

Abstract
We have identified a role for two evolutionarily related, secreted metalloproteases of the ADAMTS family, ADAMTS20 and ADAMTS9, in palatogenesis. Adamts20 mutations cause the mouse white-spotting mutant belted (bt), whereas Adamts9 is essential for survival beyond 7.5 days gestation (E7.5). Functional overlap of Adamts9 with Adamts20 was identified using Adamts9+/–;bt/bt mice, which have a fully penetrant cleft palate. Palate closure was delayed, although eventually completed, in both Adamts9+/–;bt/+ and bt/bt mice, demonstrating cooperation of these genes. Adamts20 is expressed in palatal mesenchyme, whereas Adamts9 is expressed exclusively in palate microvascular endothelium. Palatal shelves isolated from Adamts9+/–;bt/bt mice fused in culture, suggesting an intact epithelial TGFβ3 signaling pathway. Cleft palate resulted from a temporally specific delay in palatal shelf elevation and growth towards the midline. Mesenchyme of Adamts9+/–;bt/bt palatal shelves had reduced cell proliferation, a lower cell density and decreased processing of versican (VCAN), an extracellular matrix (ECM) proteoglycan and ADAMTS9/20 substrate, from E13.5 to E14.5. Vcan haploinsufficiency led to greater penetrance of cleft palate in bt mice, with a similar defect in palatal shelf extension as Adamts9+/–;bt/bt mice. Cell density was normal in bt/bt;Vcanhdf/+ mice, consistent with reduced total intact versican in ECM, but impaired proliferation persisted in palate mesenchyme, suggesting that ADAMTS-cleaved versican is required for cell proliferation. These findings support a model in which cooperative versican proteolysis by ADAMTS9 in vascular endothelium and by ADAMTS20 in palate mesenchyme drives palatal shelf sculpting and extension.
Keywords: ADAMTS, Cleft palate, Versican, Mouse
INTRODUCTION
Closure of the secondary palate (palatogenesis) separates the oral and nasal cavities, permitting compartmentalization of digestive and respiratory processes. Failure of palatogenesis leads to cleft palate, one of the commonest human developmental malformations (Cox, 2004). Although cleft palate can occur as an isolated phenomenon in humans, it is frequently associated with cleft lip or other anomalies (Cox, 2004). Palatogenesis results from complex interactions involving pharyngeal ectoderm (palatal epithelium) and mesenchyme derived primarily from craniofacial neural crest cells (NCCs) (Dudas et al., 2007; Kaufman, 1992). During mouse embryogenesis, the secondary palate develops from two sagittally oriented palatal shelves that arise from the maxillary primordia and project inferiorly on each side of the tongue at 12.5 days of gestation (E12.5). They undergo elevation to a horizontal plane at E13.5-14 and grow rapidly towards each other to meet in the midline at E14.5-E15. Upon contact, the medial edge epithelium overlying the apposed surfaces undergoes apoptosis or epithelial-mesenchymal transformation, resulting in the formation of a seamless bridge of mesenchyme by E16 (Dudas et al., 2007), within which the palatine bone forms.
Although only a few single gene mutations causing cleft palate have been described in humans (Cox, 2004; Jugessur and Murray, 2005), the inactivation of several mouse genes has led to cleft palate and identified key regulatory pathways in palatogenesis (Murray and Schutte, 2004). Inherited cleft palate in mice is always recessive, as a complete cleft of the secondary palate is incompatible with survival. Mouse genetics has clearly demonstrated how signaling by members of the TGFβ superfamily (Proetzel et al., 1995; Taya et al., 1999), FGFs (Rice et al., 2004; Trokovic et al., 2003), PDGFs (Ding et al., 2004) and EGF receptor ligands (Miettinen et al., 1999) mediates epithelial-mesenchymal interactions that drive palatal morphogenesis. In particular, midline fusion of the apposed medial epithelial edge of the palatal shelves is specifically dependent on TGFβ3 and its downstream signaling pathways (Dudas et al., 2004a; Taya et al., 1999). Extensive extracellular matrix (ECM) remodeling during palatogenesis and experimental evidence using metalloprotease inhibitors support a role for metalloproteases in palatogenesis (Blavier et al., 2001; Brown et al., 2002). However, there is only one prior report of metalloprotease mutations causing cleft palate. Combinatorial deletion of Mmp14 and Mmp16, which encode transmembrane proteases, leads to severe craniofacial dysmorphism and extensive skeletal anomalies, with cleft palate observed in 80% of double-null embryos (Shi et al., 2008). These defects were attributed to diminished turnover of the ECM protein collagen I, and cleft palate could be attributed, at least in part, to the global craniofacial anomalies (Shi et al., 2008), rather than to an exclusive and specific interference with palatal shelves.
Here, we provide unequivocal genetic evidence that two members of the ADAMTS (a disintegrin-like and metalloprotease domain with thrombospondin type 1 motif) protease family, ADAMTS9 and ADAMTS20, act locally in palate closure and cooperate in proteolysis of versican (VCAN), an abundant, large aggregating proteoglycan found in association with hyaluronan in ECM. Combinatorial genetic evidence using mice lacking these ADAMTS genes and a mouse Vcan mutant strongly suggests that defective versican proteolysis is the definitive mechanism underlying the observed cleft palate phenotype. The data further suggest that ADAMTS9 and ADAMTS20 are not required solely for versican clearance, but that they might generate versican proteolytic products that influence palate mesenchyme proliferation.
ADAMTS designates a family of secreted metalloproteases that contain thrombospondin type 1 repeats (TSRs). Of the 19 mammalian ADAMTS proteases, some are of relatively recent evolutionary origin (Huxley-Jones et al., 2005) and may have highly specialized functions (Apte, 2009). ADAMTS9 and ADAMTS20 are the only mammalian ADAMTS with a closely related non-chordate ortholog, named gon-1 in the nematode C. elegans, and are presumed to have arisen by duplication and continued evolution of a shared ancestral gene. gon-1 has an essential role in cell migration during morphogenesis of the nematode gonad (Blelloch and Kimble, 1999). Notable conserved features of GON-1, ADAMTS9 and ADAMTS20 include numerous TSRs (18 in GON-1 and 15 in ADAMTS9 and ADAMTS20) and a unique C-terminal domain (Llamazares et al., 2003; Somerville et al., 2003). gon-1 mutants are partially rescued by ADAMTS9 (Hesselson et al., 2004), suggesting that their proteolytic mechanism is conserved. Although ADAMTS9 lacks a membrane anchor, it binds to the cell surface (Somerville et al., 2003), where it is proteolytically active (Koo et al., 2006; Koo et al., 2007). Thus, it is likely to be involved in cell surface or pericellular ECM proteolysis. ADAMTS9 and ADAMTS20 belong to an ADAMTS subgroup termed the proteoglycanases that includes ADAMTS1, ADAMTS4 and ADAMTS5, the ability of which to cleave large, aggregating proteoglycans, such as versican and aggrecan, is crucial in physiological and disease processes (Apte, 2009).
Adamts20 is mutated in the recessive mouse white-spotting mutant belted (bt), which has an unpigmented belt on the lumbar torso (Rao et al., 2003). Adamts20 promotes melanoblast survival (Silver, 2008) and is expressed by dermal mesenchymal cells (Rao et al., 2003). However, the expression of Adamts20 in craniofacial mesenchyme (K. A. Jungers and S.S.A., unpublished) suggests additional developmental roles. Adamts9 is widely expressed during embryogenesis and – of relevance to the present investigation – its mRNA is found in craniofacial mesoderm and in microvascular endothelial cells of the embryo and adult organs (Jungers et al., 2005; Koo et al., 2010). Recent work suggests that Adamts9 might be anti-angiogenic in adult microvascular endothelial cells (Koo et al., 2010). We considered the possibility that mice lacking both ADAMTS9 and ADAMTS20 would show novel defects. Although Adamts9–/– mice do not survive beyond E7.5, we used them to obtain genetic evidence of cooperation with Adamts20 in closure of the secondary palate and we investigated the underlying mechanisms to identify a novel pathway operational in palatogenesis.
MATERIALS AND METHODS
Mouse strains and genotyping
All mouse work was performed under a protocol approved by the Cleveland Clinic Institutional Animal Care and Use Committee. Adamts9lacZ/+ mice (referred to here as Adamts9+/–), btbei mice (referred to here as bt) and the Vcan mutant heart defect (hdf, referred to as Vcanhdf), as well as null alleles of Adamts4 and Adamts5 and their genotyping protocols have been described previously (McCulloch et al., 2009a; McCulloch et al., 2009b; Rao et al., 2003; Silver, 2008). All mice were extensively crossed into the C57Bl/6 strain. Bmp4 and Pdgfra mutant mice in C57Bl/6 were kindly provided by Dr Jan Christian (Oregon Health and Science University, Portland, OR, USA) and Dr Phillipe Soriano (Mount Sinai Medical Center, NY, USA), respectively. For timed pregnancies the date of the vaginal plug was designated E0.5. All analyses were performed using littermate controls of the appropriate genotype. In the crosses that were used to obtain Adamts9+/–;bt/bt mice, it was not possible to obtain wild-type littermates; instead, littermates having the most appropriate alternative genotype (bt/+) were used as controls.
Micro-computed tomography (mCT) analysis and scanning electron microscopy (SEM)
Newborn mice were fixed in 10% buffered formalin for 48 hours and transferred to 70% ethanol. mCT scanning of the skull was performed as previously described (Le Goff et al., 2006). For SEM, the lower jaw was removed, the heads were fixed, sputter-coated with gold and viewed from the inferior aspect by SEM as previously described (Oblander et al., 2005). At least five newborn mice encompassing all the illustrated genotypes were examined by mCT and two litters of embryos representing the various genotypes were analyzed by SEM.
Histology, immunohistochemistry (IHC), β-gal histochemistry and in situ hybridization (ISH)
Embryos were fixed in 4% paraformaldehyde (PFA) for durations appropriate to their gestational stage, followed by embedding in paraffin and Hematoxylin and Eosin (H&E) staining. Over 20 mice of each genotype (Adamts9+/–;bt/bt and bt/+) were examined histologically at E13.5 and E14.5. pSmad2/3 immunohistochemistry of palatal shelves was performed with a polyclonal antibody (no. 3101, Cell Signaling Technology, Danvers, MA, USA) using an indirect immunoperoxidase method. Prior to pSmad2/3 staining, the sections were pretreated with 10 mM sodium citrate buffer (pH 6.0) for 10 minutes at 95°C. Cleavage of the ADAMTS-processed Glu441-Ala442 peptide bond in versican (V1 splice isoform enumeration) was detected by immunofluorescence staining using a neo-epitope antibody that specifically detects versican cleaved at this site but not intact versican (Affinity BioReagents, Golden, CO, USA) (Sandy et al., 2001), and an Alexa 488-labeled secondary antibody. For immunofluorescence, four mice of each of the Adamts9+/–;bt/bt and control (bt/+) genotypes were analyzed at each time point (E13.5 and E14.5), with consistent results.
For cell proliferation analysis, the phospho-histone H3 antibody (anti-pH3; Upstate Biotechnology-Millipore, Billerica, MA, USA) was applied using an indirect immunoperoxidase method. The proliferation index in palatal shelf mesenchyme was quantified by counting at least 500 nuclei in sections from five mice each of the Adamts9+/–;bt/bt and control (bt/+) genotypes at each time point (E13.5 and E14.5), using four sections from each mouse. Cell density within the mesenchymal component of the palate was quantified from photographic images of the shelves by overlaying them with a counting grid in the center of the palatal shelf (the area of interest is indicated in Fig. 4C), which consistently showed a difference between Adamts9+/–;bt/bt and control (bt/+) palates. Four sections each from five embryos of each genotype at each of the two time points were used in this analysis. Statistical analysis of the quantitative data for cell proliferation and cell density was performed using an unpaired, two-tailed Student's t-test.
Fig. 4.

Decreased cell proliferation and reduced cell density in Adamts9+/–;bt/bt palatal shelves. (A) Anti-pH3 immunostaining (brown) in palatal shelves. Note the blue staining (β-gal) in capillaries of Adamts9+/–;bt/bt palatal shelves. The genotype and gestational ages of the mice are indicated. The proliferation index was determined in the area between the dotted line and the medial edge of the shelf. Note the rounded shape of the Adamts9+/–;bt/bt palatal shelves. (B) Proliferation index (pH3-stained nuclei/total nuclei counted) at E13.5 and E14.5 (n=5 for each genotype at each age). A statistically significant difference was observed at both ages (*, P<0.05; #, P<0.005). (C) Cell density was quantified in the center of the palatal shelf by counting nuclei in sections from Adamts9+/–;bt/bt and bt/+ palatal shelves (n=5 for each genotype) in the area indicated by the box. (D) A statistically significant reduction in cell density (*P<0.05) at E13.5 and E14.5 was noted in Adamts9+/–;bt/bt palatal shelves. Scale bars: 50 μm.
ISH of paraffin sections from E14.5 embryos using Adamts9 and Adamts20 35S-UTP-labeled antisense cRNA probes was performed as previously described (Jungers et al., 2005; Rao et al., 2003). The corresponding sense probes did not give signal above background, as previously shown (Jungers et al., 2005; Rao et al., 2003). β-gal staining of Adamts9+/– mice followed by endomucin immunohistochemistry to identify vascular endothelium (n=6) was described previously (Koo et al., 2010).
Palatal shelf organ culture
Using a published procedure (LaGamba et al., 2005) palatal shelves were dissected from E14.5 Adamts9+/–;bt/bt embryos (n=4) and placed on the surface of a 0.9% agarose gel in DMEM/F12 supplemented with 10% fetal bovine serum (FBS) and antibiotics (100 U/ml penicillin and 100 mg/ml streptomycin), with their medial edge epithelia in contact. The medium was replaced daily. After 3 days in culture, the shelves were fixed in 10% neutral-buffered formalin and examined histologically by H&E staining of coronal sections. Affigel beads (Bio-Rad, Hercules, CA, USA) were soaked for 1 hour in conditioned medium containing the previously identified N-terminal versican fragment (G1-DPEAAE441) expressed in HEK293F cells. As a control, beads were soaked in conditioned medium from HEK293F cells transfected with vector alone. E13.5 Adamts9+/–;bt/bt maxillary arches containing the palate shelves were isolated and pre-soaked beads were inserted into the palate shelves and placed onto 0.1-μm pore size Nuclepore filters (Whatman, Florham Park, NJ, USA). The explants were cultured in a 24-well tissue culture dish at the air-liquid interface in 1:1 DMEM:Ham's F12, plus 0.1% FBS. Explants were incubated for 14 hours at 37°C in 5% CO2 followed by fixation in 4% PFA and IHC using anti-pH3 on paraffin sections.
RESULTS
Perinatal lethality and cleft palate in Adamts9+/–;bt/bt mice
In crosses between bt/bt and Adamts9+/–;bt/+ mice, or intercrosses of Adamts9+/–;bt/+ mice, we did not obtain viable Adamts9+/–;bt/bt mice upon genotyping between 8 and 10 days after birth, whereas other expected genotypes were present (Table 1). However, Adamts9+/–;bt/bt embryos were identified in E18.5 litters at the expected frequency (Table 1). A few viable Adamts9+/–;bt/bt mice were identified shortly after birth but died within 24 hours with conspicuous aerogastria (air in the stomach), suggesting a cleft palate, but were otherwise externally normal (Fig. 1A). A complete cleft of the secondary palate (Fig. 1B) was found in 100% of Adamts9+/–;bt/bt mice. By contrast, bt/bt;Adamts5–/–, bt/bt;Adamts4–/–, Adamts5–/–;Adamts9+/– and Adamts4–/–;Adamts5–/–;bt/+ mice were viable and did not have cleft palate (data not shown), indicating a specific functional association of ADAMTS20 with ADAMTS9, but not with ADAMTS4 or ADAMTS5 in palatogenesis. Consistent with these observations, Adamts4 and Adamts5 expression, as determined by β-gal staining in knockout mice (McCulloch et al., 2009a; McCulloch et al., 2009b), is undetectable in palatal shelves during development (McCulloch et al., 2009a) (data not shown). Thus, of the proteoglycanase-encoding genes tested, only Adamts9 and Adamts20 participate in palatogenesis.
Table 1.
Frequency of genotypes arising from crosses between Adamts9+/–;bt/+ and Adamts9+/+;bt/bt mice

Fig. 1.

Cleft palate in Adamts9+/–;bt/bt mice. (A) External appearance of newborn mice of the indicated genotypes. (B) Cleft palate in Adamts9+/–;bt/bt mice (right, arrows). (C) mCT of the skull in three projections shows that all skull bones are present and similar in each genotype, other than abnormal formation of the palatine (asterisk) and basi-sphenoid, the anomalous development of which is presumed secondary to abnormal palatogenesis in Adamts9+/–;bt/bt mice. The pterygoid processes of the sphenoid bone are indicated by arrows; their wider spacing in Adamts9+/–;bt/bt mice is typical of unfused palates.
mCT analysis of newborn Adamts9+/–;bt/bt mice consistently demonstrated abnormal palatine and basi-sphenoid bones (Fig. 1C), which have been previously described in mouse strains with a cleft of the secondary palate (Trokovic et al., 2003; Zhang et al., 2002) and are secondary to failed palatal closure. All other craniofacial skeletal elements were consistently present and appeared normal (Fig. 1C). The mandibles and tongue of Adamts9+/–;bt/bt mice were similar to those of littermates (Fig. 1C), arguing against mechanical interference with palatal shelf elevation (Dudas et al., 2004b).
All newborn Adamts9+/–;bt/bt mice had horizontally inclined, unfused palatal shelves that were overlaid with an intact epithelium of normal thickness. Analysis of palatogenesis in Adamts9+/–;bt/bt embryos from E12.5-15.5 illustrated that the palatal shelves were always formed correctly from the maxillary primordia by E13.5 (Fig. 2A). In Adamts9+/–;bt/bt mice, palate shelf growth towards the midline from E14.5 onwards was always retarded, and in some Adamts9+/–;bt/bt mice, palatal shelf elevation to the horizontal orientation was also delayed relative to the controls (Fig. 2A). In addition, palatal shelf sculpting (remodeling) in Adamts9+/–;bt/bt embryos was consistently abnormal as, unlike in the other genotypes obtained, the shelves remained broad, with a rounded free edge and broader base. Unlike Fgf10 and Fgfr2b deficient mice (Rice et al., 2004), adhesion of the palatal shelves to surrounding structures was not seen in Adamts9+/–;bt/bt mice.
Fig. 2.

Adamts9 and Adamts20 participate independently and cooperatively in palatal shelf extension towards the midline. (A) The palatal fusion defect in Adamts9+/–;bt/bt mice results from defective shelf elevation and extension to midline between E13.5 and E14.5. Coronal sections at various stages of palatal development are shown for the indicated genotypes. Palatal shelves are indicated by asterisks. There is reduced sculpting of the palatal shelves from bt/bt and Adamts9+/–:bt/+ mice, as well as Adamts9+/–;bt/bt mice. Scale bar: 100 μm. (B) Scanning electron microscopy of palates (inferior view) of various genotypes was obtained at the indicated gestational ages. The arrows indicate the location of the medial edge of the palatal shelf. Note the delayed closure at E14.5 in Adamts9+/–;bt/+ and bt/bt mice, and failure to close by E15.5 in Adamts9+/–;bt/bt mice.
Independent contributions of Adamts9 and Adamts20 to mouse palatogenesis
Comparative histology of the various genotypes between E12.5 and E15.5 suggested that the elevation and growth of the palatal shelves were also delayed in bt/bt and Adamts9+/–;bt/+ embryos (Fig. 2A), although their shelves did not have the abnormal shape of Adamts9+/–;bt/bt shelves. We used SEM to visualize the complete anterior-posterior extent of the palatal shelves. We confirmed the delay in shelf extension in Adamts9+/–;bt/+ and bt/bt mice and, as expected, the consistently defective palatal closure in Adamts9+/–;bt/bt mice (Fig. 2B). The delay of shelf extension affected the anterior and posterior halves equally in the affected genotypes, unlike in several other cleft palate mutants (He et al., 2008; Yu et al., 2005; Zhang et al., 2002). Palatal extension to the midline was delayed to a greater extent in bt/bt than in Adamts9+/–;bt/+ mice (Fig. 2A,B). Despite the delay, palatogenesis was successfully completed in E15.5 Adamts9+/–;bt/+ mice and bt/bt mice as viewed by SEM (Fig. 2B). Adamts9+/–;bt/+ mice survived at the predicted Mendelian frequency and did not develop cleft palate, whereas bt/bt mice were later found to have a low incidence of cleft palate (3%). These data strongly supported an independent as well as a cooperative participation of ADAMTS9 and ADAMTS20 in palatogenesis.
Palatal epithelial fusion is a TGFβ3-dependent metalloprotease-mediated process (Blavier et al., 2001; Taya et al., 1999) and defects in TGFβ3 or its downstream signaling pathway result in a specific defect in epithelial fusion (Cui et al., 2003; Cui et al., 2005). Phospho (p) Smad2/3 immunohistochemistry, which reports TGFβ receptor binding and activation of downstream Smad signaling in palatal epithelium, provided a strong nuclear signal in the prospective medial edge epithelium of Adamts9+/–;bt/bt mice at E14.5 (which corresponds to the time point at which palatal shelves normally begin fusion) despite these palatal shelves being widely separated (see Fig. S1A in the supplementary material). To determine whether Adamts9+/–;bt/bt palatal shelves were fusion competent, they were dissected at E13.5 and placed with their medial edge epithelia in contact in organ culture, where they fused successfully, with disappearance of the epithelium (see Fig. S1B in the supplementary material). Thus, a deficiency of Adamts9 and Adamts20 does not impair this key pathway in palatogenesis, and the in vitro data suggest that these palatal shelves would have fused had they been able to meet in the midline.
Since a failure of NCCs to populate the branchial arches has been described as a cause of cleft palate (Trokovic et al., 2003) and Adamts9 is expressed in branchial arches (Jungers et al., 2005), we used SEM to evaluate early branchial arch development. Externally normal branchial arches in E10.5 Adamts9+/–;bt/bt embryos (see Fig. S2 in the supplementary material) and, subsequently, the lack of abnormalities in newborn mice in the skeletal elements originating from the branchial arches (Fig. 1C), excluded abnormal branchial arch development as an underlying mechanism. Taken together with the chronological analysis of palate development from E13.5-15.5, this identified a temporally specific defect in palatal shelf growth and/or remodeling involving mesenchyme.
Adamts9 and Adamts20 mRNAs are expressed in distinct cell lineages in the palate
We determined expression of Adamts9 and Adamts20 by ISH in the extending palatal shelves of non-transgenic embryos to examine whether these genes acted locally and to identify the expressing cells. At E13.5 (not shown) and E14.5 (Fig. 3A,B), both Adamts9 and Adamts20 mRNAs were expressed in the palatal shelf mesenchyme. However, each mRNA had a distinct distribution, with Adamts9 mRNA being associated with palatal capillaries and Adamts20 being broadly expressed in mesenchyme (Fig. 3A,B). Lineage tracing of β-gal-stained Wnt1-Cre-R26R mice has demonstrated that the vast majority of palate mesenchymal cells, with the notable exception of vascular endothelium, which arises from mesoderm, are of NCC origin (Yoshida et al., 2008). β-gal staining of E13.5 and E14.5 Adamts9+/– palatal shelves followed by endomucin immunostaining (to identify vascular endothelium) demonstrated that Adamts9 was exclusively expressed by capillary endothelium (Fig. 3C,D). Thus, Adamts9 and Adamts20 cooperate in palatogenesis but are expressed by distinct cell populations with different embryonic origins. Visualization of palatal capillaries by endomucin immunostaining at E13.5 did not demonstrate a difference in palatal vasculature between Adamts9+/–;bt/bt and bt/+ (control) mice (Fig. 3E,F).
Fig. 3.
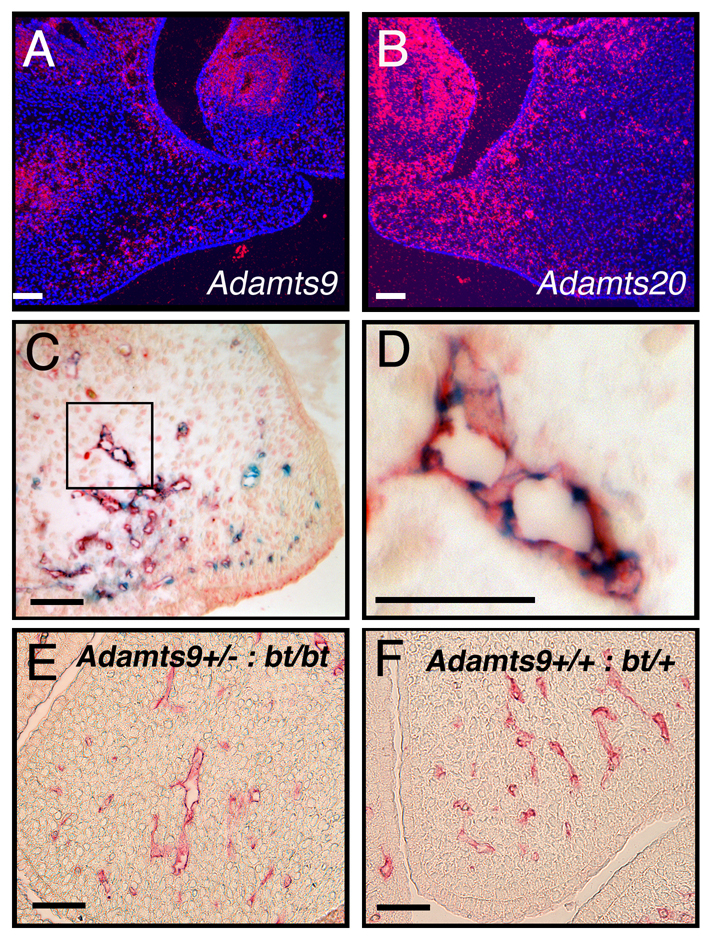
Adamts9 and Adamts20 are expressed locally in palatal shelf, but in distinct cell types. (A,B) In situ hybridization (ISH) of Adamts9 (A) and Adamts20 (B) mRNA in the palatal shelf (asterisk) at E13.5 (coronal sections). ISH signal is red and nuclei blue (DAPI). Note that expression of both Adamts9 and Adamts20 is confined to mesenchyme, but in distinct distributions. (C,D) Combined β-gal histochemistry (blue) and endomucin immunohistochemistry (red) of Adamts9+/– mice showing that Adamts9 expression is confined to microvascular endothelial cells. (C) An overview of staining in an E13.5 palatal shelf (D) Palatal capillaries (from C, boxed) at higher magnification. Note that endomucin-positive cytoplasm surrounds β-gal-stained nuclei. (E,F) Endomucin immunostaining shows comparable capillary density and distribution in Adamts9+/–;bt/bt and bt/+ palatal shelf at E13.5. Scale bars: 50 μm.
Mesenchymal cell proliferation is reduced in Adamts9+/–;bt/bt palatal shelves
Immunostaining for pH3, a marker for cycling cells (Hendzel et al., 1997; Yu et al., 2005), showed a statistically significant reduction of mesenchymal proliferation at E13.5 and E14.5 (Fig. 4A,B), the time points at which growth retardation was evident during palatogenesis in Adamts9+/–;bt/bt mice. We therefore attributed the reduced growth of the palatal shelves to decreased cell proliferation. Mesenchymal cell density was reduced in Adamts9+/–;bt/bt palatal shelves compared with those of bt/+ mice (Fig. 4C), which is consistent with reduced cell proliferation and is also suggestive of a relative excess of ECM (i.e. defective sculpting) in the mutant palatal shelves. We evaluated cell death in palatal shelves of Adamts9+/–;bt/bt mice because melanoblast survival was previously shown to be reduced in bt/bt mice and because bt/bt;Adamts5–/– mice have impaired apoptosis during interdigital web regression. However, few apoptotic cells were seen in palatal mesenchyme of Adamts9+/–;bt/bt or bt/+ (control) mice (see Fig. S3 in the supplementary material), consistent with apoptosis not being a crucial feature of palatogenesis until shelf fusion (Mori et al., 1994), nor an underlying mechanism of this cleft palate phenotype.
Versican processing is reduced in mutant palatal shelves
The relative abundance of ECM in the Adamts9+/–;bt/bt shelves led us to investigate the role of a previously identified substrate of ADAMTS9 and ADAMTS20, versican (Silver, 2008; Somerville et al., 2003), the proteolysis of which was recently implicated in ADAMTS developmental phenotypes, i.e. white spotting (Silver, 2008), soft-tissue syndactyly (McCulloch et al., 2009b) and cardiac dysmorphogenesis (Kern et al., 2007; Kern et al., 2006; Kern et al., 2010; Stankunas et al., 2008). Versican is a highly hydrated proteoglycan that forms large complexes with hyaluronan and thus has space-filling properties. Immunostaining of versican showed it to be abundant in palatal shelf mesenchyme (Fig. 5A). No difference in staining of intact versican was detected between Adamts9+/–;bt/bt and bt/+ palates (Fig. 5A). A neo-epitope antibody that specifically recognizes the cleaved Glu441-Ala442 peptide bond in the versican core protein (V1 isoform enumeration), but not intact versican (Sandy et al., 2001), demonstrated cleaved versican diffusely within wild-type and bt/+ palatal mesenchyme (Fig. 5B). However, in Adamts9+/–;bt/bt mice there was a striking decrease in the staining intensity and distribution of cleaved versican in the palatal mesenchyme. Cleaved versican was primarily detected around capillaries in Adamts9+/–;bt/bt shelves, consistent with expression of the intact Adamts9 allele in vascular endothelium and loss of ADAMTS20 in mesenchyme (Fig. 5B). These results strongly associated the cleft palate phenotype with decreased versican processing.
Fig. 5.

Versican is present normally in the palate but its processing is reduced in Adamts9+/–;bt/bt palatal shelves. (A) Immunofluorescence microscopy of intact versican using a polyclonal antibody that recognizes the GAG-β domain in the V1 and V0 isoforms shows widespread versican localization throughout the palate mesenchyme. The boxed regions are shown at higher magnification to the right. There was no discernible difference in versican localization between the two genotypes. (B) Immunofluorescence microscopy of cleaved versican (using anti-DPEAAE) illustrates its broad distribution within the growing end of the bt/+ (control) palatal shelf. There is greatly reduced staining in the intermesenchymal ECM of the Adamts9+/–;bt/bt palatal shelf, with residual staining present only around capillaries. The boxed regions are shown at higher magnification to the right. Scale bars: 50 μm.
Genetic interaction of Vcan with Adamts20 implies an active role for versican proteolysis in palatogenesis
To determine whether versican was merely a space-occupying substrate that needed clearance by ADAMTS proteases during palatal development (implying a passive role for versican), or whether versican proteolysis was required to facilitate mesenchymal proliferation (implying an active role for processed versican), we asked whether a reduction of Vcan gene dosage would rescue delayed palatal shelf growth in bt/bt mice. The Vcan mutant heart defect (hdf) dies at E10 with severe cardiac development defects, but Vcanhdf/+ mice are viable and apparently normal (Mjaatvedt et al., 1998).
Analysis of progeny resulting from crosses of bt/+;Vcanhdf/+ and bt/bt mice showed that 65% of bt/bt;Vcanhdf/+ mice developed cleft palate (Fig. 6A,B), despite having intact Adamts9 alleles. By contrast, only 3% of bt/bt mice had cleft palate (Fig. 6A). To evaluate the specificity of this genetic interaction, we crossed the Adamts9+/– and bt/bt mice to mice with inactivated Pdgfra and Bmp4. Cleft palate has been previously described in mice lacking these genes, as a consequence of impaired palatal shelf growth (Ding et al., 2004; Liu et al., 2005; Zhang et al., 2002). We obtained viable bt/bt;Pdgfra+/–, bt/bt;Bmp4+/–, Adamts9+/–;Pdgfra+/– and Adamts9+/–;Bmp4+/– mice with unimpaired palate closure (data not shown). The lack of interaction of Adamts9 and Adamts20 with Bmp4 and Pdgfra demonstrated that the genetic interaction with Vcan was specific, and suggested that cleft palate in Adamts9+/–;bt/bt mice might result from interference with a novel mechanism involving versican that was operational in the palatal mesenchyme.
Fig. 6.

High incidence of cleft palate in bt/bt;Vcanhdf/+ mice. (A) The incidence of cleft palate in crosses between bt/+;Vcanhdf/+ and bt/bt mice. (B) Complete cleft of the secondary palate in bt/bt;Vcanhdf/+ mice. In the bt/bt;Vcanhdf/+ palate, the edges of the palatal shelves are indicated by arrows. The appearance of the palate is similar to that of the Adamts9+/–;bt/bt shelves in Fig. 1B. (C) Histology of palatal closure (coronal sections stained with Hematoxylin and Eosin) in the indicated genotypes. Note the retarded growth of palatal shelves of bt/bt;Vcanhdf/+ mice, but their relatively normal shape compared with Adamts9+/–;bt/bt shelves (see Fig. 2A), and the failure to establish contact (asterisk at E16.5). Scale bar: 100 μm.
The cleft secondary palate in affected bt/bt;Vcanhdf/+ embryos (Fig. 6B) resulted from a similar delay in palatal shelf extension as in Adamts9+/–:bt/bt embryos (Fig. 6C, Fig. 2A), with no other apparent anomalies. However, bt/bt;Vcanhdf/+ palate shelves (Fig. 6C) did not have the blunt, rounded appearance of Adamts9+/–:bt/bt shelves (Fig. 2A, Fig. 4A). pH3 immunostaining demonstrated a statistically significant reduction in cell proliferation at E14.5, but not at E13.5, suggesting a similar mechanism as in Adamts9+/–;bt/bt mice (Fig. 7A,B). However, mesenchymal density was unaltered in bt/bt;Vcanhdf/+ palatal shelves compared with bt/+ shelves. Thus, unlike Adamts9+/–;bt/bt shelves, reduced cell density was not seen in bt/bt;Vcanhdf/+ shelves. This observation is consistent with a relative excess of ECM in Adamts9+/–;bt/bt mice (i.e. reduced cell density), which are expected to have normal versican production but decreased versican clearance with the deficiency of two versican-clearing proteases. Reduced versican production in bt/bt;Vcanhdf/+ shelves might well explain the lack of a difference in cell density compared with bt/+ mice because the potential accumulation of uncleaved versican could be offset by the lower production of the former. However, the higher penetrance of cleft palate in bt/bt mice with Vcan haploinsufficiency than in bt/bt mice suggested an additional, active role for proteolysed versican. Immunostaining for intact versican confirmed a reduction in versican content in the bt/bt;Vcanhdf/+ palatal shelves (Fig. 8A), and staining for cleaved versican identified considerably weaker mesenchymal and perivascular ECM staining compared with bt/+ shelves (Fig. 8B). The lack of ADAMTS20 is held to be responsible for the lack of intermesenchymal processed versican, and the persistence of versican processing around capillaries (Fig. 8B) is consistent with intact Adamts9 alleles in these mice.
Fig. 7.

Decreased cell proliferation but normal cell density in bt/bt;Vcanhdf/+ palatal shelves. (A) Anti-pH3 immunostaining in palatal shelves. The genotype and gestational ages of the mice are indicated. The proliferation index was determined in the area between the dotted line and the medial edge of the shelf. (B) Quantification of proliferation (pH3-stained nuclei/total nuclei counted) at E13.5 and E14.5. *, P<0.05 (n=4). (C) Cell density was quantified in the center of the palatal shelf by counting all nuclei in sections from bt/bt;Vcanhdf/+ and bt/+ palatal shelves in the area indicated by the box. (D) There is no significant alteration in cell density at E13.5 or E14.5 in bt/bt;Vcanhdf/+ palatal shelves (n=4 for each genotype). Scale bars: 50 μm.
Fig. 8.

Versican GAG-β and anti-DPEAAE immunofluorescence reflects both reduced Vcan gene dosage and reduced versican processing in bt/bt;Vcanhdf/+ palatal shelves. (A) Immunofluorescence microscopy of intact versican using a polyclonal antibody that recognizes the GAG-β domain in the V1 and V0 isoforms, showing reduced staining in bt/bt;Vcanhdf/+ palatal shelves, consistent with Vcan haploinsufficiency. (B) Immunofluorescence microscopy of cleaved versican (using anti-DPEAAE) illustrates greatly reduced staining in the bt/bt;Vcanhdf/+ palatal shelves. In bt/+ palatal shelves, note the bright signal around palatal capillaries and its reduction in the bt/bt;Vcanhdf/+ palatal shelf. Scale bars: 50 μm.
Taken together, the findings are consistent with a model in which decreased Vcan dosage results in a reduction of the overall amount of processed versican in bt/bt;Vcanhdf/+ mice, which has the same end result as the absence of ADAMTS20 and reduced dosage of ADAMTS9 in Adamts9+/–;bt/bt mice (Fig. 9). In this model, proteolysis of versican by ADAMTS9 (acting in endothelium) and ADAMTS20 (made by mesenchymal cells) contributes to the overall level of cleaved versican. Thus, these proteases are postulated to work cooperatively in the palatal shelf to enable mesenchymal proliferation. Interestingly, Adamts9+/–;Vcanhdf/+ mice did not develop cleft palate, suggesting that intact Adamts20 alleles and the remaining Adamts9 allele provided sufficient versican proteolysis in mesenchyme to compensate for reduced versican. To further extend this model experimentally, we introduced a bead soaked in conditioned medium containing the N-terminal versican fragment that spans residues 1-441 (G1-DPEAAE441) into palates of E13.5 Adamts9+/–;bt/bt mice. However, this fragment did not have a significant effect on cell proliferation in palatal mesenchyme compared with a control bead (see Fig. S4 in the supplementary material).
Fig. 9.

Models of versican and ADAMTS function in the mesenchyme. The models summarize the experimental observations in wild-type, Adamts9+/–;bt/bt (at E13.5 and E14.5) and bt/bt;Vcanhdf/+ (at E14.5) palatal shelves, and the proposed underlying mechanisms. In wild-type and Adamts9+/–;bt/bt mice, a deep shade of blue indicates normal versican levels in the ECM, whereas bt/bt;Vcanhdf/+ mice have half the normal amount (light-blue). The wild-type model indicates that cleaved versican is normally present in both the pericapillary and intermesenchymal ECM. In both Adamts9+/–;bt/bt and bt/bt;Vcanhdf/+ palates there is a paucity of intermesenchymal cleaved versican because ADAMTS20 is absent. In Adamts9+/–;bt/bt palates there is also a paucity of cleaved versican around capillaries because of Adamts9 haploinsufficiency. In bt/bt;Vcanhdf/+ palates there is less cleaved versican around capillaries owing to Vcan haploinsufficiency, although Adamts9 dosage is not reduced. We propose that the amount of cleaved versican affects mesenchymal cell proliferation. In addition, the model depicts reduced cell density (i.e. the cell-to-matrix ratio) in the Adamts9+/–;bt/bt palate, which results from both reduced clearance of versican (i.e. increased ECM) and decreased cell proliferation. Note that these models do not include the epithelium, as the observed mechanism seems to be wholly operational in mesenchyme.
DISCUSSION
Early embryonic lethality of the Adamts9-null mice precluded the generation of double-null mice (with bt) for complete determination of overlapping functions. Nevertheless, we examined the effect of reduced Adamts9 gene dosage in a bt/bt background by generating Adamts9+/–;bt/bt mice. These mice lack severe craniofacial anomalies other than cleft palate, suggesting that reducing the gene dosage of Adamts9 on the bt/bt background did not broadly impair craniofacial neural crest-related functions and that palatogenesis in bt/bt mice is sensitive to Adamts9 haploinsufficiency. Cleft palate in Adamts9+/–;bt/bt mice results from a temporally restricted defect that is localized to mesenchyme from E13.5-14.5. Crucial developmental events that occur before or after this interval appeared to be unaffected, as the branchial arches were morphologically normal, craniofacial skeletal patterning was unaffected, and the palatal shelves formed from the maxillary shelves and could fuse in vitro. The reduced outgrowth of the palatal shelf in the Adamts9+/–;bt/bt mutant mice resulted in part from reduced cell proliferation. A lack of sculpting of the shelves and a failure of elongation, as a likely consequence of impaired versican remodeling, are likely to be contributory factors to the failure to meet in the midline.
NCCs make a significant contribution to craniofacial mesenchyme and their interactions with ECM are believed to have a major influence on their fate (Bronner-Fraser, 1993; Francis-West et al., 1998). By contrast, the contribution of craniofacial mesoderm, which is highly concentrated in the core of developing branchial arches, and which is the source of the branchial arch vasculature and craniofacial capillaries (Yoshida et al., 2008), is less well appreciated. Adamts9 expression in the palatal shelves was restricted to microvascular endothelial cells, which are derived from mesoderm, whereas Adamts20 is expressed in NCC-derived mesenchyme.
The versican-rich ECM of the palatal shelves is typical of the provisional matrix present during embryogenesis. Versican has been implicated in the regulation of neural crest migration and cardiovascular development (Dutt et al., 2006; Henderson et al., 1997; Landolt et al., 1995; Perissinotto et al., 2000; Perris et al., 1996). Splotch, a Pax3 mutant, is believed to result in part from the overexpression of versican, which results in skin pigmentation and craniofacial defects (Henderson et al., 1997). However, once organogenesis is completed, the provisional ECM requires clearance to allow replacement by definitive ECM. ADAMTS proteases appear to be crucial for the clearance of versican in provisional ECM (Kern et al., 2007; Kern et al., 2006; Kern et al., 2010; McCulloch et al., 2009b; Stankunas et al., 2008). However, this process, and the specific proteases involved in remodeling the versican-rich ECM, have not previously been investigated in palatogenesis.
Reduced versican proteolysis in Adamts9+/–;bt/bt palatal shelves, together with altered shelf sculpting and reduced cell density, strongly implicate defective versican turnover in the cleft palate. A model of gene dosage-dependent cooperative proteolysis of versican in the palate is supported by the ability of ADAMTS9 and ADAMTS20 to cleave versican (Silver, 2008; Somerville et al., 2003), by their expression during palatal shelf extension, and by the delayed palate closure in Adamts9+/–;bt/+ and bt/bt mice. Genetic interaction with Vcan, but not Bmp4 or Pdgfra, strongly suggests that the observed interaction with Vcan was specific. Taken together, the experimental findings suggest that Adamts9 and Adamts20 act cooperatively via versican proteolysis in a novel pathway that ensures closure of the secondary palate. This work also identifies three new candidate genes for cleft palate in humans: ADAMTS9, ADAMTS20 and VCAN.
Recently, we demonstrated that Adamts9 cooperates with Adamts5 and Adamts20 in the regression of interdigital webs, a process in which versican proteolysis has a crucial role (McCulloch et al., 2009b). Specifically, combinatorial mutants of these genes led to soft-tissue syndactyly, and reduced versican processing was observed in the mutant webs (McCulloch et al., 2009b). Coincidentally, versicanolysis appears to be required in palatogenesis and web regression at approximately the same gestational age (E14.5), and bt/bt;Vcanhdf/+ mice also develop soft-tissue syndactyly (McCulloch et al., 2009b). Insertion of a bead containing the recombinant G1-DPEAAE441 versican fragment led to enhanced apoptosis in Adamts5–/–;bt/bt webs (McCulloch et al., 2009b). The present work suggests a similar bioactive function for cleaved versican, but in the maintenance of mesenchymal proliferation rather than in apoptosis. However, introduction of the G1-DPEAAE441 versican fragment into mutant palates did not affect cell proliferation. Therefore, although the genetic evidence strongly implicates versican fragmentation in regulating cell proliferation, the precise fragment or fragments that mediate the effect remain to be identified, and it is likely that the versican core protein undergoes ADAMTS-mediated proteolysis at multiple sites.
To explain the cooperative action of three ADAMTS proteases in web regression, we previously proposed a model of cooperative versicanolysis for the generation of a critical functional threshold of a bioactive fragment (McCulloch et al., 2009b). Versican is widely expressed during embryogenesis and in adult tissues, and versican-degrading ADAMTS proteases overlap considerably in their expression patterns, such as in the cardiovascular system. Previous work identified a significant role for ADAMTS1 and ADAMTS9 in versican processing during myocardial compaction and valvulogenesis (Kern et al., 2007; Kern et al., 2006; Kern et al., 2010; Stankunas et al., 2008). In addition, recent work (C. Kern, personal communication) has identified decreased versican processing associated with failed endocardial cushion remodeling in Adamts5–/– mice. In these mice, versican haploinsufficiency (i.e. Adamts5–/–;Vcanhdf/+) led to substantial rescue of the valvular defect, suggesting that the versican clearance function of ADAMTS5 is highly significant in this developmental setting. Thus, depending on the context, ADAMTS proteases may primarily mediate versican clearance or contribute to both versican clearance and the generation of bioactive versican fragments. These significant findings render it necessary to expand investigation of the biological impact of versican proteolysis by ADAMTS proteases, and to obtain additional insights into the mechanisms of versican processing.
Supplementary Material
Acknowledgments
We thank David R. Beier for providing bt mice; Dr Christine Kern, Dr Corey Mjaatvedt and Roche Pharmaceuticals for providing hdf mice; Dr Phillipe Soriano and Dr Michelle Tallquist for Pdgfra mice; Dr Jan Christian and Dr Brigid Hogan for Bmp4 mice; Dr Dietmar Vestweber for anti-endomucin antibody; Amanda Allamong and Michael Braun for technical support for histology; and Craig Bennetts for mCT. This work was supported by NIH award AR49930 (to S.S.A.). Histology and mCT analyses were supported by NIH Core Center award AR50953. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.050591/-/DC1
References
- Apte S. S. (2009). A disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin type 1 motif (ADAMTS) superfamily: functions and mechanisms. J. Biol. Chem. 284, 31493-31497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blavier L., Lazaryev A., Groffen J., Heisterkamp N., DeClerck Y. A., Kaartinen V. (2001). TGF-beta3-induced palatogenesis requires matrix metalloproteinases. Mol. Biol. Cell 12, 1457-1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blelloch R., Kimble J. (1999). Control of organ shape by a secreted metalloprotease in the nematode Caenorhabditis elegans. Nature 399, 586-590 [DOI] [PubMed] [Google Scholar]
- Bronner-Fraser M. (1993). Neural crest cell migration in the developing embryo. Trends Cell Biol. 3, 392-397 [DOI] [PubMed] [Google Scholar]
- Brown N. L., Yarram S. J., Mansell J. P., Sandy J. R. (2002). Matrix metalloproteinases have a role in palatogenesis. J. Dent. Res. 81, 826-830 [DOI] [PubMed] [Google Scholar]
- Cox T. C. (2004). Taking it to the max: the genetic and developmental mechanisms coordinating midfacial morphogenesis and dysmorphology. Clin. Genet. 65, 163-176 [DOI] [PubMed] [Google Scholar]
- Cui X. M., Chai Y., Chen J., Yamamoto T., Ito Y., Bringas P., Shuler C. F. (2003). TGF-beta3-dependent SMAD2 phosphorylation and inhibition of MEE proliferation during palatal fusion. Dev. Dyn. 227, 387-394 [DOI] [PubMed] [Google Scholar]
- Cui X. M., Shiomi N., Chen J., Saito T., Yamamoto T., Ito Y., Bringas P., Chai Y., Shuler C. F. (2005). Overexpression of Smad2 in Tgf-beta3-null mutant mice rescues cleft palate. Dev. Biol. 278, 193-202 [DOI] [PubMed] [Google Scholar]
- Ding H., Wu X., Bostrom H., Kim I., Wong N., Tsoi B., O'Rourke M., Koh G. Y., Soriano P., Betsholtz C., et al. (2004). A specific requirement for PDGF-C in palate formation and PDGFR-alpha signaling. Nat. Genet. 36, 1111-1116 [DOI] [PubMed] [Google Scholar]
- Dudas M., Nagy A., Laping N. J., Moustakas A., Kaartinen V. (2004a). Tgf-beta3-induced palatal fusion is mediated by Alk-5/Smad pathway. Dev. Biol. 266, 96-108 [DOI] [PubMed] [Google Scholar]
- Dudas M., Sridurongrit S., Nagy A., Okazaki K., Kaartinen V. (2004b). Craniofacial defects in mice lacking BMP type I receptor Alk2 in neural crest cells. Mech. Dev. 121, 173-182 [DOI] [PubMed] [Google Scholar]
- Dudas M., Li W. Y., Kim J., Yang A., Kaartinen V. (2007). Palatal fusion-where do the midline cells go? A review on cleft palate, a major human birth defect. Acta Histochem. 109, 1-14 [DOI] [PubMed] [Google Scholar]
- Dutt S., Kleber M., Matasci M., Sommer L., Zimmermann D. R. (2006). Versican V0 and V1 guide migratory neural crest cells. J. Biol. Chem. 281, 12123-12131 [DOI] [PubMed] [Google Scholar]
- Francis-West P., Ladher R., Barlow A., Graveson A. (1998). Signalling interactions during facial development. Mech. Dev. 75, 3-28 [DOI] [PubMed] [Google Scholar]
- He F., Xiong W., Yu X., Espinoza-Lewis R., Liu C., Gu S., Nishita M., Suzuki K., Yamada G., Minami Y., et al. (2008). Wnt5a regulates directional cell migration and cell proliferation via Ror2-mediated noncanonical pathway in mammalian palate development. Development 135, 3871-3879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson D. J., Ybot-Gonzalez P., Copp A. J. (1997). Over-expression of the chondroitin sulphate proteoglycan versican is associated with defective neural crest migration in the Pax3 mutant mouse (splotch). Mech. Dev. 69, 39-51 [DOI] [PubMed] [Google Scholar]
- Hendzel M. J., Wei Y., Mancini M. A., Van Hooser A., Ranalli T., Brinkley B. R., Bazett-Jones D. P., Allis C. D. (1997). Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma 106, 348-360 [DOI] [PubMed] [Google Scholar]
- Hesselson D., Newman C., Kim K. W., Kimble J. (2004). GON-1 and fibulin have antagonistic roles in control of organ shape. Curr. Biol. 14, 2005-2010 [DOI] [PubMed] [Google Scholar]
- Huxley-Jones J., Apte S. S., Robertson D. L., Boot-Handford R. P. (2005). The characterisation of six ADAMTS proteases in the basal chordate Ciona intestinalis provides new insights into the vertebrate ADAMTS family. Int. J. Biochem. Cell Biol. 37, 1838-1845 [DOI] [PubMed] [Google Scholar]
- Jugessur A., Murray J. C. (2005). Orofacial clefting: recent insights into a complex trait. Curr. Opin. Genet. Dev. 15, 270-278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungers K. A., Le Goff C., Somerville R. P., Apte S. S. (2005). Adamts9 is widely expressed during mouse embryo development. Gene Expr. Patterns 5, 609-617 [DOI] [PubMed] [Google Scholar]
- Kaufman M. H. (1992). The Atlas Of Mouse Development. London: Academic Press; [Google Scholar]
- Kern C. B., Twal W. O., Mjaatvedt C. H., Fairey S. E., Toole B. P., Iruela-Arispe M. L., Argraves W. S. (2006). Proteolytic cleavage of versican during cardiac cushion morphogenesis. Dev. Dyn. 235, 2238-2247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern C. B., Norris R. A., Thompson R. P., Argraves W. S., Fairey S. E., Reyes L., Hoffman S., Markwald R. R., Mjaatvedt C. H. (2007). Versican proteolysis mediates myocardial regression during outflow tract development. Dev. Dyn. 236, 671-683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern C. B., Wessels A., McGarity J., Dixon L. J., Alston E., Argraves W. S., Geeting D., Nelson C. M., Menick D. R., Apte S. S. (2010). Reduced versican cleavage due to Adamts9 haploinsufficiency is associated with cardiac and aortic anomalies. Matrix Biol. 29, 304-316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo B. H., Longpre J. M., Somerville R. P., Alexander J. P., Leduc R., Apte S. S. (2006). Cell-surface processing of pro-ADAMTS9 by furin. J. Biol. Chem. 281, 12485-12494 [DOI] [PubMed] [Google Scholar]
- Koo B. H., Longpre J. M., Somerville R. P., Alexander J. P., Leduc R., Apte S. S. (2007). Regulation of ADAMST9 secretion and enzymatic activity by its propeptide. J. Biol. Chem. 282, 16146-16154 [DOI] [PubMed] [Google Scholar]
- Koo B. H., Coe D. M., Dixon L. J., Somerville R. P., Nelson C. M., Wang L. W., Young M. E., Lindner D. J., Apte S. S. (2010). ADAMTS9 is a cell-autonomously acting, anti-angiogenic metalloprotease expressed by microvascular endothelial cells. Am. J. Pathol. 176, 1494-1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaGamba D., Nawshad A., Hay E. D. (2005). Microarray analysis of gene expression during epithelial-mesenchymal transformation. Dev. Dyn. 234, 132-142 [DOI] [PubMed] [Google Scholar]
- Landolt R. M., Vaughan L., Winterhalter K. H., Zimmermann D. R. (1995). Versican is selectively expressed in embryonic tissues that act as barriers to neural crest cell migration and axon outgrowth. Development 121, 2303-2312 [DOI] [PubMed] [Google Scholar]
- Le Goff C., Somerville R. P., Kesteloot F., Powell K., Birk D. E., Colige A. C., Apte S. S. (2006). Regulation of procollagen amino-propeptide processing during mouse embryogenesis by specialization of homologous ADAMTS proteases: insights on collagen biosynthesis and dermatosparaxis. Development 133, 1587-1596 [DOI] [PubMed] [Google Scholar]
- Liu W., Sun X., Braut A., Mishina Y., Behringer R. R., Mina M., Martin J. F. (2005). Distinct functions for Bmp signaling in lip and palate fusion in mice. Development 132, 1453-1461 [DOI] [PubMed] [Google Scholar]
- Llamazares M., Cal S., Quesada V., Lopez-Otin C. (2003). Identification and characterization of ADAMTS-20 defines a novel subfamily of metalloproteinases-disintegrins with multiple thrombospondin-1 repeats and a unique GON domain. J. Biol. Chem. 278, 13382-13389 [DOI] [PubMed] [Google Scholar]
- McCulloch D. R., Goff C. L., Bhatt S., Dixon L. J., Sandy J. D., Apte S. S. (2009a). Adamts5, the gene encoding a proteoglycan-degrading metalloprotease, is expressed by specific cell lineages during mouse embryonic development and in adult tissues. Gene Expr. Patterns 9, 314-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch D. R., Nelson C. M., Dixon L. J., Silver D. L., Wylie J. D., Lindner V., Sasaki T., Cooley M. A. W. S. A., Apte S. S. (2009b). ADAMTS proteases generate active versican fragments that regulate interdigital web regression. Dev. Cell 17, 687-698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miettinen P. J., Chin J. R., Shum L., Slavkin H. C., Shuler C. F., Derynck R., Werb Z. (1999). Epidermal growth factor receptor function is necessary for normal craniofacial development and palate closure. Nat. Genet. 22, 69-73 [DOI] [PubMed] [Google Scholar]
- Mjaatvedt C. H., Yamamura H., Capehart A. A., Turner D., Markwald R. R. (1998). The Cspg2 gene, disrupted in the hdf mutant, is required for right cardiac chamber and endocardial cushion formation. Dev. Biol. 202, 56-66 [DOI] [PubMed] [Google Scholar]
- Mori C., Nakamura N., Okamoto Y., Osawa M., Shiota K. (1994). Cytochemical identification of programmed cell death in the fusing fetal mouse palate by specific labelling of DNA fragmentation. Anat. Embryol. (Berl.) 190, 21-28 [DOI] [PubMed] [Google Scholar]
- Murray J. C., Schutte B. C. (2004). Cleft palate: players, pathways, and pursuits. J. Clin. Invest. 113, 1676-1678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oblander S. A., Zhou Z., Galvez B. G., Starcher B., Shannon J. M., Durbeej M., Arroyo A. G., Tryggvason K., Apte S. S. (2005). Distinctive functions of membrane type 1 matrix-metalloprotease (MT1-MMP or MMP-14) in lung and submandibular gland development are independent of its role in pro-MMP-2 activation. Dev. Biol. 277, 255-269 [DOI] [PubMed] [Google Scholar]
- Perissinotto D., Iacopetti P., Bellina I., Doliana R., Colombatti A., Pettway Z., Bronner-Fraser M., Shinomura T., Kimata K., Morgelin M., et al. (2000). Avian neural crest cell migration is diversely regulated by the two major hyaluronan-binding proteoglycans PG-M/versican and aggrecan. Development 127, 2823-2842 [DOI] [PubMed] [Google Scholar]
- Perris R., Perissinotto D., Pettway Z., Bronner-Fraser M., Morgelin M., Kimata K. (1996). Inhibitory effects of PG-H/aggrecan and PG-M/versican on avian neural crest cell migration. FASEB J. 10, 293-301 [DOI] [PubMed] [Google Scholar]
- Proetzel G., Pawlowski S. A., Wiles M. V., Yin M., Boivin G. P., Howles P. N., Ding J., Ferguson M. W., Doetschman T. (1995). Transforming growth factor-beta 3 is required for secondary palate fusion. Nat. Genet. 11, 409-414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao C., Foernzler D., Loftus S. K., Liu S., McPherson J. D., Jungers K. A., Apte S. S., Pavan W. J., Beier D. R. (2003). A defect in a novel ADAMTS family member is the cause of the belted white-spotting mutation. Development 130, 4665-4672 [DOI] [PubMed] [Google Scholar]
- Rice R., Spencer-Dene B., Connor E. C., Gritli-Linde A., McMahon A. P., Dickson C., Thesleff I., Rice D. P. (2004). Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J. Clin. Invest. 113, 1692-1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandy J. D., Westling J., Kenagy R. D., Iruela-Arispe M. L., Verscharen C., Rodriguez-Mazaneque J. C., Zimmermann D. R., Lemire J. M., Fischer J. W., Wight T. N., et al. (2001). Versican V1 proteolysis in human aorta in vivo occurs at the Glu441-Ala442 bond, a site that is cleaved by recombinant ADAMTS-1 and ADAMTS-4. J. Biol. Chem. 276, 13372-13378 [DOI] [PubMed] [Google Scholar]
- Shi J., Son M. Y., Yamada S., Szabova L., Kahan S., Chrysovergis K., Wolf L., Surmak A., Holmbeck K. (2008). Membrane-type MMPs enable extracellular matrix permissiveness and mesenchymal cell proliferation during embryogenesis. Dev. Biol. 313, 196-209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver D. L., Hou L., Somerville R., Young M. E., Apte S. S., Pavan W. J. (2008). The secreted metalloprotease ADAMTS20 is required for melanoblast survival. PLoS Genet. 4, 1-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somerville R. P., Longpre J. M., Jungers K. A., Engle J. M., Ross M., Evanko S., Wight T. N., Leduc R., Apte S. S. (2003). Characterization of ADAMTS-9 and ADAMTS-20 as a distinct ADAMTS subfamily related to Caenorhabditis elegans GON-1. J. Biol. Chem. 278, 9503-9513 [DOI] [PubMed] [Google Scholar]
- Stankunas K., Hang C. T., Tsun Z. Y., Chen H., Lee N. V., Wu J. I., Shang C., Bayle J. H., Shou W., Iruela-Arispe M. L., et al. (2008). Endocardial Brg1 represses ADAMTS1 to maintain the microenvironment for myocardial morphogenesis. Dev. Cell 14, 298-311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taya Y., O'Kane S., Ferguson M. W. (1999). Pathogenesis of cleft palate in TGF-beta3 knockout mice. Development 126, 3869-3879 [DOI] [PubMed] [Google Scholar]
- Trokovic N., Trokovic R., Mai P., Partanen J. (2003). Fgfr1 regulates patterning of the pharyngeal region. Genes Dev. 17, 141-153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T., Vivatbutsiri P., Morriss-Kay G., Saga Y., Iseki S. (2008). Cell lineage in mammalian craniofacial mesenchyme. Mech. Dev. 125, 797-808 [DOI] [PubMed] [Google Scholar]
- Yu L., Gu S., Alappat S., Song Y., Yan M., Zhang X., Zhang G., Jiang Y., Zhang Z., Zhang Y., et al. (2005). Shox2-deficient mice exhibit a rare type of incomplete clefting of the secondary palate. Development 132, 4397-4406 [DOI] [PubMed] [Google Scholar]
- Zhang Z., Song Y., Zhao X., Zhang X., Fermin C., Chen Y. (2002). Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development 129, 4135-4146 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}