

Abstract
Fragments of injured axons that detach from their cell body break down by the molecularly regulated process of Wallerian degeneration (WD). Although WD resembles local axon degeneration, a common mechanism for refining neuronal structure, several previously examined instances of developmental pruning were unaffected by WD pathways. We used laser axotomy and time-lapse confocal imaging to characterize and compare peripheral sensory axon WD and developmental pruning in live zebrafish larvae. Detached fragments of single injured axon arbors underwent three stereotyped phases of WD: a lag phase, a fragmentation phase and clearance. The lag phase was developmentally regulated, becoming shorter as embryos aged, while the length of the clearance phase increased with the amount of axon debris. Both cell-specific inhibition of ubiquitylation and overexpression of the Wallerian degeneration slow protein (WldS) lengthened the lag phase dramatically, but neither affected fragmentation. Persistent WldS-expressing axon fragments directly repelled regenerating axon branches of their parent arbor, similar to self-repulsion among sister branches of intact arbors. Expression of WldS also disrupted naturally occurring local axon pruning and axon degeneration in spontaneously dying trigeminal neurons: although pieces of WldS-expressing axons were pruned, and some WldS-expressing cells still died during development, in both cases detached axon fragments failed to degenerate. We propose that spontaneously pruned fragments of peripheral sensory axons must be removed by a WD-like mechanism to permit efficient innervation of the epidermis.
Keywords: Wallerian, Axon, Degeneration, Pruning, Regeneration, Zebrafish
INTRODUCTION
In both the peripheral and central nervous systems, axon branches that are severed from their cell bodies degenerate by a mechanistically conserved, molecularly regulated process, termed Wallerian degeneration (WD) (Coleman and Freeman, 2010; Waller, 1850). The discovery of the spontaneous mouse mutant, Wallerian degeneration slow (WldS), led to the acceptance of WD as an active, regulated process. In WldS mice, axon fragmentation is dramatically delayed compared with wild-type axons subjected to identical injury (Lunn et al., 1989). The WldS phenotype results from a genomic rearrangement that creates a fusion protein comprising the first 70 amino acids of Ube4b (N70), a multi-ubiquitylation factor, and full-length Nmnat1, an enzyme involved in the NAD salvage pathway (Conforti et al., 2000; Mack et al., 2001). Although the neuroprotective mechanism of WldS is not fully understood, both enzymatically active Nmnat and a domain at the N terminus of N70 are required (Avery et al., 2009; Conforti et al., 2009). A recent study identified the labile protein Nmnat2 as an endogenous axon survival factor for which WldS can substitute (Gilley and Coleman, 2010).
The ubiquitin/proteasome system (UPS) also acts cell-autonomously to regulate axon degeneration. Inhibiting the UPS decreases the rate of WD in mice, Drosophila and in vitro (Zhai et al., 2003). Moreover, developmentally programmed mushroom body pruning in Drosophila is inhibited by UPS loss-of-function mutations, demonstrating a role for the UPS in both injury-induced and developmentally programmed degeneration (Hoopfer et al., 2006; Watts et al., 2003). It has recently been suggested that the UPS may act in WD by degrading Nmnat2 to trigger degeneration (Gilley and Coleman, 2010).
The existence of a molecular program for axon degeneration implies that rapid removal of axon fragments provides animals with a selective advantage. Indeed, WldS mice with abnormally persistent axon fragments are compromised in their ability to recover from injury (Bisby and Chen, 1990; Brown et al., 1992), consistent with the notion that removal of axon debris facilitates regeneration. Axon debris may repel or physically block regenerating axons, but this phenomenon has not yet been directly observed. Moreover, contrary to this idea, regeneration of some axons is not impeded by axon fragments (Wang and Macagno, 1998), and some regenerating axons can even fuse with the fragment, reconnecting it to its parent arbor (Bedi and Glanzman, 2001; Ghosh-Roy et al., 2010).
A developmental function for the WD pathway could also exert selective pressure for its evolution. Many developing neurons refine their dendritic and axonal arbors via selective local degeneration (Luo and O'Leary, 2005). This type of refinement occurs as part of activity-influenced competition, developmentally programmed remodeling and sporadic spontaneous pruning. Developmentally programmed or spontaneous sporadic neuronal death is another common mechanism for refining neural circuits. For example, massive cell death is a normal feature of mammalian dorsal root ganglion (DRG) development (Bennett, 2002). Despite its potent neuroprotective activity in injured axons, and the morphological similarity between injury-induced WD and local axon degeneration, WldS expression has no effect on refinement of mouse RGC projections, pruning of cortical layer 5 axons, or remodeling of Drosophila mushroom body neurons undergoing metamorphosis (Hoopfer et al., 2006). A recent study provided the first evidence that WldS can affect development: the programmed remodeling of Drosophila peripheral sensory dendrites during metamorphosis was delayed when the WldS gene was expressed in these neurons (Schoenmann et al., 2010). However, a developmental function for WldS has not yet been shown in axons or during sporadic pruning. Similarly, although WldS expression can protect degenerating axons in some pathologically dying cells (Saxena and Caroni, 2007), a role for WldS in spontaneous developmental death has not been described.
Local axon degeneration occurs sporadically in the developing cutaneous terminal arbors of zebrafish trigeminal and Rohon-Beard peripheral sensory neurons (Sagasti et al., 2005). These local degeneration events may serve to eliminate aberrant overlap between axon branches. Death of zebrafish trigeminal neurons also occurs spontaneously and removes abnormally positioned neurons (Knaut et al., 2005). Here we studied the relationship between WD, local axon pruning and axon degeneration during spontaneous cell death in zebrafish trigeminal neurons. This model allowed us to observe the cellular progress of WD at high resolution, to investigate the involvement of molecular pathways and to determine the functional consequences of delayed WD. Overexpressing WldS or inhibiting the UPS robustly lengthened the lag phase of WD following injury. The longevity of WldS-overexpressing axon fragments allowed us to assess the consequences of failed WD on the regeneration of proximal axon branches: regenerating axons in the skin avoided persistent axon fragments. When WldS was expressed in sensory neurons, the degeneration of both spontaneously pruned axon fragments and axons of naturally dying cells was delayed. As these persistent fragments could repel sprouting and regenerating axons, rapid clearance of degenerating axons during development may be required for complete innervation of the skin.
MATERIALS AND METHODS
Imaging and axotomy
Axons were imaged in zebrafish embryos of the Tg(sensory:GFP) line (Sagasti et al., 2005). Embryos were dechorionated, anaesthetized in 0.01% tricaine and mounted in a sealed agarose chamber (O'Brien et al., 2009). A custom-built two-photon microscope equipped with a femtosecond Ti:Sapphire laser (Chameleon Ultra II, Coherent) was used to image and axotomize GFP-labeled trigeminal neurons, and images were collected using ScanImage software (Pologruto et al., 2003). Axons were visualized using a 40× water objective (0.8 NA; Olympus), a laser wavelength setting of 910 nm, and 30 mW of power at the sample. To axotomize a specified region of the axon, a single 2D scan with 180 mW at a 70× ScanImage zoom was used. For time-lapse analysis, embryos were imaged at various intervals for 1-12 hours on a confocal microscope with a 20× air objective (Zeiss LSM 510). Approximately 60 3.5 μm confocal sections were gathered at each time point, which were compiled into projections and movies with QuickTime software. Embryos were maintained at 28.5°C with a heated stage throughout imaging.
Tracing and quantification of data
Degenerating axons were digitally traced in three dimensions from confocal image stacks with NeuroLucida software (MicroBrightField). Length of axons before and after axotomy, as well as length and number of fragmented axon pieces at each time point, were quantified from these tracings with NeuroLucida Explorer software. Student's two-tailed t-tests were performed for all statistical analyses that involved comparison of two groups and one-way ANOVA was used to compare multiple groups.
Transgene construction
The Tol2/Gateway zebrafish kit developed by the lab of Chi-Bin Chien was used for construction of all plasmids (Kwan et al., 2007). The CREST3 enhancer (gift of H. Okamoto; Uemura et al., 2005) was cloned into the p5E entry vector; Gal4 and 14 repeats of the Gal4 Upstream Activating Sequence (UAS) (Koster and Fraser, 2001) were subcloned into the middle entry vector (pME); and the p3E vector contained GFP, followed by UAS sequences and WldS (gift of L. Luo). These vectors were recombined together to create a transgene for expressing GFP and WldS simultaneously in sensory neurons (CREST3:Gal4-14xUAS-GFP-14xUAS-WldS). Similarly, another vector was made to express KikGR and WldS simultaneously (14xUAS-WldS-CREST3:Gal4-14xUAS-KikGR). The UBP2 protease (Open Biosystems) was subcloned in a manner analogous to WldS so that the CREST3 enhancer drove expression of GFP and UBP2 in the same neurons (CREST3:Gal4-14xUAS-GFP-14xUAS-UBP2).
Site-directed mutagenesis of WldS
The WldS gene was mutated by changing tryptophan 258 to alanine as described previously (Conforti et al., 2009), using the Finnzymes Phusion Site-Directed Mutagenesis kit with the forward primer 5′-TGCCCAACTTGGCGAAGATGGAGG-3′ (mutation is TG to GC, mutated codon is underlined) and the non-overlapping reverse primer 5′-CGCTGAAGGACTCCAGTAAATCTGCC-3′. The mutated p3E-GFP-UAS-WldS* vector was sequenced to verify that no additional mutations were introduced. This vector was subsequently subcloned and recombined with Gateway vectors described above to yield the CREST3:Gal4-14xUAS-GFP-14xUAS-WldS* transgene.
Injection of DNA for transient transgenesis and stable lines
Approximately 15 pg of each transgene was injected into one-cell stage embryos to transiently visualize GFP in a subset of trigeminal neurons. To establish stable WldS lines, the WldS-KikGR transgene (14xUAS-WldS-CREST3:Gal4-14xUAS-KikGR) and TOL2 transposase were injected into one-cell stage embryos (Kawakami et al., 2000). The injected embryos were raised to adulthood and outcrossed to identify transgenic line founders.
Photoconversion of neurons expressing WldS and KikGR
To convert the fluorescence of trigeminal neurons expressing KikGR from green to red, a three-dimensional region of interest around their cell bodies was defined with confocal software. The 405 nm laser was used to scan through this defined volume, photoconverting the KikGR protein in the cell bodies, which then diffused throughout the axon.
RESULTS
Wallerian degeneration of peripheral sensory axons occurs in three stereotyped phases
To investigate the cellular mechanisms and developmental function of WD in peripheral sensory neurons, we developed a laser axotomy and time-lapse imaging procedure for monitoring axon degeneration in live zebrafish larvae. Single axon terminals of trigeminal neurons were severed in 54 hours post fertilization (hpf) larvae on a two-photon microscope (O'Brien et al., 2009). Following axotomy, distal, detached fragments were imaged for up to 12 hours using confocal microscopy (Fig. 1A-D). Degeneration and clearance of the detached axon fragment was always completed in less than 3 hours.
Fig. 1.

WD progresses through three phases. (A-D) Trigeminal axons were severed and time-lapse imaged at 20 minute intervals for 12 hours. Time series of confocal image stack projections, time stamps are minutes after axotomy. Arrow indicates the site of axotomy, which was performed at 54 hpf (see Movie 1 in the supplementary material). (E-H) Tracings of degenerating severed branch corresponding to A-D. (I-L) Time series of two-photon image stack projections collected every minute after axotomy to observe acute degeneration. Arrow indicates axotomy site, arrowheads indicate transient beading of severed distal fragment, asterisk marks slight fragmentation (see Movie 2 in the supplementary material). (M-P) Time series of confocal image stack projections collected every 2 minutes to capture fragmentation. Time zero in M is the last frame before fragmentation began. Broken lines outline the severed branch (see Movie 3 in the supplementary material). Scale bars: 50 μm. (Q) Quantification of total axon fragment length over time, showing three representative severed axons. (R) Quantification of number of axon fragments over time, showing three representative severed axons. Parts of the trace corresponding to the lag, fragmentation and clearance phases are indicated.
To rule out the possibility that photobleaching or the diffusion of GFP could cause the rapid disappearance of fluorescently labeled axon fragments following axotomy, we measured the rate of degeneration using different fluorescent proteins and varying time-lapse imaging intervals, both variables that could affect the rate of photobleaching. Axons imaged with soluble GFP, farnesylated GFP (which is membrane-associated and should therefore not diffuse; see Fig. S1 in supplementary material), or the photoconvertible protein KikGR (data not shown), all degenerated at the same rate. Similarly, imaging axons at 1, 2, 10 or 20 minute intervals did not alter the rate of degeneration or clearance. Finally, the fact that development and molecular manipulations could alter the rate of degeneration (see below), demonstrates that our imaging method did not limit the length of time that axon fragments could be detected.
In other systems, a dramatic acute axonal degeneration (AAD) phase has been reported to quickly remove greater than 100 μm from both ends of the severed axon immediately after axotomy (Busch et al., 2009; Kerschensteiner et al., 2005). To capture possible AAD in our model, we analyzed the first 10-20 minutes after axotomy by collecting two-photon time-lapse images every minute. These images revealed that AAD and retraction were modest, leaving a distance of only 3-9 μm between proximal and distal segments, which often both formed retraction bulbs (Fig. 1I-L). However, when neighboring keratinocytes were ablated concurrently with axotomy, fragmentation of axon branches proximal to the damaged skin cells occurred immediately (see Fig. S2 in the supplementary material).
To analyze subsequent WD, images were recorded every 20 minutes and axon fragments were traced in three dimensions (Fig. 1E-H). This analysis revealed three stereotyped phases: a lag phase, a fragmentation phase and a clearance phase. The lag phase was a latent period during which the severed fragment did not display overt signs of degeneration. This phase lasted on average about 70 minutes at 54 hpf (Fig. 1Q-R). The fragmentation phase was marked by a catastrophic breakdown of the axon into many small pieces. To determine whether fragmentation occurred in a distally- or proximally directed progressive manner, as has been observed in other systems (Beirowski et al., 2005; Gilley and Coleman, 2010; Kerschensteiner et al., 2005; Lubinska, 1977), confocal images of severed axons were collected every 2 minutes. Beading occurred simultaneously throughout the axon fragment, which preceded a non-progressive, instantaneous breakdown into axon debris within 2 minutes (Fig. 1M-P). Clearance began immediately after fragmentation. Analysis of time-lapse sequences revealed that axons fragmented into ∼1 μm pieces before disappearing. Thus, axotomy-induced degeneration of damaged trigeminal axon terminals is rapid and progresses through three stages: (1) a lag phase; (2) a rapid, non-progressive fragmentation phase; and (3) a clearance phase.
The rate of WD is regulated by developmental age and axon fragment length
Trigeminal axon arborization in zebrafish occurs from ∼18 to 36 hpf. To test whether the process of WD changes as axons mature, we compared axotomy-induced degeneration at 30, 54 and 78 hpf. At 30 hpf, when axons are still actively growing, the lag phase lasted ∼2 hours and clearance lasted ∼1 hour. The rate of the lag phase in both 54 hpf and 78 hpf embryos was significantly faster, lasting only ∼1 hour (Fig. 2A,B) (P≤0.0001). The rate of fragmentation and clearance was the same at all ages, indicating that the mechanism responsible for instigating the instantaneous breakdown specifically becomes more efficient after 2 days of development.
Fig. 2.

Developmental stage and axon fragment size regulate the rate of WD. (A) Quantification of the number of axon fragments versus time after axotomy in two 54 hpf (purple lines) and two 30 hpf (red lines) embryos. Fragmentation occurred at the peaks in the graph, which was much earlier in 54 hpf embryos. Thirty hpf embryos had a longer lag phase and fragmented into fewer axon fragments because axons were still growing and severed branches were therefore smaller. (B) The lag phase at 30 hpf was significantly longer than the lag phase in both 54 hpf (P=0.0001) and 78 hpf (P<0.0001) embryos, but the difference in lag phases between 54 hpf and 78 hpf was not significant (P=0.1578). The clearance phase occurred at the same rate at all ages examined (P≥0.2844). Error bars indicate s.e.m. (C) Quantification of lag phase length versus total axon fragment length. Scatter plot trendline indicates no correlation (r=0.09663) (n=20). (D) Quantification of clearance phase length versus total axon fragment length. Trendline indicates a modest correlation (r=0.3813) (n=20). (E) Confocal image stack projections of a 54 hpf embryo expressing GFP in all sensory neurons and RFP in a subset of sensory neurons. The entire trigeminal ganglion was ablated on the right side (left image, 2.5 hours after axotomy), and RFP-labeled debris remained 12.5 hours after axotomy (right image, higher magnification of boxed area in left image).
To determine the effect of fragment size on the rate of degeneration, we compared total axon fragment length with the rate of either the lag phase or the clearance phase. Although axon fragment size had no effect on length of the lag phase (correlation coefficient=0.09663), the length of the debris clearance phase correlated modestly with fragment size (correlation coefficient=0.3813) (Fig. 2C,D). To test this idea further, we ablated an entire trigeminal ganglion, which created dramatically more debris than severing a single axon, and found that some axon fragments persisted until at least 12 hours after ablation (Fig. 2E) (n=5). These results suggest that the capacity of the engulfment machinery is a limiting factor in the clearance phase.
Ubiquitylation specifically regulates the length of the lag phase
Previous studies have reported that inhibiting the UPS delays axon degeneration, whether injury-induced (Hoopfer et al., 2006; Zhai et al., 2003) or developmentally programmed (Hoopfer et al., 2006; Watts et al., 2003). To determine whether the UPS was involved in a specific phase of trigeminal WD, we expressed UBP2, a yeast ubiquitin protease, in peripheral sensory neurons (Fig. 3A), as was done in Drosophila mushroom body neurons (Watts et al., 2003). Ubiquitin proteases remove ubiquitin tags from substrates, thus preventing their degradation by the proteasome. We co-expressed GFP and UBP2 specifically in somatosensory neurons, axotomized their peripheral arbors at 54 hpf, and measured degeneration with time-lapse imaging. These analyses revealed that the lag phase of WD in severed UBP2-expressing axons was longer by 3.3-fold than in wild-type axons at the same age (Fig. 3B) (P<0.0001). The rate of clearance, however, was unaffected (P=0.9809), suggesting that the UPS is involved specifically in regulating the onset of fragmentation.
Fig. 3.
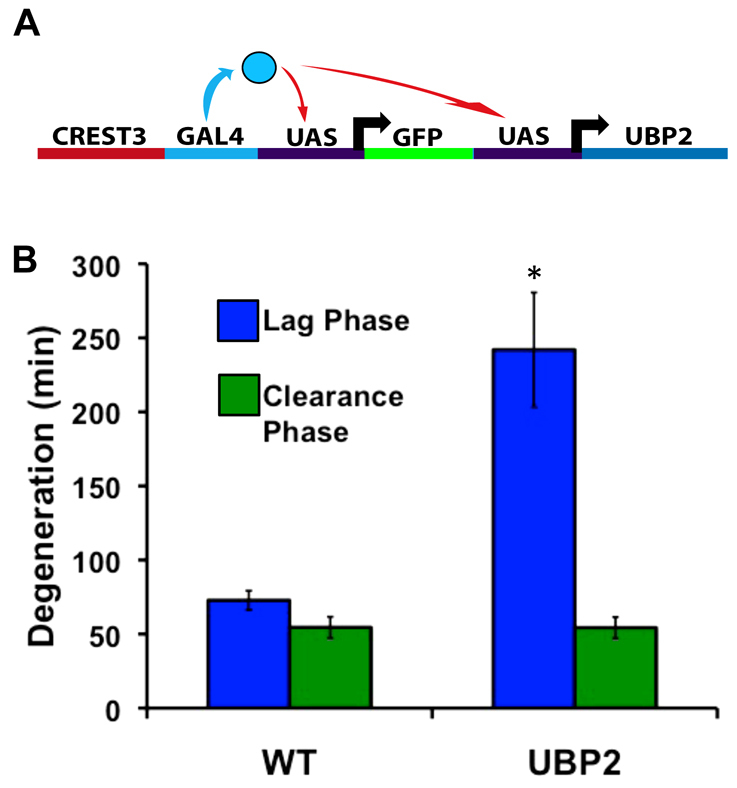
Deubiquitylation increases the length of the lag phase of WD. (A) Schematic of the transgene used to drive expression of UBP2 and GFP simultaneously in sensory neurons with the CREST3 enhancer, which drives expression in somatosensory neurons. (B) Quantification of WD in UBP2-expressing axons (n=8) compared with wild type at 54 hpf (n=12). The lag phase was significantly lengthened (242.38±38.62 minutes, P<0.0001), whereas clearance occurred at a wild-type rate (P=0.9809). Error bars indicate s.e.m.
WldS robustly delays the onset of fragmentation and requires functional Nmnat for maximal neuroprotection
The WldS fusion protein potently delays WD in injured axons in mice (Mack et al., 2001), rats (Adalbert et al., 2005) and flies (Hoopfer et al., 2006). To determine whether this protein also affects any of the phases of WD in fish trigeminal axons, we created a transgene in which a somatosensory-specific promoter drove expression of WldS and GFP (Fig. 4A). Misexpressing the WldS protein in trigeminal neurons via transient transgenesis dramatically delayed the rate of WD in severed axons. In contrast to severed axons in wild-type 30 hpf embryos, which fragment in under 2 hours, severed axons expressing WldS remained intact for at least 12 hours post-axotomy in 88% of experiments (Fig. 4B-E) (n=52). Moreover, axons that were followed for 48 hours post-axotomy remained intact (n=5). Those axons whose degeneration was not delayed (12%) may not have been expressing sufficient levels of the WldS gene, because, using our approach, GFP could occasionally be expressed without WldS (see Materials and methods; Fig. 4A). To verify that the long-lasting axon fragments had indeed been separated from the cell body, we repeated this experiment with a transgene that caused WldS and the photoconvertible protein KikGR (Tsutsui et al., 2005) to be expressed in peripheral sensory neurons, and photoconverted the cell bodies of these neurons from green to red fluorescence. In control neurons, photoconverted KikGR in the cell body spread throughout the neuronal arbor (data not shown). When we photoconverted KikGR in the cell body of severed WldS-expressing neurons, the distal region of the neuron continued to fluoresce green, thus verifying that it had been separated from the rest of the cell (Fig. 4H) (n=11).
Fig. 4.

WldS potently delays degeneration and requires Nmnat enzymatic activity. (A) Schematic of the transgene used to drive expression of WldS and GFP simultaneously in sensory neurons with the CREST3 enhancer. (B-E) Confocal time-lapse series of a trigeminal neuron expressing WldS. Image in B was collected before axotomy at 30 hpf. (C) 0.5 hours post-axotomy, arrow indicates site of axotomy. (D,E) The WldS-expressing fragment 22 hours and 48 hours post-axotomy, respectively. Arrowheads indicate the persisting severed fragment (see Movie 4 in the supplementary material). Scale bar: 50 μm. (F) Quantification of the lag phase in axons expressing WldS, WldS with a mutation compromising Nmnat enzymatic activity (WldS*) and wild type at 54 hpf. Hash marks on WldS bar indicate that the lag phase lasted at least 12 hours, as all fragments persisted throughout the 12-hour time-lapse imaging sessions. The lag phase in WldS* was significantly shorter than in WldS, but still significantly longer than wild type (P<0.0001, one-way ANOVA) (n≥12 for all groups). (G) Quantification of the lag and clearance phases in wild type and WldS*. Despite the lengthened lag phase in WldS* compared with wild type, the clearance phase occurs at a wild-type rate (P=0.1129). Error bars indicate s.e.m. (H) Axon expressing KikGR and WldS severed at ∼30 hpf. UV illumination of the cell body (white arrow) converted KikGR protein from green to red, which spread throughout the arbor but not into the severed fragment (white arrowhead). The regenerating red parent arbor did not overlap with the green fragment.
To assess whether WldS expression had developmental or physiological effects on sensory neurons, we created two stable transgenic lines expressing WldS and KikGR in somatosensory neurons. Both transgenic lines provided the same level of axon protection that we observed in the transient analysis (not shown). Similar to WldS mice, these transgenic fish grew to be normal, fertile adults. Moreover, kinematic analysis of escape responses to touch stimuli at 48 hpf showed that this somatosensory neuron-dependent behavior was indistinguishable in WldS and wild-type larvae (Fig. 5A-D). To test whether WldS expression caused developmental defects, we conducted a morphometric comparison of single WldS and wild-type peripheral axons in transient transgenics. The branching morphology of peripheral axons in WldS-expressing cells was indistinguishable from wild type (Fig. 5E-H). Together, these analyses demonstrate that WldS-expressing trigeminal neurons are normal morphologically and physiologically, despite being defective in their responses to injury.
Fig. 5.

Behavioral escape responses and peripheral axon morphology of wild-type and WldS larvae are indistinguishable. (A,A′) 48 hpf wild-type larvae touched on the left side of the head (A) escape to the right (A′). (B,B′) This behavior is similar in 48 hpf WldS larvae. (C) Quantification of turn angle in wild-type and WldS larvae. Pie chart displays a cartoon fish turning 193° in the opposite direction to the touch stimulus. On the right, a bar graph shows that wild type (blue bar, n=9) and WldS (red bar, n=24) both turn an average of 193° (P=0.9676). (D) Quantification of escape response duration in wild type and WldS. Wild-type duration (blue bar, n=9) was not significantly different from WldS duration (red bar, n=24) (P=0.4624). (E,F) Confocal image stack projections of representative 54 hpf wild-type (E, n=15) and WldS (F, n=15) peripheral axons. Single neurons were traced in three dimensions (see Materials and methods). (G) Quantification of the number of nodes (branch points) and branch ends in wild-type (blue bars) and WldS (red bars) axons were not significantly different (nodes, P=0.3260; ends, P=0.3123). (H) Quantification of total length and surface area of wild-type (blue bars) and WldS (red bars) axons revealed no significant difference between the two groups (length, P=0.6182; surface area, P=0.7416). Error bars indicate s.e.m. Scale bars: 50 μm.
WldS requires functional Nmnat to extend the rate of degeneration in other systems (Araki et al., 2004; Avery et al., 2009; Conforti et al., 2009; Sasaki et al., 2009). To test whether enzymatically active Nmnat is also necessary to delay WD in zebrafish trigeminal axons, we created a version of the WldS gene (WldS*) that encodes a protein with a single amino acid substitution, disrupting the enzymatic activity of Nmnat (Conforti et al., 2009). Expression of WldS* in zebrafish trigeminal axons only protected the severed axon for ∼3.5 hours, dramatically less than the delayed degeneration caused by WldS (Fig. 4F) (n=12, P<0.0001). However, the length of the lag phase in WldS*-expressing axons was still significantly longer than in wild type (P=0.0009), indicating that either WldS has an Nmnat-independent role in degeneration or that the mutation allows for some residual activity. In axons expressing WldS*, the rate of clearance was similar to wild type (Fig. 4G) (P=0.1129), suggesting that, like the UPS, WldS may specifically disrupt the molecular machinery that regulates the onset of fragmentation.
Regenerating axons avoid persisting fragments expressing WldS
The ability to delay WD allowed us to assess whether a regenerating severed axon can grow into the territory of a detached former branch, as has been observed in leech mechanosensory neurons (Wang and Macagno, 1998), or is instead repelled by this persistent axon fragment, consistent with the existence of a cell-surface recognition molecule that mediates axon branch repulsion. To observe the behavior of regenerating axons encountering intact axon fragments, we examined time-lapse image sequences of regenerating WldS-expressing axons. Regenerating WldS axons at 30 hpf were clearly repelled by persistent fragments (Fig. 6) (n=5), suggesting that axons recognized the severed fragment as `self'. As a further test of this phenomenon, we performed axotomies on neurons expressing WldS and KikGR and photoconverted their cell bodies at least six hours after axotomy. In all cases, the red photoconverted arbor did not overlap with the detached green fragment (n=10), even though it often closely approached it, further supporting the conclusion that regenerating axons avoid persistent axon fragments (Fig. 4H; see Fig. S3 in the supplementary material).
Fig. 6.

Regenerating axons are repelled by persistent WldS-expressing fragments. Confocal time-lapse image stack projections of two neurons expressing WldS. (A-E) A neuron axotomized at 54 hpf. (A) Before axotomy (0 hours); (B-E) time-points post-axotomy. White arrow in B indicates site of axotomy. Red arrows indicate contact points between axons. White arrowheads in B and D indicate spontaneously pruned fragments. This series also shows an example of a rare instance when a regenerating axon crossed over a fragment (blue arrow), which also sometimes occurs in encounters between sister branches (Liu and Halloran, 2005; Sagasti et al., 2005) (see Movie 5 in the supplementary material). (F-J) A neuron axotomized at 30 hpf. White arrow in F indicates site of axotomy at 0.5 hours post-axotomy. Red arrow in G indicates filopodia from both the fragment and the intact axon extending out and contacting each other. These filopodia were repelled from each other and retracted back, as seen in H. Red arrow in I also indicates a point of contact from the fragment extending out to the intact axon, which retracts back, as seen in J (see Movie 6 in the supplementary material). Scale bars: 50 μm.
Spontaneously pruned fragments and axons of dying cells expressing WldS fail to degenerate
Spontaneous local axon pruning and cell death are common in the developing zebrafish somatosensory system (Sagasti et al., 2005). To examine whether pruned or dying axons degenerate in a kinetically similar manner to WD, we time-lapse imaged uninjured axons in developing embryos between 24 and 48 hpf. In both stable and transient transgenic wild-type fish expressing GFP in trigeminal neurons, local pruning events (Fig. 7A-D) (n=9) and spontaneous cell death (Fig. 7E-H) (n=5) resulted in degeneration of the detached branch or the entire cell, respectively. Like laser-severed axon fragments, spontaneously pruned axon fragments underwent a lag phase that ranged from 60 to 140 minutes, following detachment from the parent arbor. This range of lag phase lengths is similar to the range in injured axons undergoing WD during these developmental stages (Fig. 2B). In all cases of spontaneous cell death or local pruning, fragmentation occurred instantaneously throughout the axon, as in injury-induced WD. Moreover, in instances of both spontaneous death and local pruning, debris was cleared at a similar rate as in WD (average of 55.45 minutes, P=0.9330).
Fig. 7.

Axon fragmentation following spontaneous pruning and cell death was delayed in WldS-expressing neurons. Time-lapse confocal image stack projections of WT (A-H) or WldS (I-P) neurons. (A-D) 40 hpf wild-type pruning beginning at –2 hours (A, before pruning). White arrow in B indicates the site of detachment of a spontaneously pruned branch (0 hours). Red arrows in C and D indicate fragmentation and clearance, respectively, of the pruned axon (see Movie 7 in the supplementary material). (E-H) 32 hpf wild-type spontaneous death example, beginning at –2 h (E, before apoptosis). White arrowheads in F and G indicate a dying cell body. Red arrow in H indicates axon fragmentation resulting from cell death (see Movie 8 in the supplementary material). (I-L) 48 hpf WldS pruning beginning with –1 hour (I, before pruning). White arrow in J indicates site of detachment of spontaneously pruned branch (0 h). Red arrowheads in K and L indicate a pruned branch that persists without degenerating (see Movie 9 in the supplementary material). (M-P) 36 hpf WldS cell death example beginning at –2.5 hours (M, during apoptosis). White arrowheads in M and N indicate a dying cell body. White arrow in N indicates a point of axon separation (0 hours). Red arrow in O indicates degeneration of the proximal axon nearest to the dead cell body. Red arrowhead in P indicates a large distal axon fragment that has remained intact long after cell death and proximal axon degeneration (see Movie 10 in the supplementary material). Scale bars: 50 μm.
To test whether axon degeneration resulting from spontaneous cell death or pruning can be altered by WD pathways in zebrafish trigeminal neurons, we imaged the development of uninjured neurons expressing WldS by time-lapse. Instigation of pruning and spontaneous cell death still occurred in axons expressing WldS (pruning, n=8; cell death, n=6). However, the pruned axons or branches of dying trigeminal neurons failed to degenerate completely. Pruned fragments were detached from axons but most underwent only limited degeneration throughout the imaging period (Fig. 7I-L; see Fig. S4 in the supplementary material). Photoconversion verified that a pruned branch was no longer attached to the parent arbor (Fig. S4E′ in the supplementary material). During cell death, the most proximal parts of axons degenerated, but some distal axon fragments were always persistently protected from fragmentation (Fig. 7M-P). In summary, in all 14 cases of wild-type spontaneous pruning or death captured in our time-lapse sequences, axons fragmented and were completely cleared in less than 3 hours, whereas in 14 cases of spontaneous pruning or death in WldS-expressing neurons axons persisted for the entire remainder of the time-lapse movie (∼4-14 hours). These observations revealed that WldS can delay fragmentation following developmental pruning and cell death, but does not regulate the instigation of these spontaneous events. Together with our finding that persistent axon fragments repel regenerating axons, these results suggest that rapid degeneration of spontaneously pruned axon fragments is required for comprehensive innervation of the skin by developing sensory neurons.
DISCUSSION
We have used confocal time-lapse imaging to analyze injury-induced and spontaneous degeneration of single branches of trigeminal sensory axon terminals in live zebrafish. Our method provided exceptional temporal and spatial resolution, allowing us to dissect the degeneration process into distinct phases. The degeneration we observed was particularly fast compared with what has been reported for other types of axons (Hoopfer et al., 2006; Navarro et al., 1997; Waller, 1850), perhaps reflecting the fact that we monitored the behavior of single severed axons. In most other studies of vertebrate WD, entire bundles of axons are damaged, often along with associated glia, connective tissue and blood vessels. When we ablated many cells at once, the clearance phase of WD was much longer, suggesting that non-autonomous factors can strongly influence the observed rate of WD. We also found acute degeneration to be much less dramatic than was reported by others – less than 10 μm of axon were lost following axotomy, compared with the ∼200 μm reported in other systems (Busch et al., 2009; Kerschensteiner et al., 2005). When we damaged neighboring keratinocytes, larger regions of the axons fragmented immediately after axotomy. Thus, the more robust acute phase reported by others may partially reflect damage to surrounding supportive tissues. Alternatively, different axons may be under different amounts of tension, which controls the degree of acute degeneration. Free trigeminal axon endings are held tightly between layers of epidermal cells, potentially reducing tension relative to axons in other environments.
WD in severed trigeminal axon terminals always progressed through three phases. First, the severed distal fragment persisted during a latent period, which we term the lag phase. Similar lag phases are conserved across several systems and have been proposed to correspond to the half-life of a specific survival factor, for which Nmnat2 is the best candidate (Gilley and Coleman, 2010; Lubinska, 1977). The lag phase in trigeminal neurons was shorter at later developmental stages. If the decrease of a survival factor to below a certain threshold is the molecular trigger for WD, our result suggests that the abundance or turnover of the survival factor is regulated by development. The second phase, fragmentation, may correspond to the point when the concentration of the unstable survival factor falls below a threshold, triggering the degenerative process. Within at least a 2-minute time frame, fragmentation occurred instantaneously throughout a detached trigeminal axon branch, in contrast to other systems, in which axon fragmentation occurs progressively from either the distal or proximal end (Beirowski et al., 2005; Gilley and Coleman, 2010; Kerschensteiner et al., 2005; Lubinska, 1977). The final phase of WD is clearance, the rate of which we found to depend on the amount of axon debris. This correlation highlights the fact that clearance must be an active process that can be saturated. Phagocytes have been accepted to play a role in engulfing axon fragments (Awasaki and Ito, 2004; Watts et al., 2004; Williams and Truman, 2005), but we have found that neither macrophages nor glia are involved in the clearance of trigeminal axon debris (data not shown). The skin cells between which these axons arborize might limit the access of phagocytic cells and may themselves clear debris. To determine whether the UPS and WldS/NAD pathways control specific phases of WD, we expressed a deubiquitinase and the WldS gene in trigeminal neurons. Both of these pathways delayed the lag phase of WD but did not affect clearance, suggesting that both pathways control the instigation of fragmentation.
The regulated nature of WD suggests that it is under positive natural selection in vertebrates. It has been proposed that one reason for the evolution of WD is that the removal of axon debris may facilitate axon regeneration (Vargas and Barres, 2007). Indeed, functional deficits in regeneration have been observed in WldS mice (Bisby and Chen, 1990; Brown et al., 1992). It is possible that persistent debris blocks the progress of regenerating or sprouting axons, but this phenomenon had not been previously directly observed. With time-lapse imaging and photoconversion experiments we clearly observed that regrowing axons were repelled by persistent detached axon fragments expressing WldS. Thus, at least during early larval stages, efficient WD is a prerequisite for successful regeneration. This result also demonstrates that, unlike in leech peripheral sensory neurons (Wang and Macagno, 1998), cytoplasmic continuity is not required for self-recognition and is instead probably mediated by cell-surface recognition molecules.
As precise axotomy is not likely to be a common occurrence in developing zebrafish larvae in the wild, we wondered whether there was a natural function for WD that could serve as a robust selective pressure for the evolution of such a regulated degradation process. Spontaneous local pruning and cell death are a feature of the developing zebrafish somatosensory system (Sagasti et al., 2005) and are common in many developing neurons (Bennett, 2002; Low and Cheng, 2005; Luo and O'Leary, 2005; Portera-Cailliau et al., 2005). We found that local pruning occurred during wild-type development with similar kinetics to those we observed during WD. When WldS was expressed in neurons, pruning still occurred but detached fragments did not degenerate. Moreover, spontaneous cell death of WldS-expressing cells created persistent axon fragments. Thus, WldS protects injured axons, pruned axons and axons of dying cells. Importantly, WldS did not inhibit instigation of initial detachment of axon fragments or cell death. Together with a recent study showing that WldS can delay remodeling of Drosophila sensory neurons (Schoenmann et al., 2010), our studies establish a developmental role for the WldS pathway. These observations differ from reported studies of WldS in developmentally programmed axon degeneration in mice and flies (Hoopfer et al., 2006), and thus may reflect functional differences in pruning.
Although WldS-sensitive local pruning has yet to be described in other types of neurons, it is possible that it is common in the nervous system. For example, Rohon-Beard cells, the somatosensory neurons that innervate the body early in fish and amphibian development, undergo spontaneous developmental pruning similar to trigeminal neurons (Sagasti et al., 2005). Similar sporadic pruning also occurs in axons in the central nervous system (Portera-Cailliau et al., 2005). Because we have demonstrated that persistent trigeminal axon fragments repel regenerating axons, spontaneous wild-type degeneration due to pruning or death must be fast to allow for appropriate re-innervation and thus comprehensive innervation of the skin. In peripheral sensory neurons, this may be the most functionally important role for degeneration, but in the central nervous system deficits in local pruning could lead to the persistence of unproductive synaptic contacts and consequently disrupt neural circuit function. The necessity for efficient degeneration in both the peripheral and central nervous systems thus provides a putative explanation for the evolution of a robust axon degeneration program.
Supplementary Material
Acknowledgments
We thank Larry Zipursky, Bill Lowry and members of the Sagasti laboratory for critical reading of the manuscript; Chi-Bin Chien (University of Utah) for the Tol2kit; Hitoshi Okomoto (RIKEN BSI) for the CREST3 promoter; and Liqun Luo (Stanford University) for the WldS transgene. The work was supported by predoctoral NRSA fellowships from NIDCR and NINDS to S.M.M. and G.S.O., respectively; by grants from the March of Dimes Foundation and NICHD (C.P.-C.); and by grants from the Burroughs-Wellcome Fund, the Whitehall Foundation, the Klingenstein Foundation and NIDCR (A.S.). Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.053611/-/DC1
References
- Adalbert R., Gillingwater T. H., Haley J. E., Bridge K., Beirowski B., Berek L., Wagner D., Grumme D., Thomson D., Celik A., et al. (2005). A rat model of slow Wallerian degeneration (WldS) with improved preservation of neuromuscular synapses. Eur. J. Neurosci. 21, 271-277 [DOI] [PubMed] [Google Scholar]
- Araki T., Sasaki Y., Milbrandt J. (2004). Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 305, 1010-1013 [DOI] [PubMed] [Google Scholar]
- Avery M. A., Sheehan A. E., Kerr K. S., Wang J., Freeman M. R. (2009). Wld S requires Nmnat1 enzymatic activity and N16-VCP interactions to suppress Wallerian degeneration. J. Cell Biol. 184, 501-513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awasaki T., Ito K. (2004). Engulfing action of glial cells is required for programmed axon pruning during Drosophila metamorphosis. Curr. Biol. 14, 668-677 [DOI] [PubMed] [Google Scholar]
- Bedi S. S., Glanzman D. L. (2001). Axonal rejoining inhibits injury-induced long-term changes in Aplysia sensory neurons in vitro. J. Neurosci. 21, 9667-9677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beirowski B., Adalbert R., Wagner D., Grumme D. S., Addicks K., Ribchester R. R., Coleman M. P. (2005). The progressive nature of Wallerian degeneration in wild-type and slow Wallerian degeneration (WldS) nerves. BMC Neurosci. 6, 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett M. R., Gibson W. G., Lemon G. (2002). Neuronal cell death, nerve growth factor and neurotrophic models: 50 years on. Autonomic Neuroscience: Basic and Clinical 95, 1-23 [DOI] [PubMed] [Google Scholar]
- Bisby M. A., Chen S. (1990). Delayed wallerian degeneration in sciatic nerves of C57BL/Ola mice is associated with impaired regeneration of sensory axons. Brain Res. 530, 117-120 [DOI] [PubMed] [Google Scholar]
- Brown M. C., Lunn E. R., Perry V. H. (1992). Consequences of slow Wallerian degeneration for regenerating motor and sensory axons. J. Neurobiol. 23, 521-536 [DOI] [PubMed] [Google Scholar]
- Busch S. A., Horn K. P., Silver D. J., Silver J. (2009). Overcoming macrophage-mediated axonal dieback following CNS injury. J. Neurosci. 29, 9967-9976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman M. P., Freeman M. R. (2010). Wallerian degeneration, Wld(S), and Nmnat. Annu. Rev. Neurosci. 33, 245-267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L., Tarlton A., Mack T. G., Mi W., Buckmaster E. A., Wagner D., Perry V. H., Coleman M. P. (2000). A Ufd2/D4Cole1e chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (WldS) mouse. Proc. Natl. Acad. Sci. USA 97, 11377-11382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L., Wilbrey A., Morreale G., Janeckova L., Beirowski B., Adalbert R., Mazzola F., Di Stefano M., Hartley R., Babetto E., et al. (2009). Wld S protein requires Nmnat activity and a short N-terminal sequence to protect axons in mice. J. Cell Biol. 184, 491-500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh-Roy A., Wu Z., Goncharov A., Jin Y., Chisholm A. D. (2010). Calcium and cyclic AMP promote axonal regeneration in Caenorhabditis elegans and require DLK-1 kinase. J. Neurosci. 30, 3175-3183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J., Coleman M. P. (2010). Endogenous nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 8, e1000300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoopfer E. D., McLaughlin T., Watts R. J., Schuldiner O., O'Leary D. D., Luo L. (2006). Wlds protection distinguishes axon degeneration following injury from naturally occurring developmental pruning. Neuron 50, 883-895 [DOI] [PubMed] [Google Scholar]
- Kawakami K., Shima A., Kawakami N. (2000). Identification of a functional transposase of the Tol2 element, an Ac-like element from the Japanese medaka fish, and its transposition in the zebrafish germ lineage. Proc. Natl. Acad. Sci. USA 97, 11403-11408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschensteiner M., Schwab M. E., Lichtman J. W., Misgeld T. (2005). In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat. Med. 11, 572-577 [DOI] [PubMed] [Google Scholar]
- Knaut H., Blader P., Strahle U., Schier A. F. (2005). Assembly of trigeminal sensory ganglia by chemokine signaling. Neuron 47, 653-666 [DOI] [PubMed] [Google Scholar]
- Koster R. W., Fraser S. E. (2001). Tracing transgene expression in living zebrafish embryos. Dev. Biol. 233, 329-346 [DOI] [PubMed] [Google Scholar]
- Kwan K. M., Fujimoto E., Grabher C., Mangum B. D., Hardy M. E., Campbell D. S., Parant J. M., Yost H. J., Kanki J. P., Chien C. B. (2007). The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev. Dyn. 236, 3088-3099 [DOI] [PubMed] [Google Scholar]
- Liu Y., Halloran M. C. (2005). Central and peripheral axon branches from one neuron are guided differentially by Semaphorin3D and transient axonal glycoprotein-1. J. Neurosci. 25, 10556-10563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low L. K., Cheng H. J. (2005). A little nip and tuck: axon refinement during development and axonal injury. Curr. Opin. Neurobiol. 15, 549-556 [DOI] [PubMed] [Google Scholar]
- Lubinska L. (1977). Early course of Wallerian degeneration in myelinated fibres of the rat phrenic nerve. Brain Res. 130, 47-63 [DOI] [PubMed] [Google Scholar]
- Lunn E. R., Perry V. H., Brown M. C., Rosen H., Gordon S. (1989). Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve. Eur. J. Neurosci. 1, 27-33 [DOI] [PubMed] [Google Scholar]
- Luo L., O'Leary D. D. (2005). Axon retraction and degeneration in development and disease. Annu. Rev. Neurosci. 28, 127-156 [DOI] [PubMed] [Google Scholar]
- Mack T. G., Reiner M., Beirowski B., Mi W., Emanuelli M., Wagner D., Thomson D., Gillingwater T., Court F., Conforti L., et al. (2001). Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat. Neurosci. 4, 1199-1206 [DOI] [PubMed] [Google Scholar]
- Navarro X., Verdu E., Wendelschafer-Crabb G., Kennedy W. R. (1997). Immunohistochemical study of skin reinnervation by regenerative axons. J. Comp. Neurol. 380, 164-174 [DOI] [PubMed] [Google Scholar]
- O'Brien G. S., Rieger S., Martin S. M., Cavanaugh A. M., Portera-Cailliau C., Sagasti A. (2009). Two-photon axotomy and time-lapse confocal imaging in live zebrafish embryos. J. Vis. Exp. 24, doi: 10.3791/1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pologruto T. A., Sabatini B. L., Svoboda K. (2003). ScanImage: flexible software for operating laser scanning microscopes. Biomed. Eng. Online 2, 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portera-Cailliau C., Weimer R. M., De Paola V., Caroni P., Svoboda K. (2005). Diverse modes of axon elaboration in the developing neocortex. PLoS Biol. 3, e272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagasti A., Guido M. R., Raible D. W., Schier A. F. (2005). Repulsive interactions shape the morphologies and functional arrangement of zebrafish peripheral sensory arbors. Curr. Biol. 15, 804-814 [DOI] [PubMed] [Google Scholar]
- Sasaki Y., Vohra B. P., Lund F. E., Milbrandt J. (2009). Nicotinamide mononucleotide adenylyl transferase-mediated axonal protection requires enzymatic activity but not increased levels of neuronal nicotinamide adenine dinucleotide. J. Neurosci. 29, 5525-5535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena S., Caroni P. (2007). Mechanisms of axon degeneration: from development to disease. Prog. Neurobiol. 83, 174-191 [DOI] [PubMed] [Google Scholar]
- Schoenmann Z., Assa-Kunik E., Tiomny S., Minis A., Haklai-Topper L., Arama E., Yaron A. (2010). Axonal degeneration is regulated by the apoptotic machinery or a NAD+-sensitive pathway in insects and mammals. J. Neurosci. 30, 6375-6386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui H., Karasawa S., Shimizu H., Nukina N., Miyawaki A. (2005). Semi-rational engineering of a coral fluorescent protein into an efficient highlighter. EMBO Rep. 6, 233-238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura O., Okada Y., Ando H., Guedj M., Higashijima S., Shimazaki T., Chino N., Okano H., Okamoto H. (2005). Comparative functional genomics revealed conservation and diversification of three enhancers of the isl1 gene for motor and sensory neuron-specific expression. Dev. Biol. 278, 587-606 [DOI] [PubMed] [Google Scholar]
- Vargas M. E., Barres B. A. (2007). Why is Wallerian degeneration in the CNS so slow? Annu. Rev. Neurosci. 30, 153-179 [DOI] [PubMed] [Google Scholar]
- Waller A. (1850). Experiments on the section of the glossopharyngeal and hypoglossal nerves of the frog, and observations of the alterations produced thereby in the structure of their primitive fibres. Philos. Trans. R. Soc. London 140, 423-429 [PMC free article] [PubMed] [Google Scholar]
- Wang H., Macagno E. R. (1998). A detached branch stops being recognized as self by other branches of a neuron. J. Neurobiol. 35, 53-64 [DOI] [PubMed] [Google Scholar]
- Watts R. J., Hoopfer E. D., Luo L. (2003). Axon pruning during Drosophila metamorphosis: evidence for local degeneration and requirement of the ubiquitin-proteasome system. Neuron 38, 871-885 [DOI] [PubMed] [Google Scholar]
- Watts R. J., Schuldiner O., Perrino J., Larsen C., Luo L. (2004). Glia engulf degenerating axons during developmental axon pruning. Curr. Biol. 14, 678-684 [DOI] [PubMed] [Google Scholar]
- Williams D. W., Truman J. W. (2005). Cellular mechanisms of dendrite pruning in Drosophila: insights from in vivo time-lapse of remodeling dendritic arborizing sensory neurons. Development 132, 3631-3642 [DOI] [PubMed] [Google Scholar]
- Zhai Q., Wang J., Kim A., Liu Q., Watts R., Hoopfer E., Mitchison T., Luo L., He Z. (2003). Involvement of the ubiquitin-proteasome system in the early stages of wallerian degeneration. Neuron 39, 217-225 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}