

Abstract
Mutations in the human Notch ligand jagged 1 (JAG1) result in a multi-system disorder called Alagille syndrome (AGS). AGS is chiefly characterized by a paucity of intrahepatic bile ducts (IHBD), but also includes cardiac, ocular, skeletal, craniofacial and renal defects. The disease penetration and severity of the affected organs can vary significantly and the molecular basis for this broad spectrum of pathology is unclear. Here, we report that Jag1 inactivation in the portal vein mesenchyme (PVM), but not in the endothelium of mice, leads to the hepatic defects associated with AGS. Loss of Jag1 expression in SM22α-positive cells of the PVM leads to defective bile duct development beyond the initial formation of the ductal plate. Cytokeratin 19-positive cells are detected surrounding the portal vein, yet they are unable to form biliary tubes, revealing an instructive role of the vasculature in liver development. These findings uncover the cellular basis for the defining feature of AGS, identify mesenchymal Jag1-dependent and -independent stages of duct development, and provide mechanistic information for the role of Jag1 in IHBD formation.
Keywords: Notch, Liver development, Endothelium, Vascular smooth muscle, Vasculature, Mouse
INTRODUCTION
The Notch pathway is a conserved signaling system used extensively throughout embryonic development that continues to function in a cell context-dependent manner in the maintenance of tissues and stem cells in adults (Artavanis-Tsakonas et al., 1999; Ehebauer et al., 2006; Weinmaster, 2000). A hallmark of Notch signaling is the requirement for direct cell-to-cell contact, as both the Notch ligands and receptors are integral membrane proteins (D'Souza et al., 2008). Given the widespread use of Notch signaling, it is not surprising that mutations in receptors and ligands of the Notch pathway have been implicated in the onset of several human pathologies, including: cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), T-cell acute lymphoblastic leukemia (T-ALL) and Alagille syndrome (AGS) (Gridley, 2007; Hansson et al., 2004; Hofmann and Iruela-Arispe, 2007b; Weng and Lau, 2005). In particular, AGS is an autosomal-dominant disorder linked to the Notch ligand jagged 1 (JAG1) (Alagille et al., 1987; Li et al., 1997; Oda et al., 1997b) and, to a lesser extent, to the Notch 2 receptor (McDaniell et al., 2006). The disease is primarily characterized by paucity of intrahepatic bile ducts (IHBD) in the liver, but also includes a variety of cardiac, skeletal, renal, vascular and ophthalmologic abnormalities (Alagille et al., 1987; Li et al., 1997; Oda et al., 1997a). Although Jag1 mutations produce pleiotropic effects, the initiating cellular and molecular alterations responsible for the wide spectrum of AGS phenotypes have not been clearly defined. Our findings suggest that the initial onset of AGS is probably rooted during early developmental stages.
The development of IHBDs occurs in a series of sequential steps that can be molecularly and histologically identified (Lemaigre, 2003; Si-Tayeb et al., 2010). A subpopulation of hepatoblasts initiates expression of biliary-specific genes, transcription factors (such as Sox9) and a subset of cytokeratins. This event segregates the biliary precursor cells (cytokeratin-positive) from cells destined to become hepatocytes (cytokeratin-negative) (Lemaigre, 2003; Raynaud et al., 2009). These biliary progenitor cells further differentiate into (BECs) or cholangiocytes. At later stages in development (E15.5-E16.5), BECs organize into a single-layered sheet called the ductal plate, which exclusively surrounds the portal vein. Biliary morphogenesis proceeds with the sequential and asymmetrical differentiation of a second ductal plate layer from hepatoblasts on the parenchymal side to form lumenized mature bile ducts (Antoniou et al., 2009; Si-Tayeb et al., 2010).
The paucity of bile ducts and histological features of AGS may occur through several distinct mechanisms: interruption of cholangiocyte fate, poor proliferation, increased cell death or defects in terminal differentiation. Although it is clear that JAG1 and NOTCH2 contribute to the disease, it has been difficult to identify the ligand-expressing cell, the onset and mechanistic origin of the phenotype, and the specific cellular events that are impaired in AGS (Lemaigre, 2008; Raynaud et al., 2009).
Upon investigation of the contributions of Jag1 to vascular smooth muscle in mice, we unexpectedly encountered the major hepatic phenotype of AGS. Specifically, deletion of Jag1 in SM22α-expressing cells of the developing portal vein mesenchyme (PVM) resulted in jaundice, liver failure and paucity of IHBDs. These findings delineate a crucial role for the vasculature in providing specific and irreversible instructional information via Jag1 that is essential for IHBD development.
MATERIALS AND METHODS
Mouse lines
All strains of mice have been previously described: VE-Cadherin-Cre (Alva et al., 2006), SM22-Cre (TagCre) (Holtwick et al., 2002) and Jag1lox/lox provided by G. Radtke (Mancini et al., 2005). These mice were crossed to reporter lines Rosa26R LacZ (Soriano, 1999) or Rosa26R eYFP (Srinivas et al., 2001), and were backcrossed six generations on a C57BL/6J background. For assessment of survival of SM22-Cre;Jag1 mice, 108 J1SMKO and 287 J1WT+SMHet mice were sampled. All studies were conducted in accordance with Animal Research Committee guidelines at UCLA.
Tissue collection and staining
Standard protocols for collecting, staining, immunohistochemistry, β-galactosidase (β-Gal) were performed as previously described (Alva et al., 2006). For non-fluorescent immunostaining, tissues were incubated with primary antibodies and biotinylated secondary antibodies (Jackson ImmunoResearch Laboratories), treated with a Vectastain ABC kit (Vector Labs) and DAB substrate (Vector Labs).
Antibodies, lectins and nuclear imaging
Antibodies for immunohistochemistry included: rabbit wide-spectrum cytokeratins (Dako) (1:800), rat cytokeratin 8 [Troma I; Developmental Studies Hybridoma Bank (DHSB)] (1:300), rat cytokeratin 19 (Troma III; DSHB) (1:300), goat polyclonal jagged 1 (R&D Systems) (1:25); anti-Jag1 (1:200, Santa Cruz Biotechnology) Type IV Collagen (Cosmo Bio) (1:1000), anti-laminin (Sigma) (1:800), rabbit Sox9 (Chemicon) (1:500), goat HNF1β (Santa Cruz Biotechnology) (1:25), goat HNF4α (Santa Cruz Biotechnology) (1:50), rabbit Hes1 (a gift from Ben Stanger, University of Pennsylvania, Philadelphia, PA, USA) (1:1500), rabbit PECAM1 (a gift from Joseph Madri, Yale, New Haven, CT, USA) (1:250), Texas-Red-Phalloidin (Molecular Probes) (1:50) and rat PECAM1 (BD Pharmingen) (1:250). For Sox9, Hes1, HNF1β and HNF4α immunostaining, tyramide signal amplification was performed using the TSA Kit (Molecular Probes). Antibodies used for western blots included cytokeratin 8 (Troma I; DSHB), cytokeratin 19 (Troma III; DSHB), E-cadherin (Cell Signaling), Notch2 (Santa Cruz Biotechnology; Cell Signaling; R&D Systems) (1:500), Jagged1 (Santa Cruz Biotechnology) (1:800), α-tubulin, fibronectin and α-enolase. Fluorescein-labeled Dolichos biflorus agglutinin (DBA) (Vector Laboratories) was used for FACs. For nuclear counterstaining, we used DAPI (Invitrogen) (1:1000) and TOPRO-3 (Molecular Probes) (1:500). Alexa Fluor (Invitrogen) or FITC-conjugated (Jackson ImmunoResearch Laboratories) fluorescent secondaries were used for confocal evaluation on a Zeiss LSM multiphoton microscope.
Blood serum analysis
Blood was collected immediately post-mortem from three litters of P25 mice and standard liver panels were performed by the UCLA Department of Laboratory Animal Medicine.
FACS analysis, cell sorting, western blot analysis and immunoprecipitation
Livers from E16.5, P0, P2 and P10-11 J1WT, J1SMHet and J1SMKO mice were processed for FACS analysis, as previously described (Zovein et al., 2010). DBA was used for BEC identification and negatively gated using APC-conjugated CD45 antibodies (BD Pharmingen) for neonatal livers. All samples were stained for viability with 7-AAD (BD Pharmingen) and analyzed on a FACSCaliber with the appropriate isotype controls. E16.5 and P2 BECs for western blot analysis were counted and protein levels were quantified. For immunoprecipitation, lysates of embryonic were quantified for protein concentration (DC protein, BioRad). A total of 800 μg per sample was used in a volume of 400 μl. Samples were pre-cleared with beads bound to secondary antibodies and incubated overnight with anti-Jag1. Recovery of the immunocomplexes and western blot were carried out as previously described (Chen et al., 2010).
Tissue culture
BECs were collected from J1WT or J1SMKO E16.5 or P0-P2 mice, as already described, and cells were plated on collagen-coated dishes. For mesenchyme isolation, whole E16.5 and P0-P2 livers were cut, treated with 0.5 mg/ml collagenase and plated on fibronectin-coated plates. Non-attached cells were removed 1-2 hours after initial plating by washing the plates with DMEM. Isolation of PVM was confirmed by eYFP. Dermal fibroblasts were collected by a similar procedure. For 2D cultures, BECs and PVM were mixed and cultured on collagen-coated dishes. For 3D cultures, a matrigel and fibronectin (50 μg/ml) gel was mixed gently with equal number of epithelial and fibroblast/PVM cells. The mixture was polymerized in 24-well plates for 30 minutes at 37°C and media was added. Three dimensional cultures were fixed in 1:1 acetone/methanol at –20°C, washed with 25% sucrose, immunostained and were imaged on a confocal microscope. For Notch inhibition, cells were treated with 50-100 μM DAPT (N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester), changed every 48 hours. Cultures were grown for 7-14 days and spheroid formation was assessed. For Notch2 pre-activation, Jag1 was immobilized on 100 mm2 Petri dishes as previously described (Varnum-Finney et al., 2000). Briefly, plates were coated with anti-human IgG (Fcg-specific) at 20 μg/ml (Jackson Immuno Research) for 30 minutes, washed and blocked with 2% BSA for 2 hours. Subsequently, Jag1 (10 μg/ml) (R&D Systems) was bound to the treated plates for 2 hours at 37°C. Controls included plates treated in an identical manner, but without Jag1. BECs were plated on Jag1 and Fc-coated plates overnight, removed by trypsinization and lysed or mixed with PVM for 3D cultures.
RESULTS
Vascular smooth muscle deletion of Jag1 recapitulates the liver phenotype of AGS
To investigate the role of Jag1 within distinct cell populations of the vasculature, we developed two separate mouse models that allowed for conditional Jag1 deletion in either vascular smooth muscle or the endothelium. Jag1 deletion in smooth muscle cells was investigated in SM22-Cre mice crossed to Jag1 floxed mice (Fig. 1A) to create Jag1-smooth muscle knockout mice (J1SMKO). To generate Jag1-endothelial cell knockout mice (J1ECKO), we bred VE-Cadherin Cre mice with Jag1 floxed mice (Fig. 1A). Both lines yielded viable pups (Fig. 1B,C), although J1SMKO mice unexpectedly exhibited a high degree of jaundice and growth retardation, obvious at birth (Fig. 1B; see Fig. S1 in the supplementary material). These features were absent in Cre-negative Jag1 floxed mice (J1WT), Jag1 smooth muscle heterozygous (J1SMHet) littermates and J1ECKO mice (Fig. 1C). The J1SMKO mice were born at appropriate Mendelian frequencies; however, the majority of J1SMKO neonates died within the first 2 days and only a small subset (9/136 total mice) survived beyond 4 weeks (Fig. 1D; Table 1). J1SMKO mice that survived beyond a week continued to exhibit growth retardation, hair loss and lethargy that worsened over time (Fig. 1E; see Fig. S1 in the supplementary material). These features recapitulate symptoms displayed by AGS patients and are clinically related to liver dysfunction (Emerick et al., 1999; Krantz et al., 1997).
Fig. 1.

Smooth muscle deletion of Jag1, but not endothelial deletion, leads to jaundice, cholestasis and bile duct paucity. (A) Mating schema for generating J1SMKO and J1ECKO mice. (B) J1SMKO exhibited jaundice and growth retardation from birth. (C) J1ECKO mice were viable and normal in size and appearance. (D) Kaplan-meier survival analysis of newborn J1SMKO mice. (E) Weight (mean, in grams) of J1SMKO, J1ECKO and J1WT+Het mice. Bars indicate s.e.m. (F) (Top) Livers of J1SMKO mice showed hemorrhage (chevrons) and bile deposits (open arrows). (Middle) Hematoxylin and Eosin staining of P7 livers revealed bile ducts in J1WT and J1ECKO mice (arrows), and a lack of patent bile ducts (arrowhead), as well as necrosis (broken white line, open arrow), in J1SMKO mice. (Bottom) Cytokeratin immunostaining of P25 livers indicated bilayered lumenized bile ducts in J1WT and J1ECKO (arrows) and non-lumenized ducts in J1SMKO (open arrows) mice. gb, gall bladder; pv, portal vein. Scale bars: top row, 20 mm; middle and bottom rows, 100 μm.
Table 1.
Mendelian analysis of mice with smooth muscle Jag1 deletion

Upon gross examination, the livers of P25 J1SMKO mice exhibited large patches of bile accumulation within the parenchyma (Fig. 1F, open arrows) and multiple hemorrhagic foci (Fig. 1F, chevrons), in contrast to the appearance of J1ECKO and J1WT mice (Fig. 1F). Histological examination of J1SMKO livers also revealed bile deposits and areas of necrosis (Fig. 1F, white outlines and open arrows), hemorrhage, and either absent or incompletely formed bile ducts near the portal veins (Fig. 1F, arrowhead), when compared to the ducts (Fig. 1F, arrows) in J1WT and J1ECKO livers. Blood chemistry profiles of J1SMKO mice additionally revealed increased serum levels of liver enzymes (Table 2), indicative of extensive liver dysfunction and a likely cause of the high lethality in the J1SMKO population.
Table 2.
Blood chemistry analysis

Identification of BECs using cytokeratin demonstrated that J1SMKO BECs were unable to organize into tubes (Fig. 1F; see Fig. S2 in the supplementary material). BECs were, however, located mostly in a single layer adjacent to the portal vein in J1SMKO mice. By contrast, J1WT and J1ECKO mice had fully remodeled, lumenized ducts (Fig. 1F, arrows). The structures in J1SMKO mice resembled the epithelial sheets of the ductal plate that form during early IHBD development. Extracellular matrix molecules were expressed at similar levels between the mice (see Fig. S2 in the supplementary material).
Loss of Jag1 in the periportal mesenchyme affects completion of bile duct morphogenesis
Although expression of Jag1 in blood vessels is most frequently confined to arteries (Hofmann and Iruela-Arispe, 2007a; Villa et al., 2001), the portal vein is an exception to the rule (Fig. 2B) (Loomes et al., 2007). As a result, much debate has ensued over whether Jag1 in cells that make up the portal vein (endothelium or mesenchyme) could be responsible for interacting with Notch2 expressed by BECs to promote bile duct morphogenesis (Geisler et al., 2008; Kodama et al., 2004; Lemaigre, 2008; Loomes et al., 2007; Lozier et al., 2008). Using immunocytochemistry during the stages of IHBD development, Jag1 was found in both the endothelium of the portal vein and the PVM (Fig. 2A-C, arrows and asterisks), and in BECs later in duct development (Fig. 2B,C,I-J′). In J1SMKO mice, Jag1 expression was predominantly in the endothelium and was absent in perivascular mesenchyme (Fig. 2D-F). Jag1 immunoprecipitation from whole-liver extracts revealed that loss of Jag1 in J1SMKO livers occurred progressively after E14.5, during ductal plate morphogenesis (Fig. 2G). We further isolated endothelium, epithelium and mesenchyme from P3 J1SMKO and littermate controls (Fig. 2H). All three cell types express Jag1 (Fig. 2H-J′), but only loss of Jag1 in the mesenchyme (Fig. 2H,K-L′, Fig. 6A) elicited defects in biliary duct formation and AGS-like features.
Fig. 2.

Loss of Jag1 in portal vein mesenchyme results in defective bile duct morphogenesis. (A-F) During IHBD development, Jag1 was found in the endothelium (arrows) of both mice, but only in the mesenchyme (asterisks) of J1WT mice. BECs (arrowheads) labeled with wide-spectrum cytokeratin (ws-CK) form a ductal plate alongside Jag1-positive PVM, while the J1SMKO liver had not yet formed this structure (E). At P3, the J1WT liver displayed a well-formed bile duct and Jag1 expression (C, arrows) remained in the endothelium, PVM and BECs. J1SMKO mice lack patent duct formation and showed a single layer of ws-CK positive cells that resembled the primitive ductal plate (pdp) (F, arrowheads). (G) Immunoprecipitation of Jag1 in whole liver lysates from J1WT and J1SMKO mice at E14.5, E16.5 and E18.5. Ns, non-specific band. (H) Western blot of isolated endothelium, BECs and PVM from P3 J1WT and J1SMKO livers. (I-L′) Jag1 expression (in red) alone in the endothelium and PVM, and co-localized with ws-CK (green) in BECs (I′,J′, arrowheads) of E18.5 and P3 J1WT livers. (J′,J′) Jag1-positive PVM (asterisks) at P3 (J′ is a higher magnification of J′). (K,K′) J1SMKO livers maintain Jag1 expression in the endothelium, but no staining was seen in the PVM or the BECs. (L′,L′) At P3, J1SMKO livers showed Jag1 colocalized with ws-CK in the disorganized BECs (arrowheads), but absent from PVM (L′ is a higher magnification of L′, bracket). bd, bile duct; bec, biliary epithelial cell; dp, ductal plate; en, endothelium; ha, hepatic artery; pdp, primitive ductal plate; pv, portal vein; pvm, portal vein mesenchyme. Scale bars: 50 μm.
Fig. 6.

Jag1 in the PVM is required for BEC spheroid formation. (A) Western blot of Jag1 in PVM cultures from mutant and control mice. (B) Two-dimensional co-cultures containing BECs and PVM do not form spheroids, but 3D co-cultures with PVM, not with dermal fibroblasts (DF) induce clustering of BECs (arrowhead). (C) Wild-type BECs cultured with J1WT PVM form spheroids (arrows), but failed to compact when co-cultured with J1SMKO PVM. (D) Quantification of spheroids from multiple cultures (n=5 independent cultures). *P≤0.001; error bars represent ±s.e.m. (E-G′) Immunostaining of the 3D cultures showed expression of BEC differentiation markers (ws-CK or CK19, in green). Wild-type BECS cultured with J1SMKO PVM did not form large spheroids, but did show smaller cell aggregates that expressed ws-CK (open arrow), similar to the clumps of cells around the portal vein seen in vivo (arrows). Scale bars: 100 μm in B,C,F; 50 μm in E-G,G′; 10 μm in E′,F′.
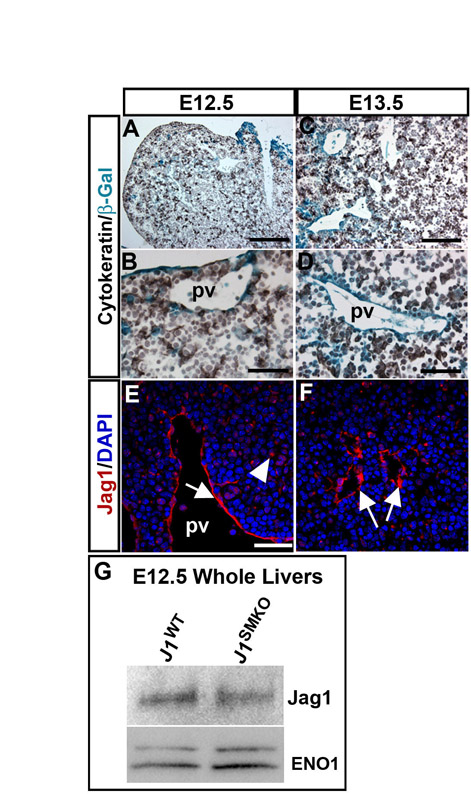
Jag1-positive cells in the PVM (Fig. 2I-J′) were adjacent to BECs during ductal plate formation and morphogenesis. Furthermore, Jag1 expression in the hepatic artery was also evident in neonatal J1WT livers (Fig. 2C). Cre-mediated deletion of Jag1 was examined in the endothelium and PVM, and further confirmed by evaluation of protein expression (Fig. 2D-F,G,H,K-L; see Figs S2 and S3 in the supplementary material). Jag1 expression in the BECs of J1SMKO mice was not apparent until P3, which was slightly delayed compared with Jag1-positive BECs in E18.5 J1WT mice (Fig. 2I-L′; arrowheads). Evaluation of BECs at P3 revealed that J1SMKO mice retained the non-lumenized structure of the ductal plate in a stage analogous to E18.5. This occurred despite expression of Jag1 in BECs (Fig. 2L,L′; arrowheads). Loss of Jag1 was evident in the PVM of P3 J1SMKO mice (Fig. 2L′; bracket) in comparison with J1WT mice (Fig. 2J′; asterisks).
The unexpected hepatic phenotype of J1SMKO mice prompted us to examine the temporal and cellular distribution of SM22-Cre in the context of IHBD development. As IHBD development has been well characterized (Lemaigre, 2003; Raynaud et al., 2009), we chose representative time-points encompassing the main stages of differentiation including: biliary specification (∼E12.5-E14.5), ductal plate formation (∼E15.5), ductal plate morphogenesis/lumen initiation (∼E16-birth) and the emergence of differentiated ducts (birth-P14) (Fig. 3A-D′; see Fig. S3 in the supplemental material). Cre activity, tracked by the Rosa26 reporter mouse line, was found in vascular elements, including the PVM, and in some endothelium, a caveat of this Cre line (Zovein et al., 2008) (Fig. 3). Both SM22-Cre and Jag1 expression during early liver development (E12.5-E13.5) were mostly confined to the endothelium and some cells in the liver parenchyma as cytokeratin-positive BECs were emerging (see Fig. S3 in the supplemental material). Later, during ductal plate formation and differentiation, BECs were embedded within Cre-recombined mesenchymal cell populations (Fig. 3B-D′). The developmental window corresponds to the absence of Jag1 within the PVM in J1SMKO mice (Fig. 2D-F,K-L′). Cre protein expression was evaluated throughout these stages by western blot and was found as early as E12.5 (Fig. 3E). Cre levels increased at E16.5-E18.5, during ductal plate morphogenesis (Fig. 3E), but Cre was clearly absent from BECs (Fig. 3F,F′).
Fig. 3.

Specificity of Cre expression in developing livers. (A-D) SM22-Cre;R26R double transgenic mice were used to trace Cre activity (β-gal staining, blue) during bile duct morphogenesis (cytokeratin, black). At E15.5, BECs (B,B′, arrowheads) aggregate near β-gal-positive mesenchymal cells that form the primitive ductal plate. By E18.5, a few lumenized bile ducts (C,C′, arrows) form within the mesenchyme (red bracket) and continue to develop at P3 (D,D′, arrows). (E) Western blot for Cre on liver lysates from SM22-Cre;R26R and control mice. Non-specific bands (arrowhead) indicate loading. (F) Sequential sections of SM22Cre;Rosa26R E18.5 (F,F′) livers with and without ws-CK staining. Arrowheads indicate BECs. (G-I) Mesenchymal layer thickness was decreased in J1SMKO livers compared with heterozygous littermates (brackets), and BEC organization was similar to the primitive ductal plate (H, arrowhead). In J1ECKO livers, the endothelium (I, open arrows), but not the mesenchyme, was β-gal positive. (J) SM22-Cre;R26R adults show persistence of β-gal staining in both endothelial and smooth muscle cells (open arrows), as well as the PVM (red bracket) surrounding mature bile ducts (arrows). ha, hepatic artery; pv, portal vein; Std, molecular weight standard. Scale bars: 100 μm in A-D; 50 μm in A′-D′,F-J.
We compared Cre recombination within the PVM of J1SMHet, J1SMKO and J1ECKO neonatal livers and found that J1SMHet and J1ECKO mice exhibited normal mesenchymal expansion (Fig. 3G,I) and differentiated bile ducts (arrows). By contrast, J1SMKO PVM was reduced (Fig. 3H, bracket), and the organization of the BECs (arrowheads) resembled ductal plate formation at E16.5. Overall, the findings suggest a lack of progression of biliary duct morphogenesis beyond the initial formation of the ductal plate. Additionally, J1SMHet mice showed abnormally organized cytokeratin-positive cells at 3 months of age (see Fig. S2 in the supplementary material), similar to the livers of surviving J1SMKO mice. This may suggest a gene-dose effect of Jag1 protein on proper bile duct assembly, as previously reported in Jag1 conditional/null mice (Loomes et al., 2007). SM22-Cre;Rosa26R adult mice (wild-type levels of Jag1 expression) exhibited bile duct incorporation within the expanded mesenchyme that also encompasses the vascular smooth muscle population surrounding the hepatic artery (Fig. 3J) (Lemaigre, 2008).
The PVM probably includes other cell types that may have at one time expressed SM22α, including hepatic stellate cells (Suzuki et al., 2008) and myofibroblasts (Libbrecht et al., 2002). However, the majority of SM22-Cre expression was localized to mesenchymal tissue, and not BECs or hepatocytes. There was minimal leakage of β-gal expression into the BEC lineage of adult SM22-Cre:R26R mice, and Jag1 expression was maintained in the BECs of J1SMKO mice (Fig. 2L), supporting the notion that epithelial-expressed Jag1 was not responsible for the observed biliary defects. Although the SM22-Cre-mediated deletion might also include a small subset of endothelial cells within the liver, the levels are minimal (Figs 2, 3). More importantly, full deletion of Jag1 in the endothelium (Fig. 3I, open arrows; see Fig. S2 in the supplementary material) does not recapitulate the hepatic defects of AGS. Instead, our findings indicate that only the loss of Jag1 in the PVM leads to paucity of bile ducts.
Deletion of Jag1 in the PVM halts BEC tubulogenesis and disrupts Sox9 expression
Several recently published studies have characterized the expression of differentiation markers in BECs during the various stages of bile duct development and morphogenesis, and have suggested that tubulogenesis occurs through a process of transient asymmetry (Antoniou et al., 2009; Zong et al., 2009). Thus, we examined the expression patterns of several BEC markers in J1SMKO livers to determine whether they were affected by the loss of Jag1. In particular, Sox9 has been shown to control the timing of duct maturation, and has been identified as a Notch target gene (Antoniou et al., 2009; Zong et al., 2009). At E18.5, BECs of J1WT mice expressed Sox9 contiguously around the portal vein and in both layers of lumenized ducts (Fig. 4A,C, arrows); by contrast, J1SMKO mice lacked Sox9 expression (Fig. 4B,D). In J1WT neonates, Sox9 expression became increasingly confined to maturing ducts (Fig. 4E,G,I, arrows). In J1SMKO mice, Sox9 was only found in a few epithelial cells between P1 and P10 (Fig. 4F,H,J, arrows), comparable with expression at E18.5 in wild-type mice (Fig. 4A,C). These findings support the notion that Sox9 is a direct target of Jag1-Notch signaling (Antoniou et al., 2009; Zong et al., 2009).
Fig. 4.

Expression of Sox9 in J1SMKO livers is affected by the loss of Jag1. (A-J) Immunostaining revealed delayed onset and reduced numbers of Sox9-positive cells (arrows) in J1SMKO mice in embryonic and neonatal livers compared with J1WT mice. (K-R) Hes1 expression (arrows) at E16.5 (N) was present in both J1WT and J1SMKO CK19-positive populations, but was still in the single layer in the disorganized BECs of E18.5 J1SMKO livers (R). bd, bile duct; pv, portal vein.
We also examined the expression of HNF1β and HNF4α, two genes involved in liver differentiation (Tanimizu and Miyajima, 2004), at E18.5 (see Fig. S4 in the supplemental material). HNF1β, which plays an essential role in epithelial differentiation (Coffinier et al., 2002), was expressed in a single layer of ductal cells in J1SMKO mice, unlike the circumferential expression in the ducts of J1WT mice. The pattern of expression of HNF4α, a hepatocyte-specific gene that regulates the development and adhesion of the epithelium (Battle et al., 2006; Parviz et al., 2003), appeared closer to the portal vein in J1SMKO livers, probably owing to the loss of PVM, yet the levels were comparable with wild type (see Fig. S4 in the supplemental material). Similar to homolog of hairy/enhancer of split-1 (Hes1) knockout mice, J1SMKO mice appeared to have normal differentiation of BECs from hepatoblasts, but defective duct morphogenesis (Kodama et al., 2004). Additionally, the loss of Sox9 has been shown to disturb Hes1 expression (Antoniou et al., 2009). This prompted us to evaluate Hes1 expression in J1WT and J1SMKO livers (Fig. 4K-R). At E16.5, Hes1 was present in the single, cytokeratin-positive epithelial layer of the ductal plate in both mutant and control mice (Fig. 4K-N). By E18.5, the disorganized BECs of J1SMKO mice maintained Hes1 expression but still in a single layer and lacked the formation of the second layer noted in the control mice (Fig. 4O-R). Expression levels also appeared reduced (Fig. 4R).
As the specification and differentiation of BECs are associated with expression of a cohort of cytokeratins, we examined the pattern of several markers in the absence of PVM Jag1. Although the expression level of cytokeratin 8 (CK8), an early BEC marker (Shiojiri, 1997), was similar between J1WT and J1SMKO mice, differences in the distribution and cellular organization of this protein were observed (Fig. 5). At P10, CK8-positive BECs of wild-type mice were arranged into a lumenized duct within the PVM (arrows, Fig. 5A,A′,E,F), whereas CK8-positive J1SMKO BECs were disorganized along the portal vein (Fig. 5B,B′,F). To examine whether the number of BECs was affected in the J1SMKO mice, we used FACS analysis of P0 and P10-P11 livers, and compared kinetics of BEC expansion. At P0, the number of BECs in J1SMKO mice was equivalent to J1WT and J1SMHet littermates, but the J1SMKO BEC population decreased significantly by P10-P11 (Fig. 5C). The BECs did not appear to progress beyond the formation of the second layer of the ductal plate, similar to AGS-associated paucity of bile ducts (Lemaigre, 2008; Li et al., 1997; Oda et al., 1997b). Furthermore, the differences in numbers of BECs at P10 indicate that in the absence of PVM-Jag1 these cells either stop proliferating or undergo apoptosis.
Fig. 5.

Loss of Jag1 halts biliary duct tubulogenesis. (A-B′) Cytokeratin 8 (CK8) in P10 J1WT and J1SMKO livers (higher magnifications in A′,B′). CK8-positive BECs formed ducts in J1WT livers (arrows), while J1SMKO BECs (arrowheads) did not. (C) FACS analysis of P0 livers from J1WT, J1SMHet and J1SMKO mice show a decrease number of BECs in J1SMKO mice at P10-11. Bars represent mean±s.e.m. *P<0.05 (n=3-7 animals per time point). (D) Western blot of E16.5 and P0 sorted BECs from J1WT and J1SMKO livers. E-cadherin (E-cad), CK8 and cytokeratin 19 (CK19) were increased in both mice at P0 in relation to E16.5. α-Enolase was used for loading control. (E) Fluorescent immunostaining of CK8 (red) and TOPRO3 (blue) in J1WT livers at E18.5 and P10. CK8 expression was initially present along the portal side (arrowheads), but expanded when both epithelial layers were present. Keratin filaments in BECs were bundled and visible at E18.5 (open arrows). By P10, BECs were organized into ducts and CK8 expression circumscribed the cells (arrows). (F,G) Ws-CK, (green) colocalized with CK8 and CK19 in J1WT P10 livers (arrows, fourth panels). By contrast, CK8 expression was widespread and did not fully overlap with ws-CK (F, arrowheads) in J1SMKO livers. CK19 expression in J1SMKO livers was present (G) on the portal side of the BEC layer and not on the parenchymal layer where ws-CK was expressed (arrowheads). CK8 and CK19 keratin filaments were less compacted and organized in J1SMKO BECs (open arrows). pv, portal vein; nt, necrotic tissue. Scale bars: 100 μm in A,B; 50 μm in A′,B′; 50 μm in E (first and third panels); 10 μm in E (second and fourth panels); 50 μm in F,G; 10 μm in F,G (fourth panels).
Using sorted BECs from J1WT and J1SMKO mice, we examined the expression of BEC markers during two stages of IHBD development: E16.5 and P0. We found increased expression of two duct-specific cytokeratins, as well as E-cadherin in BECs at P0 (in both mice) when compared with E16.5 (Fig. 5D). The persistence of CK19, a marker of later BEC differentiation (Shiojiri, 1997), in ductal cells has been reported previously in hepatoblast-specific Notch-deficient mice, but could also be a result of BECs not part of the developing bile ducts becoming incompetent to receive signals for elimination (Geisler et al., 2008; Sparks et al., 2009). To closely examine the cellular distribution of cytokeratins during embryonic and postnatal stages, we evaluated CK8 in E18.5 and P10 J1WT livers. At E18.5, CK8 was initially expressed in BECs along the portal side of the ductal plate, and extended to the cells on the parenchymal side upon formation of the second layer of the ductal plate (arrowheads, Fig. 5E). This asymmetric distribution was similar to that reported for CK19 (Zong et al., 2009) and is indicative of the establishment of polarity. In addition, keratin filaments were tightly bundled (open arrows, Fig. 5E) in these columnar cells. By P10, CK8-positive BECs were cuboidal and incorporated within ducts, with CK8 ringing the nucleus (arrows, Fig. 5E).
We then compared expression patterns of CK8 and CK19 in conjunction with a wide-spectrum cytokeratin (ws-CK) antibody in postnatal J1WT and J1SMKO livers. CK8 and CK19 overlapped with the ws-CK in J1WT BECs (Fig. 5F,G, arrows) and higher magnification showed the ring-like distribution of these keratins in the mature ducts. By contrast, CK8 in the J1SMKO BECs did not fully overlap with ws-CK expression (Fig. 5F, arrowheads). Higher magnification revealed disorganized keratin fibers (Fig. 5F, open arrows) and columnar shapes typically seen at E18.5 (Fig. 5E). Similarly, CK19 in J1SMKO mice was expressed along the portal side, but did not extend to the parenchymal side that was positive for ws-CK (Fig. 5G, arrowheads). Disorganized keratin filaments were also evident (Fig. 5F, open arrow). The expression pattern of CK19 resembled that of the wild type at an earlier more immature stage of development (Fig. 4E) (Zong et al., 2009). Thus, the BECs of J1SMKO mice appear to arrest at an intermediate stage of IHBD development when cytokeratin expression is still asymmetrical.
Lack of epithelial aggregation in the absence of Jag1 from the portal vein mesenchyme
To further clarify the specific contribution of Jag1 in the PVM during IHBD development, we isolated epithelial and mesenchymal cells from J1WT and J1SMKO mice at different stages of duct development and explored their interactions in vitro. BECs at E16.5, as well as neonatal stages of J1WT and J1SMKO livers were isolated by FACS (see Fig. S5 in the supplementary material). BECs from both genotypes grew equally well in culture and PVM cells were isolated and cultured from dissociated livers of J1WT and J1SMKO mice (see Fig. S5 in the supplementary material). We examined the deletion of Jag1 in cultured J1SMKO PVM using western blots (Fig. 6A), and then employed an in vitro co-culture system to evaluate the effect of Jag1 loss on BECs. In 2D co-cultures, BECs and PVM were able to interact; however, in 3D matrix gels, BECs bound to and grew on PVM cells and eventually compacted to form spheroids (Fig. 6B, arrowhead). Interestingly, compaction requires PVM, as co-cultures of BECs with dermal fibroblasts at the same developmental stage do not induce the formation of these structures (Fig. 6B). Wild-type E16.5 BECs were then cultured with either J1WT or J1SMKO PVM to compare the effects of Jag1 loss. The design of this 3D co-culture system allowed for the direct use of the Jag1-depleted PVM from J1SMKO mice at specific stages of duct development. Other ductal culture systems require the use of Jag1-positive hepatic progenitor lines to differentiate into the distinct hepatic lineages for ductal development (Tanimizu et al., 2009).
The formation of cytokeratin-positive spheroids (arrows in Fig. 6C-F) occurred progressively over several weeks using J1WT PVM. The spheroids also expressed CK19 in culture (Fig. 6G), and even formed lumenal structures (see Fig. S5 in the supplementary material). By contrast, spheroids were rare in J1SMKO PVM/wild-type BEC co-cultures (Fig. 6C-F). The BECs differentiated and expressed ws-CK (open arrow, Fig. 6F′); however, they were unable to form the compact spheres shown in J1WT cultures (Fig. 6E,E′, arrows). When J1SMKO BECs were grown on J1WT PVM, spheroids formed, indicating that Jag1 from the BEC lineage was not sufficiently instructive for the formation of spheroids (Fig. 6G′). Together, these data suggest that BECs require instructional information from the PVM, which is specifically mediated by Jag1 in order to initiate epithelial aggregation and complete duct morphogenesis.
A narrowly defined window of mesenchymally derived Jag1 signaling during early liver development promotes irreversible changes in BEC differentiation
Using the SM22-Cre line, our data indicated that mesenchymal Jag1 was not necessary for the initial formation of the ductal plate nor for the expression of cytokeratins by cholangiocytes, but nonetheless was required for assembly of mature and patent bile ducts. Thus, to better define the temporal window of Jag1 signaling during later BEC tubulogenesis, we used pharmacological blockade of Notch activity (Fig. 7). In the 3D co-culture system, Notch signaling was blocked using the pan-Notch inhibitor DAPT in wild-type PVM-epithelial cultures isolated at E16.5 and P2. Although E16.5 control cultures were able to form duct-like spheroid structures, exposure to DAPT effectively blocked compaction of epithelial cells (Fig. 7A) without affecting viability. By contrast, Notch inhibition did not affect spheroid formation in P2 BEC cultures (Fig. 7A), suggesting that P2 cultures have the potential to form spheroids in the absence of Notch signaling. A possible explanation for these differences is that an irreversible change in BEC differentiation (probably driven by Notch activation) had occurred in P2 epithelial cells that had not yet occurred in E16.5 cells (Fig. 7A). To test this hypothesis, we pre-activated E16.5 wild-type BECs with exogenous Jag1 ligand and then co-cultured them with J1SMKO PVM (Fig. 7B). Remarkably, the Jag1-pre-treated BECs enhanced detection of a cleaved form of Notch2 (Fig. 7B), indicative of Notch2 activation. As previously shown, E16.5 untreated control BECs did not form spheroids on J1SMKO PVM. By contrast, Jag1-activated BECs formed these structures despite being cultured with J1SMKO mesenchyme (Fig. 7B). The findings indicate that the Jag1 signaling axis gives an instructive cue for spheroid formation during a small, but critical, time window and is dispensable thereafter. The sum of these mechanistic experiments identifies both PVM-Jag1-dependent and -independent events during bile duct morphogenesis and has further expanded our understanding of the temporal and spatial involvement of this Notch ligand during the process of IHBD development (Fig. 8).
Fig. 7.

Temporal requirement of Jag1 signaling for morphogenesis of bile ducts. (A) Notch signaling blockade in wild-type co-cultures of PVM and BECs (at E16.5 and P2) with DAPT prevents spheroid formation (arrows) at E16.5 but not at P2. (B) Western blot demonstrates Notch2 activation within BECs after binding to Jag1 peptide (Fc, control). Black arrow indicates intact Notch2; black arrowhead indicates cleaved Notch2. When cultured with J1SMKO PVM, E16.5 BECs previously activated with soluble Jag1 peptide regained the ability to form spheroids (arrows), whereas BECs treated with Fc control peptide did not. Scale bars: 100 μm in A,B.
Fig. 8.

Spatial and temporal requirement of Jag1 during IHBD development. BEC specification and initial ductal plate formation occurs in a mesenchymal Jag1-independent manner (steps 1 and 2). Jag1 expression in the PVM controls the complete formation of the second layer of the ductal plate, allowing the BECs to eventually remodel into a lumenized duct. Expansion of PVM also appears to be dependent on Jag1 signaling (steps 3 and 4). Loss of Jag1 expression in the PVM causes duct development to stall midway during ductal plate morphogenesis (step 3), leading to a paucity of bile ducts.
DISCUSSION
Absence of Jag1 in the portal vein mesenchyme results in paucity of bile ducts and reveals the vascular roots of Alagille syndrome
The data presented in this study provide detailed insight into the contribution of the Notch ligand Jag1 in the vasculature to liver morphogenesis and pathology. Our findings indicate that Jag1, specifically in the PVM, is essential for biliary duct development, and is required within a critical embryonic timeframe. Initial BEC specification and the formation of the first layer of ductal plate appear to be PVM-Jag1 independent using both Cre deletion models. Importantly, Jag1-dependent signaling in the PVM is essential for the morphogenesis of BECs, expansion of the mesenchyme and the organization of mature bile ducts. Loss of developmental cues initiated by the Jag1-Notch2 signaling axis is known to be associated with paucity of bile ducts (one of the most prominent diagnostic indicators of AGS). The J1SMKO mouse line described herein provides a new liver-specific AGS model where conditional deletion of Jag1 in the PVM is sufficient to recapitulate the hepatic defects of AGS. Although other vascular anomalies associated with AGS were observed in both the J1SMKO and J1ECKO mice, further investigation is beyond the scope of the current study.
The connection between how impaired Notch signaling directly translates to bile duct paucity in AGS has not been fully mechanistically explained. Although previous work in mice has uncovered Notch2 requirements for BECs (Geisler et al., 2008; Tchorz et al., 2009), it remained unknown whether IHBD paucity is a result of insufficient BEC specification or abnormal ductal plate morphogenesis. It was also unclear which Jag1-expressing cell was responsible for the IHBD defects and when Notch activation by Jag1 was required.
Jag1 expression in the PVM, not the endothelium or biliary epithelium, directs bile duct tubulogenesis
A puzzling aspect to AGS is that it encompasses a broad spectrum of organ variability. Although many reports have subsequently focused on uncovering the role of Notch2 and other factors throughout the stages of biliary differentiation, the contribution of Jag1 has been less clear. Investigations of Jag1 have involved expression analysis or cell-specific loss-of-function studies that, despite much effort, have failed to replicate the disease (Crosnier et al., 2000; Flynn et al., 2004; Jones et al., 2000; Kodama et al., 2004; Loomes et al., 2007; Loomes et al., 2002; Louis et al., 1999; Nijjar et al., 2001; Xue et al., 1999). Only Jag1;Notch2 double heterozygous mice (McCright et al., 2002) and mice with a missense mutation of Jag1 (Vrijens et al., 2006) were able to reproduce the major defects of AGS, leaving much controversy over which Jag1-expressing cell controlled IHBD development. Full deletion of Jag1 results in death at E10.5 before IHBD development is initiated, whereas, unlike humans, Jag1 heterozygotes do not exhibit obvious liver defects (Xue et al., 1999). Ablation of Jag1 in hepatoblasts also did not show evidence of bile duct paucity (Loomes et al., 2007).
Recently, several groups have reported endothelial-specific Jag1 deletions in mice. However, most animals either died before the onset of IHBD development or the surviving mice did not display obvious liver defects (Benedito et al., 2009; High et al., 2008). Additional data from zebrafish cloche mutants, in which bile ducts develop normally despite lacking endothelial cells, also support the idea that endothelial cells are not required for IHBD development (Lorent et al., 2004). Together with our findings, we suggest that the endothelium is not responsible for IHBD development. The PVM had been proposed to be the Jag1-presenting cell population, but no definitive experiments had been carried out to investigate this further (Lemaigre, 2008; Si-Tayeb et al., 2010).
Defects in the organization and dilation of the second ductal layer, as revealed in the J1SMKO mice (Figs 2, 3, 4, and 5), clearly identifies Jag1 in the PVM as a regulator of biliary morphogenesis and tubulogenesis. In this regard, we noted a persistence of CK19-positive cells around the portal vein of surviving J1SMKO mice and also found that continuous Jag1 signaling is not required for duct development. Although a Notch inhibitor suppressed spheroid formation when E16.5 epithelium was used, the same inhibitor did not prevent neonatal wild-type BECs from organizing spheroids in vitro on Jag1-null mesenchyme (Fig. 7). It is possible that Jag1 within BECs could be activating Notch2 at the neonatal stage; however, the levels of Jag1 in E16.5 and P2 epithelium are equivalent and much lower than those in PVM (see Fig. S5 in the supplementary material). Thus, it is unlikely that epithelial Jag1 is playing a role exclusively at the neonatal stage.
The communication required for proper orchestration of duct morphogenesis, revealed by the heterotypic interactions of the PVM and BECs, serve to highlight the vascular roots of AGS and to define the role of Jag1 in bile duct formation (Fig. 8). Although other genes such as Tgfb and Hes1 have been implicated in IHBD development with similar biliary defects as the J1SMKO mice, only JAG1 and NOTCH2 have been linked to AGS (Antoniou et al., 2009; Clotman et al., 2002). Examination of Hes1 during early stages of IHBD development (E16.5) in J1SMKO revealed essentially no differences in expression or distribution compared with wild type, suggesting that Hes1 is not a target of mesenchymal Jag1. By contrast, during ductal plate morphogenesis we found that expression of Sox9 was dependent on the presence of Jag1 in the PVM.
Crosstalk between the portal vein mesenchyme and biliary epithelium
Our findings support the previously proposed hypothesis for crossregulation between IHBD morphogenesis and the portal vein vasculature (Coffinier et al., 2002; Lemaigre, 2003; Libbrecht et al., 2005; Raynaud et al., 2009). Just as the mesenchyme provides instructive signals to the developing BECs (Shiojiri and Koike, 1997), the BECs likely exert control over expansion of the PVM, as well as the development of the hepatic arteries (Lemaigre, 2008; Raynaud et al., 2009). Two factors important for early BEC differentiation, HNF1β and HNF6, are expressed in BECs but not the vasculature; yet, the absence of either leads to vascular defects (Clotman et al., 2002; Clotman et al., 2003; Coffinier et al., 2002; Lemaigre, 2003). Hepatic arteries were scarce in J1SMKO livers (Figs 1, 2; see Fig. S2 in the supplementary material), similar to HNF1β or HNF6 knockout mice (Clotman et al., 2003; Coffinier et al., 2002), and the expression of HNF1β itself was delayed in BECs around the portal vein. The thickness of the PVM layer in the J1SMKO mice was reduced (Figs 2, 3), whereas reports on Hnf6 knockout mice also note abnormal mesenchyme (Clotman et al., 2003; Lemaigre, 2003). Thus, BECs may signal back to the PVM in a heterotypic manner. In the J1SMKO mice, the defect in BEC tubulogenesis probably prevents this crosstalk from occurring.
The new information included in this study answers several long-standing questions about the spatial and temporal involvement of Jag1 in IHBD development and identifies crucial heterotypic interactions between BECs and blood vessels. In a broader developmental sense, the insights gleaned from this study underscore the essential and instructive role of the vasculature in the organization and differentiation of liver parenchyma. This instructive contribution has been highlighted in the pancreas and liver using elegant genetic models that either eliminate or perturb the endothelium (Cleaver and Melton, 2003; Coultas et al., 2005; Lammert et al., 2003; Matsumoto et al., 2001; Sakaguchi et al., 2008). Our studies with a single gene deletion provide further support for this notion. Collectively, these findings indicate that, in addition to providing oxygen and nutrients, blood vessels are active contributors to signaling events that drive organogenesis.
Supplementary Material
Acknowledgments
The authors thank Stephanie Lauw for outstanding technical help, Liman Zhao for support with animal husbandry, and the contribution of the Tissue Procurement Core Laboratory Shared Resource, the Jonsson Cancer Center Flow Cytometry Core and the Department of Laboratory Animal Medicine at UCLA. This work was supported by a grant from the National Institutes of Health (RO1 HL085618 to M.L.I.-A.) and by fellowships from The American Heart Association (AHA-0715053Y to J.J.H.), NICHD K12-HD00850 Pediatric Scientist & CIRM Fellowships (A.C.Z.) and the Leducq Foundation. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.052118/-/DC1
References
- Alagille D., Estrada A., Hadchouel M., Gautier M., Odievre M., Dommergues J. P. (1987). Syndromic paucity of interlobular bile ducts (Alagille syndrome or arteriohepatic dysplasia): review of 80 cases. J. Pediatr. 110, 195-200 [DOI] [PubMed] [Google Scholar]
- Alva J. A., Zovein A. C., Monvoisin A., Murphy T., Salazar A., Harvey N. L., Carmeliet P., Iruela-Arispe M. L. (2006). VE-Cadherin-Cre-recombinase transgenic mouse: a tool for lineage analysis and gene deletion in endothelial cells. Dev. Dyn. 235, 759-767 [DOI] [PubMed] [Google Scholar]
- Antoniou A., Raynaud P., Cordi S., Zong Y., Tronche F., Stanger B. Z., Jacquemin P., Pierreux C. E., Clotman F., Lemaigre F. P. (2009). Intrahepatic bile ducts develop according to a new mode of tubulogenesis regulated by the transcription factor SOX9. Gastroenterology 136, 2325-2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artavanis-Tsakonas S., Rand M. D., Lake R. J. (1999). Notch signaling: cell fate control and signal integration in development. Science 284, 770-776 [DOI] [PubMed] [Google Scholar]
- Battle M. A., Konopka G., Parviz F., Gaggl A. L., Yang C., Sladek F. M., Duncan S. A. (2006). Hepatocyte nuclear factor 4alpha orchestrates expression of cell adhesion proteins during the epithelial transformation of the developing liver. Proc. Natl. Acad. Sci. USA 103, 8419-8424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedito R., Roca C., Sorensen I., Adams S., Gossler A., Fruttiger M., Adams R. H. (2009). The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell 137, 1124-1135 [DOI] [PubMed] [Google Scholar]
- Chen T. T., Luque A., Lee S., Anderson S. M., Segura T., Iruela-Arispe M. L. (2010). Anchorage of VEGF to the extracellular matrix conveys differential signaling responses to endothelial cells. J. Cell Biol. 188, 595-609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleaver O., Melton D. A. (2003). Endothelial signaling during development. Nat. Med. 9, 661-668 [DOI] [PubMed] [Google Scholar]
- Clotman F., Lannoy V. J., Reber M., Cereghini S., Cassiman D., Jacquemin P., Roskams T., Rousseau G. G., Lemaigre F. P. (2002). The onecut transcription factor HNF6 is required for normal development of the biliary tract. Development 129, 1819-1828 [DOI] [PubMed] [Google Scholar]
- Clotman F., Libbrecht L., Gresh L., Yaniv M., Roskams T., Rousseau G. G., Lemaigre F. P. (2003). Hepatic artery malformations associated with a primary defect in intrahepatic bile duct development. J. Hepatol. 39, 686-692 [DOI] [PubMed] [Google Scholar]
- Coffinier C., Gresh L., Fiette L., Tronche F., Schutz G., Babinet C., Pontoglio M., Yaniv M., Barra J. (2002). Bile system morphogenesis defects and liver dysfunction upon targeted deletion of HNF1beta. Development 129, 1829-1838 [DOI] [PubMed] [Google Scholar]
- Coultas L., Chawengsaksophak K., Rossant J. (2005). Endothelial cells and VEGF in vascular development. Nature 438, 937-945 [DOI] [PubMed] [Google Scholar]
- Crosnier C., Attie-Bitach T., Encha-Razavi F., Audollent S., Soudy F., Hadchouel M., Meunier-Rotival M., Vekemans M. (2000). JAGGED1 gene expression during human embryogenesis elucidates the wide phenotypic spectrum of Alagille syndrome. Hepatology 32, 574-581 [DOI] [PubMed] [Google Scholar]
- D'Souza B., Miyamoto A., Weinmaster G. (2008). The many facets of Notch ligands. Oncogene 27, 5148-5167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehebauer M., Hayward P., Arias A. M. (2006). Notch, a universal arbiter of cell fate decisions. Science 314, 1414-1415 [DOI] [PubMed] [Google Scholar]
- Emerick K. M., Rand E. B., Goldmuntz E., Krantz I. D., Spinner N. B., Piccoli D. A. (1999). Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology 29, 822-829 [DOI] [PubMed] [Google Scholar]
- Flynn D. M., Nijjar S., Hubscher S. G., de Goyet Jde V., Kelly D. A., Strain A. J., Crosby H. A. (2004). The role of Notch receptor expression in bile duct development and disease. J. Pathol. 204, 55-64 [DOI] [PubMed] [Google Scholar]
- Geisler F., Nagl F., Mazur P. K., Lee M., Zimber-Strobl U., Strobl L. J., Radtke F., Schmid R. M., Siveke J. T. (2008). Liver-specific inactivation of Notch2, but not Notch1, compromises intrahepatic bile duct development in mice. Hepatology 48, 607-616 [DOI] [PubMed] [Google Scholar]
- Gridley T. (2007). Notch signaling in vascular development and physiology. Development 134, 2709-2718 [DOI] [PubMed] [Google Scholar]
- Hansson E. M., Lendahl U., Chapman G. (2004). Notch signaling in development and disease. Semin. Cancer Biol. 14, 320-328 [DOI] [PubMed] [Google Scholar]
- High F. A., Lu M. M., Pear W. S., Loomes K. M., Kaestner K. H., Epstein J. A. (2008). Endothelial expression of the Notch ligand Jagged1 is required for vascular smooth muscle development. Proc. Natl. Acad. Sci. USA 105, 1955-1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann J. J., Iruela-Arispe M. L. (2007a). Notch expression patterns in the retina: An eye on receptor-ligand distribution during angiogenesis. Gene Expr. Patterns 7, 461-470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann J. J., Iruela-Arispe M. L. (2007b). Notch signaling in blood vessels: who is talking to whom about what? Circ. Res. 100, 1556-1568 [DOI] [PubMed] [Google Scholar]
- Holtwick R., Gotthardt M., Skryabin B., Steinmetz M., Potthast R., Zetsche B., Hammer R. E., Herz J., Kuhn M. (2002). Smooth muscle-selective deletion of guanylyl cyclase-A prevents the acute but not chronic effects of ANP on blood pressure. Proc. Natl. Acad. Sci. USA 99, 7142-7147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones E. A., Clement-Jones M., Wilson D. I. (2000). JAGGED1 expression in human embryos: correlation with the Alagille syndrome phenotype. J. Med. Genet. 37, 658-662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama Y., Hijikata M., Kageyama R., Shimotohno K., Chiba T. (2004). The role of notch signaling in the development of intrahepatic bile ducts. Gastroenterology 127, 1775-1786 [DOI] [PubMed] [Google Scholar]
- Krantz I. D., Piccoli D. A., Spinner N. B. (1997). Alagille Syndrome. J. Med. Genet. 34, 152-157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammert E., Cleaver O., Melton D. (2003). Role of endothelial cells in early pancreas and liver development. Mech. Dev. 120, 59-64 [DOI] [PubMed] [Google Scholar]
- Lemaigre F. P. (2003). Development of the biliary tract. Mech. Dev. 120, 81-87 [DOI] [PubMed] [Google Scholar]
- Lemaigre F. P. (2008). Notch signaling in bile duct development: new insights raise new questions. Hepatology 48, 358-360 [DOI] [PubMed] [Google Scholar]
- Li L., Krantz I. D., Deng Y., Genin A., Banta A. B., Collins C. C., Qi M., Trask B. J., Kuo W. L., Cochran J., et al. (1997). Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat. Genet. 16, 243-251 [DOI] [PubMed] [Google Scholar]
- Libbrecht L., Cassiman D., Desmet V., Roskams T. (2002). The correlation between portal myofibroblasts and development of intrahepatic bile ducts and arterial branches in human liver. Liver 22, 252-258 [DOI] [PubMed] [Google Scholar]
- Libbrecht L., Spinner N. B., Moore E. C., Cassiman D., Van Damme-Lombaerts R., Roskams T. (2005). Peripheral bile duct paucity and cholestasis in the liver of a patient with Alagille syndrome: further evidence supporting a lack of postnatal bile duct branching and elongation. Am. J. Surg. Pathol. 29, 820-826 [DOI] [PubMed] [Google Scholar]
- Loomes K. M., Taichman D. B., Glover C. L., Williams P. T., Markowitz J. E., Piccoli D. A., Baldwin H. S., Oakey R. J. (2002). Characterization of Notch receptor expression in the developing mammalian heart and liver. Am. J. Med. Genet. 112, 181-189 [DOI] [PubMed] [Google Scholar]
- Loomes K. M., Russo P., Ryan M., Nelson A., Underkoffler L., Glover C., Fu H., Gridley T., Kaestner K. H., Oakey R. J. (2007). Bile duct proliferation in liver-specific Jag1 conditional knockout mice: effects of gene dosage. Hepatology 45, 323-330 [DOI] [PubMed] [Google Scholar]
- Lorent K., Yeo S. Y., Oda T., Chandrasekharappa S., Chitnis A., Matthews R. P., Pack M. (2004). Inhibition of Jagged-mediated Notch signaling disrupts zebrafish biliary development and generates multi-organ defects compatible with an Alagille syndrome phenocopy. Development 131, 5753-5766 [DOI] [PubMed] [Google Scholar]
- Louis A. A., Van Eyken P., Haber B. A., Hicks C., Weinmaster G., Taub R., Rand E. B. (1999). Hepatic jagged1 expression studies. Hepatology 30, 1269-1275 [DOI] [PubMed] [Google Scholar]
- Lozier J., McCright B., Gridley T. (2008). Notch signaling regulates bile duct morphogenesis in mice. PLoS ONE 3, e1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini S., Mantei N., Dumortier A., Suter U., MacDonald H., Radtke F. (2005). Jagged1-dependent Notch signaling is dispensable for hematopoietic stem cell self-renewal and differentiation. Blood 105, 2340-2342 [DOI] [PubMed] [Google Scholar]
- Matsumoto K., Yoshitomi H., Rossant J., Zaret K. S. (2001). Liver organogenesis promoted by endothelial cells prior to vascular function. Science 294, 559-563 [DOI] [PubMed] [Google Scholar]
- McCright B., Lozier J., Gridley T. (2002). A mouse model of Alagille syndrome: Notch2 as a genetic modifier of Jag1 haploinsufficiency. Development 129, 1075-1082 [DOI] [PubMed] [Google Scholar]
- McDaniell R., Warthen D. M., Sanchez-Lara P. A., Pai A., Krantz I. D., Piccoli D. A., Spinner N. B. (2006). NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am. J. Hum. Genet. 79, 169-173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijjar S. S., Crosby H. A., Wallace L., Hubscher S. G., Strain A. J. (2001). Notch receptor expression in adult human liver: a possible role in bile duct formation and hepatic neovascularization. Hepatology 34, 1184-1192 [DOI] [PubMed] [Google Scholar]
- Oda T., Elkahloun A. G., Meltzer P. S., Chandrasekharappa S. C. (1997a). Identification and cloning of the human homolog (JAG1) of the rat Jagged1 gene from the Alagille syndrome critical region at 20p12. Genomics 43, 376-379 [DOI] [PubMed] [Google Scholar]
- Oda T., Elkahloun A. G., Pike B. L., Okajima K., Krantz I. D., Genin A., Piccoli D. A., Meltzer P. S., Spinner N. B., Collins F. S., et al. (1997b). Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat. Genet. 16, 235-242 [DOI] [PubMed] [Google Scholar]
- Parviz F., Matullo C., Garrison W. D., Savatski L., Adamson J. W., Ning G., Kaestner K. H., Rossi J. M., Zaret K. S., Duncan S. A. (2003). Hepatocyte nuclear factor 4alpha controls the development of a hepatic epithelium and liver morphogenesis. Nat. Genet. 34, 292-296 [DOI] [PubMed] [Google Scholar]
- Raynaud P., Carpentier R., Antoniou A., Lemaigre F. P. (2009). Biliary differentiation and bile duct morphogenesis in development and disease. Int. J. Biochem. Cell Biol. Epub ahead of print [DOI] [PubMed] [Google Scholar]
- Sakaguchi T. F., Sadler K. C., Crosnier C., Stainier D. Y. (2008). Endothelial signals modulate hepatocyte apicobasal polarization in zebrafish. Curr. Biol. 18, 1565-1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiojiri N. (1997). Development and differentiation of bile ducts in the mammalian liver. Microsc. Res. Tech. 39, 328-335 [DOI] [PubMed] [Google Scholar]
- Shiojiri N., Koike T. (1997). Differentiation of biliary epithelial cells from the mouse hepatic endodermal cells cultured in vitro. Tohoku J. Exp. Med. 181, 1-8 [DOI] [PubMed] [Google Scholar]
- Si-Tayeb K., Lemaigre F., Duncan S. A. (2010). Organogenesis and development of the Liver. Dev. Cell 18, 175-189 [DOI] [PubMed] [Google Scholar]
- Soriano P. (1999). Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21, 70-71 [DOI] [PubMed] [Google Scholar]
- Sparks E. E., Huppert K. A., Brown M. A., Washington M. K., Huppert S. S. (2009). Notch signaling regulates formation of the three-dimensional architecture of intrahepatic bile ducts in mice. Hepatology 51, 1391-1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas S., Watanabe T., Lin C. S., William C. M., Tanabe Y., Jessell T. M., Costantini F. (2001). Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 1, 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K., Tanaka M., Watanabe N., Saito S., Nonaka H., Miyajima A. (2008). p75 Neurotrophin receptor is a marker for precursors of stellate cells and portal fibroblasts in mouse fetal liver. Gastroenterology 135, 270-281e3 [DOI] [PubMed] [Google Scholar]
- Tanimizu N., Miyajima A. (2004). Notch signaling controls hepatoblast differentiation by altering the expression of liver-enriched transcription factors. J. Cell Sci. 117, 3165-3174 [DOI] [PubMed] [Google Scholar]
- Tanimizu N., Miyajima A., Mostov K. E. (2009). Liver progenitor cells fold up a cell monolayer into a double-layered structure during tubular morphogenesis. Mol. Biol. Cell 20, 2486-2494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchorz J. S., Kinter J., Muller M., Tornillo L., Heim M. H., Bettler B. (2009). Notch2 signaling promotes biliary epithelial cell fate specification and tubulogenesis during bile duct development in mice. Hepatology 50, 871-879 [DOI] [PubMed] [Google Scholar]
- Varnum-Finney B., Wu L., Yu M., Brashem-Stein C., Staats S., Flowers D., Griffin J. D., Bernstein I. D. (2000). Immobilization of Notch ligand, Delta-1, is required for induction of notch signaling. J. Cell Sci. 113, 4313-4318 [DOI] [PubMed] [Google Scholar]
- Villa N., Walker L., Lindsell C. E., Gasson J., Iruela-Arispe M. L., Weinmaster G. (2001). Vascular expression of Notch pathway receptors and ligands is restricted to arterial vessels. Mech. Dev. 108, 161-164 [DOI] [PubMed] [Google Scholar]
- Vrijens K., Thys S., De Jeu M. T., Postnov A. A., Pfister M., Cox L., Zwijsen A., Van Hoof V., Mueller M., De Clerck N. M., et al. (2006). Ozzy, a Jag1 vestibular mouse mutant, displays characteristics of Alagille syndrome. Neurobiol. Dis. 24, 28-40 [DOI] [PubMed] [Google Scholar]
- Weinmaster G. (2000). Notch signal transduction: a real rip and more. Curr. Opin. Genet. Dev. 10, 363-369 [DOI] [PubMed] [Google Scholar]
- Weng A. P., Lau A. (2005). Notch signaling in T-cell acute lymphoblastic leukemia. Future Oncol. 1, 511-519 [DOI] [PubMed] [Google Scholar]
- Xue Y., Gao X., Lindsell C. E., Norton C. R., Chang B., Hicks C., Gendron-Maguire M., Rand E. B., Weinmaster G., Gridley T. (1999). Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum. Mol. Genet. 8, 723-730 [DOI] [PubMed] [Google Scholar]
- Zong Y., Panikkar A., Xu J., Antoniou A., Raynaud P., Lemaigre F., Stanger B. Z. (2009). Notch signaling controls liver development by regulating biliary differentiation. Development 136, 1727-1739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zovein A. C., Hofmann J. J., Lynch M., French W. J., Turlo K. A., Yang Y., Becker M. S., Zanetta L., Dejana E., Gasson J. C., et al. (2008). Fate tracing reveals the endothelial origin of hematopoietic stem cells. Cell Stem Cell 3, 625-636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zovein A. C., Luque A., Turlo K. A., Hofmann J. J., Yee K. M., Becker M. S., Fassler R., Mellman I., Lane T. F., Iruela-Arispe M. L. (2010). beta1 integrin establishes endothelial cell polarity and arteriolar lumen formation via a Par3-dependent mechanism. Dev. Cell 18, 39-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}