

Abstract
Recombinant human tissue plasminogen activator (rPA) is a truncated version of tissue plasminogen activator (tPA), which contains nine disulfide bonds and is prone to forming inactive inclusion bodies when expressed in bacteria. To obtain functional rPA expression, we displayed the rPA on the surface of polyhydroxybutyrate (PHB) granules using phasin as the affinity tag. rPA was fused to the N terminus of the phasin protein with a thrombin cleavage site as the linker. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblot analysis showed that rPA fusion was successfully displayed on the surface of PHB granules. An activity assay indicated that the rPA fusion is active. The in vivo surface display strategy for functional rPA expression in Escherichia coli is distinct for its efficient folding and easier purification and may be expanded to the expression of other eukaryotic proteins with complex conformation.
Tissue plasminogen activator (tPA) derives from a fibrinolytic system of blood vessel endothelial cells, activates plasminogen to form plasmin, and is an effective drug for thrombolytic therapy. Native tPA is composed of 527 amino acid residues with five structural domains and 17 disulfide bonds (19). Recombinant human tissue plasminogen activator (rPA) is a variant version of tPA with nine disulfide bonds, consisting of kringle 2 and serine protease domain (12). rPA was confirmed to possess enhanced capability for thrombolysis compared with that of tPA. Therefore, rPA is more beneficial for the treatment of acute myocardial infarction (17, 26, 28).
Heterologous expression of tPA as well as rPA in Escherichia coli often results in the formation of the insoluble aggregates known as inclusion bodies due to the multidisulfide bonds (3). The refolding of the inclusion bodies in vitro is a long and difficult task, especially for proteins with complex conformation and multiple disulfide bonds. In order to obtain directly the functional rPA from recombinant E. coli, many approaches have been utilized: expressing the rPA gene in E. coli trxB gor ahpC* mutant strains, of which the cytoplasm is highly oxidized; fusing the rPA gene with gpIII of ΦM13 and linking to the OmpA signal sequence, through which rPA is secreted into the medium; exploiting the novel twin-arginine translocation (Tat) pathway to obtain active rPA in the periplasmic space based on its inherent properties; and cosecreting of rPA with chaperones and adding low-molecular-size medium additives to promote the formation of disulfide bonds (6, 11, 15, 25). However, the successful expression of rPA in its soluble or active form gives rise to another task: separation and purification of soluble active rPA from large amounts of other proteins in cytoplasm or medium.
Normal protein purification typically involves several chromatographic steps. Each step can be costly and time-consuming (4). The development of simple and reliable methods for protein purification, which can be applied to arbitrary products, is therefore an important goal in bioseparation technology developments. One method that was recently developed is the addition of an affinity tag sequence to the target protein gene (13). It was demonstrated that heterologous proteins can be displayed actively on the surface of biopolyester granules in E. coli by fusing to the polyhydroxyalkanoate (PHA) synthase (PhaC), which serves as an affinity tag of PHA granules (21). PHA granules are carbon inclusions produced intracellularly by bacteria for coping with changing, often oligotrophic environments (1). These inclusions are composed of a hydrophobic polyester core and hydrophilic phospholipid membrane with many embedded proteins (24). Besides PhaC, phasins (namely PhaPs) are the main proteins tightly attached to the surface of polyhydroxybutyrate (PHB) granules, which can stabilize and prevent coalescence of separate PHB granules (22). Due to the inherent properties, PhaP has been used as the affinity tag in vivo to display recombinant proteins on the surface of PHB granules (5).
In this study, we fused the rPA gene to the N terminus of phaP. A thrombin cleavage site was introduced between them to release rPA from PHB granules. The fusion gene was then expressed in engineered E. coli, which was conferred with the PHB production pathway by cloning the PHB biosynthesis genes. We confirmed that recombinant rPA fusion was able to be actively expressed in vivo on the surface of PHB granules.
MATERIALS AND METHODS
Strains and materials.
E. coli strain DH5α [F− endA1 hsdR17(rK− mK+) supE44 thi-l λ− recA1 gyrA96 ΔlacU169 (φ80dlacZΔM15)] was used as the host for recombinant DNA manipulation. E. coli strain XL1-Blue [recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac F′ proAB lacIqZΔM15 Tn10 (Tetr)] from Stratagene was used for recombinant protein expression. Plasmid pHBS01, carrying the phbCAB operon for PHB biosynthesis, was constructed previously in our laboratory (9). Plasmid pBluescript SK− from Stratagene was used to construct the fusion expression vector. Restriction endonucleases and T4 ligase were purchased from MBI Fermentas (Canada). The rPA standard was obtained from IND Pharm Co. Ltd. (Beijing, People's Republic of China). Other reagents for the rPA activity assay were obtained from Sigma (USA).
Construction of the fusion expression vector.
The gene phaP encoding phasin was amplified from genomic DNA of Ralstonia eutropha H16 (kind gift of Alexander Steinbüechel, Germany) using primers phaP-F (5′-GCGGAATTCATGATCCTCACCCCGGAACAAG-3′) (EcoRI site in italics) and phaP-R (5′-ACAAAGCTTGGCAGCCGTCGTCTTCTTTGCCGT-3′) (HindIII site in italics). The PCR conditions were as follows: denaturation at 94°C for 5 min, followed by 29 cycles of 94°C for 30 s, 58°C for 30 s, and 72°C for 40 s, with a final elongation at 72°C for 10 min. The PCR fragment was then digested with EcoRI/HindIII and ligated into plasmid pBluescript SK−. The resulting plasmid, named pSKPA, was then ligated with the rPA gene amplified from a stocked plasmid containing the rPA gene. The forward primer was 5′-ACCAAGCTTCTGGTTCCGCGTGGATCCTCTTATCAGGGAAACAGTGACT-3′ (where the HindIII site is in italics and the thrombin cleavage site is underlined), and the reverse primer was 5′-AAACTCGAGTCACGGTCGCATGTTGTCACGAA-3′ (XhoI site in italics). The PCR conditions consisted of a 5-min incubation at 94°C, followed by 29 cycles of 94°C for 30s, 58°C for 30s, and 72°C for 70s, and a final extension at 72°C for 10 min. The PCR product was then digested and subcloned into the HindIII/XhoI sites of the pSKPA plasmid. The final plasmid, named pSPAr, containing the rPA gene and the thrombin cleavage site between PhaP and rPA, was transformed into E. coli XL1-Blue for expression. All plasmids constructed in the study were confirmed by DNA sequencing.
Expression of the fusion protein.
Recombinant E. coli XL1-Blue cells containing plasmids pHBS01and pSPAr were selected on a Luria-Bertani (LB) agar plate containing 100 μg/ml spectinomycin and 100 μg/ml ampicillin. A single clone was precultured in 5 ml Luria-Bertani medium (1% tryptone, 0.5% yeast extract, and 1% NaCl) at 37°C and 200 rpm overnight. Then, 1 ml overnight cells were inoculated into 100 ml Luria-Bertani medium supplemented with 5 g/liter glucose (wt/vol) and antibiotics. The culture was first cultivated at 37°C until glucose was almost fully consumed. IPTG (isopropyl-β-d-thiogalactopyranoside) was then added to a final concentration of 1 mM. Cells were incubated at 30°C and 180 rpm for an additional 12 h and harvested by centrifugation at 5,000 × g at 4°C for 10 min.
SDS-PAGE analysis.
Harvested cells were washed three times with PBS buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, 0.05% Tween 20, pH 7.4) and resuspended in the same buffer, followed by ultrasonication at 4°C (40% output, 10 min). After centrifugation at 14,000 × g at 4°C for 30 min, the sediments were resuspended in 1 ml PBS buffer, and the PHB granules were isolated by ultracentrifugation (2). The collected PHB granules and/or cell lysate were analyzed by 12% (vol/vol) SDS-PAGE. The gels were stained with Coomassie Brilliant G250.
Western blot analysis.
After SDS-PAGE, the protein bands on the gel were transferred onto a polyvinylidene difluoride (PVDF) membrane. After being washed with TBST (10 mM Tris, pH 8.0, 150 mM NaCl, 0.05% Tween 20), the membrane was blocked with 3% (wt/vol) bovine serum albumin (BSA) for 1 h at room temperature and then incubated with tissue plasminogen activator antibody (ab27418; ABCam) overnight at 4°C. The horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibody (diluted 1:50,000) was then added, and the mixture was incubated at room temperature for 1 h. The membrane was washed three times with 20 ml TBST for 10 min each. The blots were detected with DAB (3,3′-diaminobenzidine tetrahydrochloride)-NiCl solution in darkness followed by the addition of distilled water to stop the reaction.
Activity assay for the recombinant protein.
Plasminogen activation of rPA was detected on an agarose-fibrin plate according to a slightly modified method described previously (8). The agarose-fibrin plate was prepared as follows. A total of 1.4 IU thrombin, 140 μg plasminogen, and 13.2 mg human fibrinogen was added into 20 ml of 0.8% agarose gels dissolved in normal saline at 55°C to 60°C. The mixture was incubated at room temperature for 30 min. The sample was then loaded, and the plate was incubated at 37°C. To quantify rPA in samples, the standard is serially diluted and spotted onto the fibrin/plate for activity assay. A logarithmic curve is then constructed with the diameter of the lytic zone as the ordinate and the activity as the abscissa (based on the formula Y = a+bX, where Y is the logarithm of the diameter, X is the logarithm of the activity, a is the intercept of Y, and b is the slope). The concentration of rPA is calculated from the standard curve.
Release of rPA from the PHB granules.
To release rPA from PHB granules, the isolated PHB granules with PhaP-rPA fusion protein were treated with thrombin (Novagen) at 20°C in PBS buffer for 12 h. After the reaction, the mixture was centrifuged at 14,000 × g at 4°C for 30 min. The supernatant was analyzed by 12% SDS-PAGE.
RESULTS
Selection of PHB accumulation strains.
For PHB granule biosynthesis, the phbCAB operon from R. eutropha H16 containing the three essential genes, which encode PHB synthase (PhbC), β-ketothiolase (PhbA), and acetoacetyl-coenzyme A (acetoacetyl-CoA) reductase (PhbB), was inserted into a low-copy-number plasmid pSCP (9) to form the plasmid pHBS01, where the phbCAB operon was under the control of a stress-induced promoter. The stress-induced promoter was developed previously in our lab and allowed the recombinant E. coli to begin to accumulate PHB at the late exponential growth phase without the addition of any inducer (10). In order to obtain suitable PHB accumulation for functional rPA gene expression, E. coli BL21(DE3) and E. coli XL1-Blue were selected as the expression host candidates. The strains exhibited different PHB accumulation when they were cultivated in LB medium plus 5 g/liter glucose for 12 h (Fig. 1 A). E. coli XL1-Blue/pHBS01 accumulated 25% PHB of the cell dry weight, whereas E. coli BL21(DE3)/pHBS01 accumulated only about 9% (wt/wt). Moreover, the PHB granules in E. coli XL1-Blue were relatively small and well proportioned (Fig. 1B). Moderate PHB accumulation with small granule size will offer maximum surface area and will be beneficial to the heterologous protein expression. Besides, it has been reported that PhaP can affect the size and morphology of PHB granules, with overexpression of PhaP resulting in many small PHB granules (22). The final biomass of E. coli XL1-Blue/pHBS01 was nearly twice as much as that of E. coli BL21(DE3)/pHBS01, which is also important for heterologous protein expression (Fig. 1A).
FIG. 1.

Cell growth and PHB accumulation of recombinant E. coli. The strains were cultivated in LB medium supplemented with 0.5% glucose. (A) Comparison of PHB content and cell mass in E. coli BL21/pHBS01 and XL1-Blue/pHBS01. (B) Fluorescence micrograph of E. coli XL1-Blue/pHBS01 cultivated for 12 h. Arrows indicate the small PHB granules in cells. Scale bar, 5 μm.
Display of rPA on the surface of intracellular PHB granules.
To display rPA on the surface of intracellular PHB granules, the rPA gene was fused to the N terminus of phaP with a linker-including thrombin cleavage site. PhaP is a noncatalytic protein consisting of a hydrophobic domain, which associates with the PHB granule surface, and of a hydrophilic domain exposed to the cytoplasm of the cell (23). The thrombin cleavage site enables the easy and specific removal of rPA from the fusion protein after PHB granule isolation. The resulting fusion gene was introduced into recombinant E. coli XL1-Blue already harboring the PHB production system. Then the engineered E. coli XL1-Blue (pHBS01/pSPAr) was cultivated in LB medium supplied with 5 g/liter glucose. The expression of Phap-rPA fusion was subjected to SDS-PAGE analysis (Fig. 2). An additional protein band at 60 kDa, which is the predicted molecular size of the fusion protein, was clearly visible, whereas it could not be detected in the control. Isolation of the PHB granules by ultracentrifugation showed that this additional band was in the insoluble fraction, indicating that it was anchored on the intracellular PHB granules. The harvested PHB granules were then analyzed by Western blotting using the tissue plasminogen activator antibody (Fig. 3). The cross-reaction at the corresponding region confirmed that the additional protein anchored on the PHB granule surface is a Phap-rPA fusion.
FIG. 2.
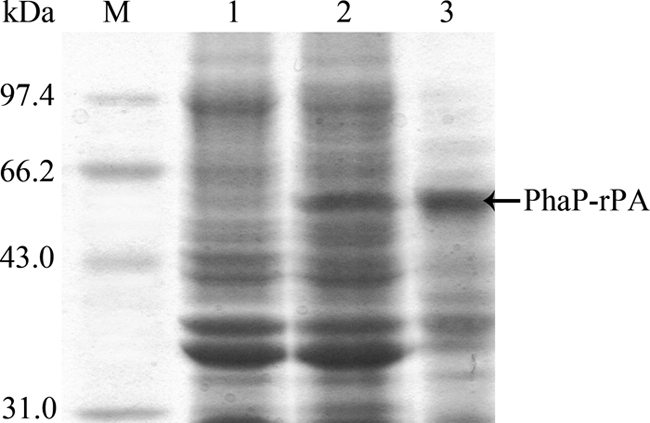
SDS-PAGE analysis of the PhaP-rPA fusion protein. Lane M, protein marker; lane 1, control; lane 2, lysate of E. coli XL1-Blue (pHBS01/pSPAr); lane 3, PHB granules isolated from E. coli XL1-Blue (pHBS01/pSPAr).
FIG. 3.
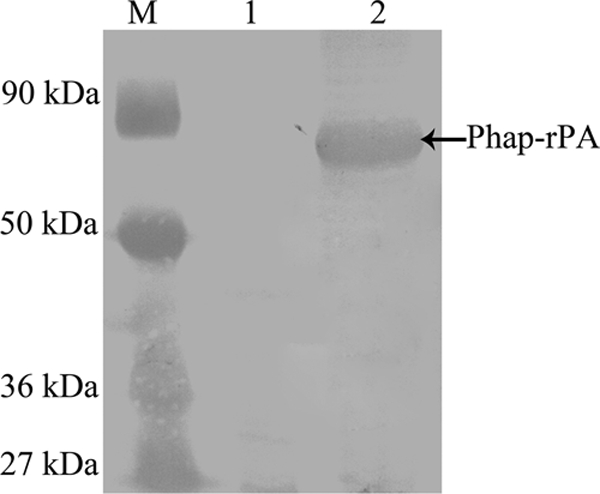
Western blot analysis of the PhaP-rPA fusion protein. Lane M, protein marker; lane 1, control; lane 2, PHB granules isolated from E. coli XL1-Blue (pHBS01/pSPAr).
Activity evaluation of the Phap-rPA fusion.
Functional expression of protein with multiple disulfide bonds, such as rPA, in E. coli has proved to be a challenge. To see if our constructed rPA in the fusion which attached to the surface of PHB granules is properly folded and to see if it is active, the fibrin plate analysis method was used. Plasminogen was used as the substrate of rPA in the medium of the plate. Active rPA can convert plasminogen into plasmin, which will digest the fibrin, resulting in a clear lytic zone on the fibrin/agar plate. To do this, we applied the isolated PHB granules with Phap-rPA fusion on the fibrin plates and incubated them at 37°C for phenotype observation. As indicated in Fig. 4, there is a clear lytic zone on the plate where isolated PHB granules were supplied, indicating the degradation of the fibrin, whereas there is no such lytic zone on the control. The results confirmed that the Phap-rPA fusion attached to the PHB granule surface was active and was able to convert the inactive plasminogen to active plasmin. Our control experiment showed that E. coli expressing only the rPA gene could not obtain functional rPA in the cell lysate.
FIG. 4.

Fibrin plate analysis of rPA activity. Equal amounts of the samples were spotted on the fibrin plate. (A) Cell debris from E. coli XL1-Blue (pSCP/pSPAr). (B) Isolated PHB granules from E. coli XL1-Blue (pHBS01/pSPAr).
Release of rPA from the PHB granule.
To determine if the thrombin site is readily accessible, the PHB granules with PhaP-rPA fusion were incubated in phosphate buffer with thrombin for 12 h. After incubation, the reaction mixture was centrifuged and the supernatant was analyzed with SDS-PAGE. As shown in Fig. 5, a clear band of about 39 kDa, corresponding to the molecular mass of rPA, can be clearly observed. As we expected, the released rPA is quite pure with only a very faint contamination band (Fig. 5). Fibrin plate analysis showed that this released rPA from PHB granules has activity comparable with that of standard rPA. The results demonstrated that correctly folded rPA as a fusion with PhaP can be completely removed by thrombin treatment with retained activity. In addition, the rPA preparation process can be simplified by thrombin treatment of the washed cell debris. No PHB granule isolation process is needed (data not shown).
FIG. 5.
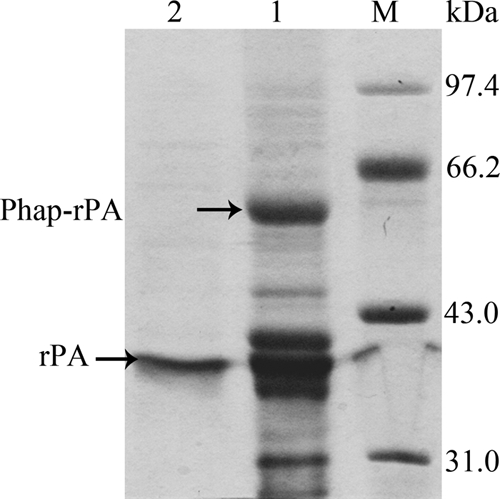
SDS-PAGE analysis of rPA released from the PHB granules. Lane M, protein marker; lane 1, isolated PHB granules with Phap-rPA fusions; lane 2, supernatant after cleavage with thrombin for 12 h.
Optimization of rPA expression.
Since the PHB accumulation in our strains is induced automatically when the cells entered the late exponential growth phase, the induction time for Phap-rPA expression was critical. We compared the different induction times for Phap-rPA fusion expression (Fig. 6). Induction at the late exponential growth phase was the optimal condition. The rPA activity induced at this time can achieve nearly twice that induced at the early exponential phase, reaching 760 IU/g cdw (cell dry weight) (Fig. 6). GC analysis showed that PHB accumulation in the cells is changed with different rPA induction times. When expression was induced at the early exponential growth phase, the intracellular PHB accumulation was at a very low level, corresponding to about 2.5% of the cell dry weight. When expression was induced at the late exponential growth phase, the PHB accumulation reached 23% (wt/wt), which is close to the maximum accumulation (Fig. 1A). These results also agreed well with previous results that the PHB biosynthesis genes are maximally induced at the late exponential growth phase (10). According to standard rPA, we assessed that the yield of rPA was about 53.8 μg/g cells.
FIG. 6.

Relationship of rPA activity and PHB accumulation. (A) rPA gene expression was induced at the early exponential growth phase (4 h). (B) rPA gene expression was induced at the midexponential growth phase (8 h). (C) rPA gene expression was induced at the late exponential growth phase (12 h). E. coli XL1-Blue (pHBS01/pSPAr) was grown in LB medium plus 0.5% glucose and induced at indicated time points. After induction, the cells were cultivated for a further 10 h. rPA activity was expressed as IU per gram of cell dry weight (IU/g cdw).
DISCUSSION
Under physiological conditions, the reductive cytoplasmic environment of wild-type E. coli is the bottleneck of the oxidative folding of heterologous proteins with multiple disulfide bonds. Expression of rPA in E. coli is a typical example of this phenomenon, often resulting in unfolded or misfolded inactive protein (12). Although many strategies have been applied to express heterologous proteins with multiple disulfide bonds in E. coli (16), it is thorny to seek out an appropriate pathway to achieve active rPA expression in wild-type E. coli. Previous studies have focused on improving the oxidation status of the cytoplasm of E. coli or choosing the periplasmic space, which is a more oxidized space, for active rPA expression (6, 18). However, because of the narrow periplasmic space, especially the complicated and expensive purification process, these methods are confined to small-scale study in the laboratory.
In the study, we presented a novel method for functional rPA expression. By fusion to the affinity tag of PhaP protein from R. eutropha H16, we expressed the functional PhaP-rPA fusion on the intracellular surface of the PHB granules coupled with in vivo PHB accumulation. In general, the oxidation of cysteine thiols in cytoplasmic proteins is strongly disfavored for both thermodynamic and kinetic reasons, because the thiol-disulfide redox potential of the cytoplasm is too low to provide a sufficient driving force for the formation of stable disulfide (6). The biosynthesis of PHB granules in vivo is a NADPH-dependent pathway (14) and therefore may serve as an electron sink for the reducing power (7). During the PHB biosynthesis process, electrons are no longer able to traverse the electron transfer chain to oxygen at the same rate and thus are diverted to the reductive step of polymer synthesis. Thus, we suggested that PHB biosynthesis in vivo provides the cytoplasm a suitable and possibly more oxidized environment for disulfide bond formation (Fig. 7). Both PhaP-rPA synthesis and PHB accumulation happening near the PHB granule may also enhance this effect. Meanwhile, efficient folding of rPA may also be due to the surface display, which provides a space for protein folding and reduces the opportunity for proteins to interact with each other. The PhaP-rPA proteins are anchored onto the surface of PHB granules as soon as they are synthesized, which facilitates the proper formation of disulfide bonds and proper folding of the protein.
FIG. 7.

Schematic illustration of in vivo surface display on the PHB granules. A PHB biosynthesis pathway is shown to explain NADPH consumption.
Several reports have been published based on the development of the PHB granule-based expression system (4, 20, 27). Phasin, as the major protein located on the surface of PHB granules, binds tightly to the granules. Display of heterologous protein on the intracellular PHB granule surface using phasin as the affinity tag was confirmed to be a successful and efficient strategy. This method is distinct due to the simple recovery of the target protein and self-contained nature of the system. We found that the system is especially useful for expression of complex proteins with multiple disulfide bonds, such as rPA. The simultaneous PHB biosynthesis and protein expression provide the system inimitable advantages over other strategies.
Conclusions.
In summary, we described a novel method for functional rPA expression in E. coli. To our knowledge, it is the first successful example of displaying active rPA on the intracellular PHB granule surface. PHB biosynthesis and granule display provided a suitable environment for the expression and proper folding of rPA. The self-contained nature of the system and the simple recovery of the target protein are other key advantages of the system. This system may be applied to the heterologous expression of other eukaryotic proteins in E. coli.
Acknowledgments
This work was financially supported by a grant from the National Natural Science Foundation of China (30870022).
Footnotes
Published ahead of print on 10 September 2010.
REFERENCES
- 1.Anderson, A. J., and E. A. Dawes. 1990. Occurrence, metabolism, metabolic role, and industrial uses of bacterial polyhydroxyalkanoates. Microbiol. Rev. 54:450-472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Backstrom, B. T., J. A. Brockelbank, and B. H. Rehm. 2007. Recombinant Escherichia coli produces tailor-made biopolyester granules for applications in fluorescence activated cell sorting: functional display of the mouse interleukin-2 and myelin oligodendrocyte glycoprotein. BMC Biotechnol. 7:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baneyx, F., and M. Mujacic. 2004. Recombinant protein folding and misfolding in Escherichia coli. Nat. Biotechnol. 22:1399-1408. [DOI] [PubMed] [Google Scholar]
- 4.Banki, M. R., T. U. Gerngross, and D. W. Wood. 2005. Novel and economical purification of recombinant proteins: intein-mediated protein purification using in vivo polyhydroxybutyrate (PHB) matrix association. Protein Sci. 14:1387-1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barnard, G. C., J. D. McCool, D. W. Wood, and T. U. Gerngross. 2005. Integrated recombinant protein expression and purification platform based on Ralstonia eutropha. Appl. Environ. Microbiol. 71:5735-5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bessette, P. H., F. Aslund, J. Beckwith, and G. Georgiou. 1999. Efficient folding of proteins with multiple disulfide bonds in the Escherichia coli cytoplasm. Proc. Natl. Acad. Sci. U. S. A. 96:13703-13708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dawes, E. A. 1988. Polyhydroxybutyrate: an intriguing biopolymer. Biosci. Rep. 8:537-547. [DOI] [PubMed] [Google Scholar]
- 8.Granelli-Piperno, A., and E. Reich. 1978. A study of proteases and protease-inhibitor complexes in biological fluids. J. Exp. Med. 148:223-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang, Z., Y. P. Geng, Y. Z. Xia, J. H. Kang, and Q. S. Qi. 2009. Engineering Escherichia coli for an efficient aerobic fermentation platform. J. Biotechnol. 144:58-63. [DOI] [PubMed] [Google Scholar]
- 10.Kang, Z., Q. Wang, H. Zhang, and Q. Qi. 2008. Construction of a stress-induced system in Escherichia coli for efficient polyhydroxyalkanoates production. Appl. Microbiol. Biotechnol. 79:203-208. [DOI] [PubMed] [Google Scholar]
- 11.Kim, J. Y., E. A. Fogarty, F. J. Lu, H. Zhu, G. D. Wheelock, L. A. Henderson, and M. P. DeLisa. 2005. Twin-arginine translocation of active human tissue plasminogen activator in Escherichia coli. Appl. Environ. Microbiol. 71:8451-8459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kohnert, U., R. Rudolph, J. H. Verheijen, E. J. Weening-Verhoeff, A. Stern, U. Opitz, U. Martin, H. Lill, H. Prinz, M. Lechner, et al. 1992. Biochemical properties of the kringle 2 and protease domains are maintained in the refolded t-PA deletion variant BM 06.022. Protein Eng. 5:93-100. [DOI] [PubMed] [Google Scholar]
- 13.LaVallie, E. R., and J. M. McCoy. 1995. Gene fusion expression systems in Escherichia coli. Curr. Opin. Biotechnol. 6:501-506. [DOI] [PubMed] [Google Scholar]
- 14.Lee, I. Y., M. K. Kim, Y. H. Park, and S. Y. Lee. 1996. Regulatory effects of cellular nicotinamide nucleotides and enzyme activities on poly(3-hydroxybutyrate) synthesis in recombinant Escherichia coli. Biotechnol. Bioeng. 52:707-712. [DOI] [PubMed] [Google Scholar]
- 15.Manosroi, J., C. Tayapiwatana, F. Gotz, R. G. Werner, and A. Manosroi. 2001. Secretion of active recombinant human tissue plasminogen activator derivatives in Escherichia coli. Appl. Environ. Microbiol. 67:2657-2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Messens, J., and J. F. Collet. 2006. Pathways of disulfide bond formation in Escherichia coli. Int. J. Biochem. Cell Biol. 38:1050-1062. [DOI] [PubMed] [Google Scholar]
- 17.Nordt, T. K., and C. Bode. 2003. Thrombolysis: newer thrombolytic agents and their role in clinical medicine. Heart 89:1358-1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Obukowicz, M. G., M. E. Gustafson, K. D. Junger, R. M. Leimgruber, A. J. Wittwer, T. C. Wun, T. G. Warren, B. F. Bishop, K. J. Mathis, D. T. McPherson, et al. 1990. Secretion of active kringle-2-serine protease in Escherichia coli. Biochemistry 29:9737-9745. [DOI] [PubMed] [Google Scholar]
- 19.Pennica, D., W. E. Holmes, W. J. Kohr, R. N. Harkins, G. A. Vehar, C. A. Ward, W. F. Bennett, E. Yelverton, P. H. Seeburg, H. L. Heyneker, D. V. Goeddel, and D. Collen. 1983. Cloning and expression of human tissue-type plasminogen activator cDNA in E. coli. Nature 301:214-221. [DOI] [PubMed] [Google Scholar]
- 20.Peters, V., and B. H. A. Rehm. 2006. In vivo enzyme immobilization by use of engineered polyhydroxyalkanoate synthase. Appl. Environ. Microbiol. 72:1777-1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peters, V., and B. H. A. Rehm. 2005. In vivo monitoring of PHA granule formation using GFP-labeled PHA synthases. FEMS Microbiol. Lett. 248:93-100. [DOI] [PubMed] [Google Scholar]
- 22.Pötter, M., M. H. Madkour, F. Mayer, and A. Steinbüchel. 2002. Regulation of phasin expression and polyhydroxyalkanoate (PHA) granule formation in Ralstonia eutropha H16. Microbiology 148:2413-2426. [DOI] [PubMed] [Google Scholar]
- 23.Pötter, M., and A. Steinbüchel. 2005. Poly(3-hydroxybutyrate) granule-associated proteins: impacts on poly(3-hydroxybutyrate) synthesis and degradation. Biomacromolecules 6:552-560. [DOI] [PubMed] [Google Scholar]
- 24.Rehm, B. H. A., and A. Steinbüchel. 1999. Biochemical and genetic analysis of PHA synthases and other proteins required for PHA synthesis. Int. J. Biol. Macromol. 25:3-19. [DOI] [PubMed] [Google Scholar]
- 25.Schaffner, J., J. Winter, R. Rudolph, and E. Schwarz. 2001. Cosecretion of chaperones and low-molecular-size medium additives increases the yield of recombinant disulfide-bridged proteins. Appl. Environ. Microbiol. 67:3994-4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smalling, R. W. 1997. Pharmacological and clinical impact of the unique molecular structure of a new plasminogen activator. Eur. Heart J. 18(suppl. F):F11-F16. [DOI] [PubMed] [Google Scholar]
- 27.Wang, Z., H. Wu, J. Chen, J. Zhang, Y. Yao, and G. Q. Chen. 2008. A novel self-cleaving phasin tag for purification of recombinant proteins based on hydrophobic polyhydroxyalkanoate nanoparticles. Lab Chip 8:1957-1962. [DOI] [PubMed] [Google Scholar]
- 28.Wooster, M. B., and A. B. Luzier. 1999. Reteplase: a new thrombolytic for the treatment of acute myocardial infarction. Ann. Pharmacother. 33:318-324. [DOI] [PubMed] [Google Scholar]