


Abstract
DNA breaks occur during many processes in mammalian cells, including recombination, repair, mutagenesis and apoptosis. Here we report a simple and rapid method for assaying DNA breaks and identifying DNA breaksites. Breaksites are first tagged and amplified by ligation-mediated PCR (LM-PCR), using nested PCR primers to increase the specificity and sensitivity of amplification. Breaksites are then mapped by batch sequencing LM-PCR products. This allows easy identification of multiple breaksites per reaction without tedious fractionation of PCR products by gel electrophoresis or cloning. Breaksite batch mapping requires little starting material and can be used to identify either single- or double-strand breaks.
INTRODUCTION
DNA breaks are generated as intermediates in recombination, repair, mutagenesis and apoptosis. To study the molecular mechanisms and dynamics of these processes, a quick and accurate method for mapping breaksites is required. The method commonly used for identification of breaks in a defined region of genomic DNA is known as ligation-mediated PCR (LM-PCR). LM-PCR involves ligation of a linker of defined sequence to the breaksite, facilitating PCR amplification. Typically, breaksites are mapped by sequencing products of the LM-PCR reaction that have been isolated by gel electrophoresis or cloning. LM-PCR was initially developed for sensitive in vivo footprinting and sequencing (1,2). Subsequent optimization by Riggs and co-workers increased the sensitivity and specificity of LM-PCR, so that breaksites could be identified in samples containing as little as several hundred nanograms of template DNA (3–5).
We have devised a method for rapidly mapping multiple DNA breaks at heterogeneous sites within a defined region of the genome. In this method, breaksites are tagged and amplified by LM-PCR, using nested PCR primers to increase sensitivity and specificity of amplification. Following amplification, reaction products are directly sequenced in batch to identify breaksites. Batch sequencing can accurately identify multiple breaks in a single PCR reaction. Because batch sequencing eliminates the need for DNA fractionation by gel electrophoresis or cloning, the speed and throughput of analysis are high. Batch mapping is also very sensitive: we present an example of identification of multiple single-strand breaks in a sample containing 250 cell-equivalents (a few nanograms) of mammalian genomic DNA. Batch mapping can be applied to analysis of either single- or double-strand breaks.
MATERIALS AND METHODS
Linkers and primers for batch mapping breaksites in the murine λ1 gene
Genomic DNA was quantified in PCR reactions using primers LF7 (TCTCATGGAGAAGGAAAACC ) and LR4a (TATGCCTTCTGGGTACAAG). A duplex DNA linker with one blunt and one staggered end was generated by annealing LL3 (CGAGTTCAGTCCGTAGACCATGGAGATCTGAATTC) and LP2 (GAATTCAGATCTCC), each at 20 µM, in 250 mM Tris–Cl pH 7.7. The linker was stored at –20°C. The oligonucleotides LL4 (CGAGTTCAGTCCGTAGAC) and LL2 (GTAGACCATGGAGATCTGAATTC) were used as linker-specific primers for the first and second rounds of nested PCR, respectively. In some cases, the 5′-biotinylated oligonucleotide, LRB3 (BBBBGAATGTTCTGTGCTCTC; B, biotin), was used in the primer extension step for detection of breaks on the top strand; and the λ1-specific primers used for first and second rounds of nested PCR following extension with LRB3 were LR4d (CTGTGTCTCTCTCTATGAC) and LR4e (GTTTTCCTTCTCCATGAGATAGC). A second primer set used for detection of top strand breaks was LRB (BBBBATGCTCTTGCTGTCAGG), LR2 (GTAGAAATCAGTGATCGTAC) and LR3 (ACAGGGTGACTGATGGCGAAG). LFB2 (BBBBGATAGTGGGTGTTTATG) was used in primer extension for detection of breaks on the bottom strand and, following extension with LFB2, wLF2 (ACTCTGGATAAGCCTGAAC) and wLF3 (GATGATTAATGCCCCTGAGCTC) were used for nested PCR. LR3a (CATCTTTCAGTAGCAATCCTGG) was kinase-labeled and used as the probe for hybridization analysis when the primer set LRB, LR2 and LR3 was used. LR5b (GAGATTAGACATGAAAGGCTACAG) and LR6 (TGGTTGCTGTACCATAGAG) were used for sequence analysis.
Genomic DNA preparation
Genomic DNA was prepared by suspending 104–106 cells in 140 µl of lysis buffer (50 mM Tris–Cl pH 8.0, 100 mM EDTA, 100 mM NaCl, 1% SDS) containing 1 mg/ml proteinase K that had been pre-incubated at 37°C for 30 min in the lysis buffer. Following incubation at 55°C for 3 h and addition of 320 µl water, the lysates were very gently extracted, once with phenol, then once with 1:1 phenol–chloroform. To ensure efficient DNA recovery, 10 µg of DNase-free glycogen (Roche) was added as carrier to samples that contained <5 × 104 cells. DNA was precipitated with 0.1 vol of 3.0 M sodium acetate pH 5.2 and 2 vol of 100% ethanol. The DNA pellet was dissolved at a concentration equivalent to 1000 cells/µl by incubation at 4°C overnight.
DNA quantification
DNA recovery from small numbers of cells was quantified by PCR amplification of a 472 bp fragment from the Jλ1–Cλ1 intron, using primers LF7 and LR4a. PCR was carried out in a total volume of 50 µl containing 1× PCR buffer (Perkin Elmer), 3 mM MgCl2, 200 nM each primer, 0.25 mM dNTPs and 0.8 µl of AmpliTaqGOLD DNA polymerase (Perkin Elmer). PCR cycles were: 1× (95°C for 10 min, 48°C for 4 min, 72°C for 3 min), 30–32× (94°C for 40 s, 48°C for 40 s, 72°C for 40 s) and 1× (72°C for 7 min). Sample DNAs (∼500 cell-equivalents) were amplified in parallel with a standard produced by amplifying DNA from the murine lymphoma J558L at serial dilutions ranging from 50 to 2000 cell-equivalents. The PCR products were resolved on an agarose gel and the DNA concentration in each sample was estimated according to the intensity of the band in comparison to the standard.
Primer extension
For primer extension, 0.6 pmol biotinylated primer was added to 2000 cell-equivalents of genomic DNA in 1× Sequenase buffer (50 mM NaCl, 40 mM Tris–Cl pH 7.7) in a total volume of 15 µl, and the DNA denatured at 95°C for 3 min and then annealed at 43°C for 30 min. Following addition of 7.5 µl of an ice-cold freshly made solution containing 20 mM MgCl2, 20 mM DTT, 0.25 mM each of four dNTPs and 1.5 µl of Sequenase 2.0 (USB) freshly diluted 1:4 in ice-cold 10 mM Tris–Cl pH 7.7, primer extension was carried out at 45°C for 20–30 min. Then 6 µl of cold 0.3 M Tris–Cl pH 7.7 was added and the Sequenase was inactivated by incubation at 70°C for 15 min.
Linker ligation
Linker ligation was carried out by adding 45 µl of a cold, freshly made ligation mixture containing 13.3 mM MgCl2, 30 mM DTT, 83 µg/ml BSA, 1.66 mM ATP, 100 pmol of pre-annealed LL3/LP2 linker in 250 mM Tris–Cl pH 7.7 and 400 U of T4 DNA ligase (New England Biolabs) to products of the extension reaction, and ligation was carried out at 16°C overnight. Extension–ligation products were recovered by addition of 300 µg of pre-washed M280 streptavidin beads (Dynal), suspended in 75 µl of 2× bind/wash buffer (10 mM Tris–Cl pH 7.3, 1 mM EDTA, 2 M NaCl, 0.02% Tween-20) and the tubes were incubated at 43°C for 1 h with brief mixing every 5 min. The beads were then pelleted with a magnet, and washed twice with 2× bind/wash buffer and once with 1× PCR buffer containing 2.5 mM MgCl2.
Nested PCR
For nested PCR, the washed beads carrying extension–ligation products were aliquoted so that each tube received DNA corresponding to ∼250 cell-equivalents for analysis of the single-copy endogenous λ1 gene and ∼50 cell-equivalents for analysis of a λ1 transgene present in 6–8 copies per cell (6). Both rounds of nested PCR were carried out in 50 µl reactions containing 1× PCR buffer, 3 mM MgCl2, 0.25 mM dNTPs and 0.8 µl of AmpliTaq DNA polymerase (Perkin Elmer). Primer concentration was 100 nM in the first round of PCR, and 400 nM in the second round. PCR tubes were kept cold and inserted into the block when its temperature reached at least 70°C. For the first PCR, bead-bound extension products were added to the reaction mixture and, following limited PCR [1× (95°C for 2 min, 52°C for 4 min, 72°C for 3 min), 2× (94°C for 45 s, 52°C for 1 min, 72°C for 2 min) and 1× (72°C for 7 min)], beads were removed, reaction products transferred to new PCR tubes and PCR continued as follows: 1× (94°C for 4 min), 28× (94°C for 45 s, 52°C for 1 min, 72°C for 2 min) and 1× (72°C for 7 min). A 2 µl aliquot of the products of the first round of PCR was used for the second round of amplification, which was carried out in a 50 µl reaction, as follows: 1× (94°C for 4 min), 35× (94°C for 45 s, 62°C for 1 min, 72°C for 2 min) and 1× (72°C for 30 min). For analysis by gel electrophoresis, 15 µl of the PCR reaction was resolved by electrophoresis on 1.2% agarose gels. For hybridization, DNA was transferred to Zeta-probe membrane in 0.4 N NaOH and probed with a complementary oligonucleotide that had been 32P-labeled with T4 polynucleotide kinase (New England Biolabs).
Sequencing of PCR products
For sequence analysis, PCR products were purified using the PCR Purification Kit (Qiagen) and then directly sequenced in batch on an ABI 377 sequencer using a primer complementary to genomic sequence. The break position was identified as the junction between the genomic DNA sequence and the linker sequence on the sequencing profile.
RESULTS
Figure 1 presents an overview of the amplification and sequencing protocol. In the first step, a small amount of denatured genomic DNA (2000 cell-equivalents) is annealed to a biotinylated primer specific for the gene of interest and extended with Sequenase. Extension terminates at the site of a break to produce a blunt end. This newly-formed blunt end is tagged by ligation to a linker with one blunt and one staggered end, the extension–ligation products recovered on magnetic streptavidin beads and diluted to a level shown in control experiments to produce approximately four PCR product bands per reaction. The ligation products are then amplified in two rounds of nested PCR, purified using the PCR Purification Kit (Qiagen) and the column eluate directly sequenced on an ABI 377 sequencer. Breaksites are identified as positions at which the characteristic linker sequence appears in the sequencing readout. Multiple breaksites can be readily identified in a single sequencing reaction.
Figure 1.
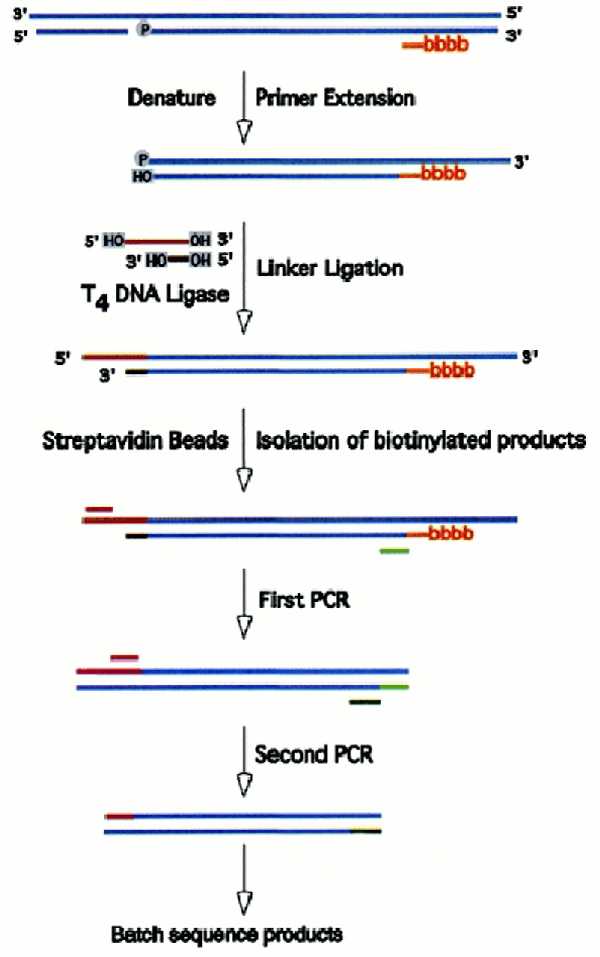
Identification of DNA breaks. DNA is denatured in the presence of a 5′-biotinylated (BBBB) primer specific for the gene of interest and the primer is extended with Sequenase. Synthesis terminates at the 5′-end of the template to produce a duplex with a blunt end. This duplex is ligated to a linker, and biotinylated DNAs are recovered by binding to streptavidin-coated magnetic beads. Following dilution, the extension products are amplified by nested PCR and breaksites identified by batch sequencing. See Materials and Methods for details.
To demonstrate that this method specifically and sensitively detects DNA breaks, control experiments were carried out using DNA from the murine B cell line J558L, which carries a rearranged immunoglobulin λ1 light chain gene. DNA was digested with SpeI to create a double-strand break within the λ1 variable region and the break was tagged with a linker (LL3/LP2) and amplified using a primer set (LRB3, LR4d and LR4e) designed to detect breaks in the top (non-transcribed) strand of the DNA. Figure 2A shows the products produced in duplicate amplification reactions. In the absence of either the input genomic DNA (lanes 1 and 2) or the linker (lanes 3 and 4) there were no visible bands on the agarose gel. Control reactions in which the undigested DNA was used as template for extension produced faint and heterogeneous bands (lanes 5 and 6). Reactions in which the SpeI-digested genomic DNA sample was used as template produced a product of the predicted size, 610 bp (lanes 7 and 8), and sequence analysis showed that the linker had ligated to the 5′-end of the unique SpeI site in the λ1 gene (data not shown). A unique band was also generated when the primer set LFB2, wLF2 and wLF3 was used to identify breaks on the bottom strand (data not shown). Note that faint and heterogeneous products are evident in reactions with either undigested (lanes 5 and 6) or digested (lanes 7 and 8) DNA. We suspect that these products identify sites of nicks that were either present in the living cell or inadvertently introduced during preparation and storage of the DNA template. Consistent with this, the heterogeneous background increases upon prolonged storage or repeated freezing–thawing of the template DNA (data not shown).
Figure 2.

Specificity of amplification and batch sequence analysis. (A) Agarose gel electrophoresis of amplification products. Amplification reactions used the λ1-specific primer set LRB3, LR4d and LR4e to detect top strand breaks. Extension reactions were carried out in the absence of template DNA (lanes 1 and 2) or linker (lanes 3 and 4). Template for extension was genomic DNA (2000 cell-equivalents) from the murine B cell line J558L, either undigested (lanes 3–6) or digested with SpeI (lanes 7 and 8). After the linker ligation step, one-eighth of each extension reaction (250 cell-equivalents of DNA) was amplified by nested PCR and products resolved by agarose gel electrophoresis and stained with ethidium bromide. Lane pairs represent duplicate reactions. M is the 100 bp ladder (Gibco BRL) marker and the intense band is 600 bp. (B) Breaks were amplified in reactions that used as template DNA from primary murine B cells and the primer set LRB, LR2 and LR3. Products were resolved by agarose gel electrophoresis, visualized by ethidium bromide staining (left), then blotted, probed with the 32P-labeled λ1-specific oligonucleotide, LR3a, and autoradiographed (right). (C) A 472 bp fragment from the Jλ1–Cλ1 intron was amplified using primers LF7 and LR4a. The amount of J558L genomic DNA was, from left to right, 2000, 1000, 500, 200, 100 and 50 cell-equivalents. The sample in the right hand lane (B6) was amplified using template genomic DNA from a C57BL/6 mouse, and was estimated to correspond to 500 cell-equivalents by FACS. (D) Products of a single amplification reaction analyzed by gel electrophoresis and ethidium bromide staining. Template DNA was isolated from B cells from mice that carry a λ1 transgene (6) and amplified using the primer set LRB3, LR4d and LR4e. M is the 100 bp ladder (Gibco BRL) marker. Bands a–d correspond to sequenced regions shown in the corresponding panels of Figure 2E. (E) Batch sequence analysis of products of the amplification reaction shown in Figure 2D. Sequence analysis was carried out on an ABI 377 Sequencer, using oligonucleotide LR5b as the primer. The four regions of the sequence trace correspond to bands a–d in Figure 2D, which are 78, 132, 243 and 543 bp in length, respectively, as determined by sequence analysis. The arrows mark the positions at which the linker sequence interrupts the λ1 gene sequence. The linker sequence, GAATTCAGATCTCCATGGTCTAC, can be read easily in the regions of the trace shown in panels c and d, where the linker sequence is shown to the right of the arrow.
Figure 2B demonstrates that the amplification protocol is specific. In this experiment, template DNA was purified from primary murine B cells, extended and amplified using primer set LRB, LR2 and LR3, resolved by agarose gel electrophoresis, blotted and probed with the λ1-specific oligonucleotide, LR3a. Comparison of the profiles produced by ethidium bromide staining (left) or autoradiography (right) shows that essentially every fragment produced in the reaction hybridized to the λ1-specific probe. In the assay shown, DNA concentration was estimated based upon the number of cells in the initial sample. In order to compare levels of breaks in different samples, the starting DNA was quantified relative to a standard as described in Materials and Methods. An example of the PCR quantification is shown in Figure 2C.
Batch sequence analysis was used to identify breaksites in the LM-PCR products. Despite the fact that the LM-PCR products are heterogeneous in length, they all share the identical genomic sequence up to the position at which the linker was inserted. Thus, when sequenced with a primer complementary to the genomic sequence, the products of the sequencing reaction will be identical to one another except at sites where breaks were present in the starting sample, which are tagged by the linkers and easily identified in the sequence trace. In the example shown in Figure 2D and E, DNA breaksites were amplified as described above and analyzed by batch sequencing. Figure 2D shows the products of one amplification reaction analyzed by gel electrophoresis and Figure 2E shows results of batch sequencing of those products on an ABI 377 sequencer. The breaksites corresponding to the 78, 132, 243 and 543 bp LM-PCR products visible in the stained gel (Fig. 2D) are indicated by arrows in the sequence trace (Fig. 2E).
DISCUSSION
Batch mapping is a rapid and quantitative method for assay and identification of DNA breaksites in the mammalian genome. We describe its use in mapping single-strand DNA breaks, but a very similar approach can be used to map double-strand DNA breaks. Two features of this method represent valuable refinements of the LM-PCR protocols in general use. First, the use of nested PCR primers increases specificity of DNA amplification. Second, direct sequencing of PCR products in batch allows precise determination of multiple break points in each reaction without tedious and costly steps of gel purification or cloning. This improves the speed and throughput of the assay.
This method requires very little DNA template. DNA was routinely isolated from 10 000 cells, initial extension reactions were carried out on DNA from 2000 cell-equivalents and bead-bound extension products aliquoted into reactions containing 250 cell-equivalents for PCR amplification. Batch mapping of breaksites can therefore be carried out on cell types that are of low abundance but can be recovered by microdissection or by cell sorting. We have ourselves used this method to identify breaksites in immunoglobulin genes of hypermutating B cells, isolated by fluorescence-activated cell sorter We have ourselves used this method to identify breaksites in immunoglobulin genes of hypermutating B cells isolated by fluorescence activated cell sorting (FACS) (7). In these experiments, we used the primers and conditions described herein to show that there is a 2- to 3-fold excess of breaks in λ1 genes of hypermutating B cells relative to non-hypermutating B cells, and that 1.3% of germinal center B cells contain breaks in the λ1 genes that are associated with hypermutation. Batch mapping should also be applicable to breaksites in other cell types carrying out active recombination or repair.
Acknowledgments
ACKNOWLEDGEMENT
This research was supported by NIH grant R01 GM41712.
References
- 1.Pfeifer G.P., Steigerwald,S.D., Mueller,P.R., Wold,B. and Riggs,A.D. (1989) Genomic sequencing and methylation analysis by ligation mediated PCR. Science, 246, 810–813. [DOI] [PubMed] [Google Scholar]
- 2.Mueller P.R. and Wold,B. (1989) In vivo footprinting of a muscle specific enhancer by ligation mediated PCR. Science, 246, 780–786. [DOI] [PubMed] [Google Scholar]
- 3.Steigerwald S.D., Pfeifer,G.P. and Riggs,A.D. (1990) Ligation-mediated PCR improves the sensitivity of methylation analysis by restriction enzymes and detection of specific DNA strand breaks. Nucleic Acids Res., 18, 1435–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tormanen V.T., Swiderski,P.M., Kaplan,B.E., Pfeifer,G.P. and Riggs,A.D. (1992) Extension product capture improves genomic sequencing and DNase I footprinting by ligation-mediated PCR. Nucleic Acids Res., 20, 5487–5488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pfeifer G.P. and Riggs,A.D. (1993) Genomic footprinting by ligation-mediated polymerase chain reaction. Methods Mol. Biol., 15, 153–168. [DOI] [PubMed] [Google Scholar]
- 6.Kong Q., Zhao,L., Subbaiah,S. and Maizels,N. (1998) A λ 3′ enhancer drives active and untemplated somatic hypermutation of a λ1 transgene. J. Immunol ., 161, 294–301. [PubMed] [Google Scholar]
- 7.Kong Q. and Maizels,N. (2001) DNA breaks in hypermutating immunoglobulin genes: evidence for a break and repair pathway of somatic hypermutation. Genetics, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]