. J. Theor. Biol. 217:137-148. [DOI] [PubMed] [Google Scholar]
- 10.Hanpithakpong, W., B. Kamanikom, A. M. Dondorp, P. Singhasivanon, N. J. White, N. P. Day, and N. Lindegardh. 2008. A liquid chromatographic-tandem mass spectrometric method for determination of artesunate and its metabolite dihydroartemisinin in human plasma. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 876:61-68. [DOI] [PubMed] [Google Scholar]
- 10a.Hatz, C., S. Abdulla, R. Mull, D. Schellenberg, I. Gathmann, P. Kibatala, H. P. Beck, M. Tanner, and C. Royce. 1998. Efficacy and safety of CGP 56697 (artemether and benflumetol) compared with chloroquine to treat acute falciparum malaria in Tanzanian children aged 1-5 years. Trop. Med. Int. Health 3:498-504. [DOI] [PubMed] [Google Scholar]
- 11.Holford, N. 2005. A degenerative predictive check, abstr. 738, p. 14. 14th Annu. Meet. Pop. Appr. Group Eur., Pamplona, Spain.
- 12.Hoshen, M. B., R. Heinrich, W. D. Stein, and H. Ginsburg. 2000. Mathematical modelling of the within-host dynamics of Plasmodium falciparum. Parasitology 121:227-235. [DOI] [PubMed] [Google Scholar]
- 13.Jafari, S., J. Le Bras, O. Bouchaud, and R. Durand. 2004. Plasmodium falciparum clonal population dynamics during malaria treatment. J. Infect. Dis. 189:195-203. [DOI] [PubMed] [Google Scholar]
- 13a.Jafari-Guemouri, S., C. Boudin, N. Fievet, P. Ndiaye, and P. Deloron. 2006. Plasmodium falciparum genotype population dynamics in asymptomatic children from Senegal. Microbes Infect. 8:1663-1670. [DOI] [PubMed] [Google Scholar]
- 14.Jonsson, E. N., and M. O. Karlsson. 1999. Xpose—an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput. Methods Programs Biomed. 58:51-64. [DOI] [PubMed] [Google Scholar]
- 15.Kabanywanyi, A. M., A. Mwita, D. Sumari, R. Mandike, K. Mugittu, and S. Abdulla. 2007. Efficacy and safety of artemisinin-based antimalarial in the treatment of uncomplicated malaria in children in southern Tanzania. Malar. J. 6:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lefevre, G., P. Carpenter, C. Souppart, H. Schmidli, J. M. Martin, A. Lane, C. Ward, and D. Amakye. 2002. Interaction trial between artemether-lumefantrine (Riamet) and quinine in healthy subjects. J. Clin. Pharmacol. 42:1147-1158. [DOI] [PubMed] [Google Scholar]
- 17.Lindbom, L., P. Pihlgren, and E. N. Jonsson. 2005. PsN-Toolkit—a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput. Methods Programs Biomed. 79:241-257. [DOI] [PubMed] [Google Scholar]
- 18.Martensson, A., B. Ngasala, J. Ursing, M. Isabel Veiga, L. Wiklund, C. Membi, S. M. Montgomery, Z. Premji, A. Farnert, and A. Bjorkman. 2007. Influence of consecutive-day blood sampling on polymerase chain reaction-adjusted parasitological cure rates in an antimalarial-drug trial conducted in Tanzania. J. Infect. Dis. 195:597-601. [DOI] [PubMed] [Google Scholar]
- 19.Mårtensson, A., J. Stromberg, C. Sisowath, M. I. Msellem, J. P. Gil, S. M. Montgomery, P. Olliaro, A. S. Ali, and A. Bjorkman. 2005. Efficacy of artesunate plus amodiaquine versus that of artemether-lumefantrine for the treatment of uncomplicated childhood Plasmodium falciparum malaria in Zanzibar, Tanzania. Clin. Infect. Dis. 41:1079-1086. [DOI] [PubMed] [Google Scholar]
- 20.Mordi, M. N., S. M. Mansor, V. Navaratnam, and W. H. Wernsdorfer. 1997. Single dose pharmacokinetics of oral artemether in healthy Malaysian volunteers. Br. J. Clin. Pharmacol. 43:363-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20a.Na Bangchang, K., J. Karbwang, C. G. Thomas, A. Thanavibul, K. Sukontason, S. A. Ward, and G. Edwards. 1994. Pharmacokinetics of artemether after oral administration to healthy Thai patients with acute, uncomplicated falciparum malaria. Br. J. Clin. Pharmacol. 37:249-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Piola, P., C. Fogg, F. Bajunirwe, S. Biraro, F. Grandesso, E. Ruzagira, J. Babigumira, I. Kigozi, J. Kiguli, J. Kyomuhendo, L. Ferradini, W. Taylor, F. Checchi, and J. P. Guthmann. 2005. Supervised versus unsupervised intake of six-dose artemether-lumefantrine for treatment of acute, uncomplicated Plasmodium falciparum malaria in Mbarara, Uganda: a randomised trial. Lancet 365:1467-1473. [DOI] [PubMed] [Google Scholar]
- 22.Simpson, J. A., L. Aarons, W. E. Collins, G. M. Jeffery, and N. J. White. 2002. Population dynamics of untreated Plasmodium falciparum malaria within the adult human host during the expansion phase of the infection. Parasitology 124:247-263. [DOI] [PubMed] [Google Scholar]
- 23.Svensson, U. S., H. Alin, M. O. Karlsson, Y. Bergqvist, and M. Ashton. 2002. Population pharmacokinetic and pharmacodynamic modelling of artemisinin and mefloquine enantiomers in patients with falciparum malaria. Eur. J. Clin. Pharmacol. 58:339-351. [DOI] [PubMed] [Google Scholar]
- 24.Teja-Isavadharm, P., F. Nosten, D. E. Kyle, C. Luxemburger, F. Ter Kuile, J. O. Peggins, T. G. Brewer, and N. J. White. 1996. Comparative bioavailability of oral, rectal, and intramuscular artemether in healthy subjects: use of simultaneous measurement by high performance liquid chromatography and bioassay. Br. J. Clin. Pharmacol. 42:599-604. [DOI] [PubMed] [Google Scholar]
- 25.Udomsangpetch, R., B. Pipitaporn, K. Silamut, R. Pinches, S. Kyes, S. Looareesuwan, C. Newbold, and N. J. White. 2002. Febrile temperatures induce cytoadherence of ring-stage Plasmodium falciparum-infected erythrocytes. Proc. Natl. Acad. Sci. U. S. A. 99:11825-11829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Agtmael, M. A., S. Cheng-Qi, J. X. Qing, R. Mull, and C. J. van Boxtel. 1999. Multiple dose pharmacokinetics of artemether in Chinese patients with uncomplicated falciparum malaria. Int. J. Antimicrob. Agents 12:151-158. [DOI] [PubMed] [Google Scholar]
- 27.van Agtmael, M. A., V. Gupta, C. A. van der Graaf, and C. J. van Boxtel. 1999. The effect of grapefruit juice on the time-dependent decline of artemether plasma levels in healthy subjects. Clin. Pharmacol. Ther. 66:408-414. [DOI] [PubMed] [Google Scholar]
- 28.White, N. J. 1997. Assessment of the pharmacodynamic properties of antimalarial drugs in vivo. Antimicrob. Agents Chemother. 41:1413-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.White, N. J., D. Chapman, and G. Watt. 1992. The effects of multiplication and synchronicity on the vascular distribution of parasites in falciparum malaria. Trans. R. Soc. Trop. Med. Hyg. 86:590-597. [DOI] [PubMed] [Google Scholar]
- 30.White, N. J., M. van Vugt, and F. Ezzet. 1999. Clinical pharmacokinetics and pharmacodynamics and pharmacodynamics of artemether-lumefantrine. Clin. Pharmacokinet. 37:105-125. [DOI] [PubMed] [Google Scholar]
- 31.WHO. 2006. WHO guidelines for the treatment of malaria. World Health Organization, Geneva, Switzerland.