

Abstract
Toxoplasma gondii is an obligate intracellular protozoan parasite that invades and replicates within most nucleated cells of warm-blooded animals. The basis for this wide host cell tropism is unknown but could be because parasites invade host cells using distinct pathways and/or repertoires of host factors. Using synchronized parasite invasion assays, we found that host microtubule disruption significantly reduces parasite invasion into host cells early after stimulating parasite invasion but not at later time points. Host microtubules are specifically associated with the moving junction, which is the site of contact between the host cell and the invading parasite. Host microtubules are specifically associated with the moving junction of those parasites invading early after stimulating invasion but not with those invading later. Disruption of host microtubules has no effect on parasite contact, attachment, motility, or rate of penetration. Rather, host microtubules hasten the time before parasites commence invasion. This effect on parasite invasion is distinct from the role that host microtubules play in bacterial and viral infections, where they function to traffic the pathogen or pathogen-derived material from the host cell's periphery to its interior. These data indicate that the host microtubule cytoskeleton is a structure used by Toxoplasma to rapidly infect its host cell and highlight a novel function for host microtubules in microbial pathogenesis.
Toxoplasma gondii is an obligate intracellular protozoan parasite that is capable of causing disease in fetuses and immunocompromised individuals (23). The parasite infects a wide range of nucleated cells of most warm-blooded animals. The mechanisms underlying this wide tropism are not known but could be due to either the parasite infecting cells using a ubiquitously expressed host receptor and associated machinery, inserting its own receptor into the host cell's plasma membrane, or using multiple host cell receptors/machinery (5).
Toxoplasma invasion is a multistep, complex process consisting of parasite contact to host cells, intimate attachment, parasite motility, and then penetration (5). Host cell contact is a loose, low-affinity interaction that is mediated by parasite surface antigens. An unknown signal then triggers the release of proteins from a specialized secretory organelle called micronemes whose contents include proteins that function as adhesins. This is then followed by parasite gliding motility on the host cell surface. At some point, proteins from a second secretory organelle, named rhoptries, are exocytosed. Among these rhoptry proteins, several (RON2, RON4, RON5, and RON8) are part of a preformed complex that binds the previously secreted AMA1 microneme protein (1, 2, 20, 33). Together, these proteins form the moving junction complex, which defines the parasite entry site on the host cell plasma membrane. Parasite penetration occurs by the parasite propelling itself forward, via acto-myosin-dependent motility, into the host plasma membrane (35). This causes an invagination of the plasma membrane resulting in the formation of the parasitophorous vacuole (PV), which is the compartment that the parasite resides in throughout its time in the host cell. However, host plasma membrane-associated proteins are selectively incorporated into the developing PV such that glycosylphosphatidylinositol (GPI)-linked proteins are included, while single-pass transmembrane proteins are excluded (7, 24).
In contrast to parasite molecules that function during invasion, few host cell components involved in this process are known. A notable exception is the finding that host Arp2/3-dependent actin polymerization promotes Toxoplasma invasion (11). Nevertheless, how actin or other host molecules function during invasion remains to be determined. The host microtubule cytoskeleton has been widely studied for its role during receptor-mediated endocytosis, as well as in bacterial and viral infections, where microtubules act to facilitate cargo transport from the host cell periphery to the interior (8, 15, 27, 29, 40). Consistent with this role in cargo transport, host microtubules also promote trafficking of rhoptry proteins secreted into the host cell (12). However, whether this host cell structure functions during parasite invasion per se is unknown.
Here, we tested the hypothesis that host microtubules are used by Toxoplasma tachyzoites to penetrate into its host cell. Using synchronized parasite invasion assays, we find that disruption of host microtubules significantly reduces parasite invasion into host cells early after stimulating parasite invasion but not at later time points. Host microtubules are localized to the moving junction but, unlike their previously described role in pathogen invasion, host microtubules promote tachyzoite invasion by hastening the time that parasites initiate invasion.
MATERIALS AND METHODS
Cell lines and parasites.
Human foreskin fibroblasts (HFFs), HeLa, Vero, and CHO cells were obtained from the American Type Culture Collection (ATCC; Manassas, VA). LLCPK-1 cells stably expressing green fluorescent protein (GFP)-tubulin were from Patricia Wadsworth (University of Massachusetts). CHO cells stably expressing N-terminal hemagglutinin (HA)-tagged CD59GPI (HA-CD59GPI) were previously described (19). Transmembrane N-terminal HA-tagged CD59 (HA-CD59) was synthesized by Genscript (Piscataway, NJ) by replacing the C-terminal GPI anchor site of CD59 with the transmembrane domain from human CD142 (amino acid sequence EFREIFYIIGAVVFVVIILVIILAISLHK with the underlined residues representing the membrane-spanning domain). HA-CD59 was cloned into pCDNA3.1 (Invitrogen, Carlsbad, CA), transfected into CHO cells, and stable transformants selected with G418. The HA antibody was used to fluorescence-activated cell sort for cell populations equally expressing high levels of HA-CD59GPI and HA-CD59. All cells (except CHO cells) were grown at 37°C at 5% CO2 in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), glutamine, and antibiotics. CHO cells were maintained in Ham F-12 medium with 10% FBS, glutamine, and antibiotics. The RH Toxoplasma strain was used for all experiments and maintained in HFFs as described previously (30). Tomato red fluorescent protein (tRFP+) parasites were from Michael Reese and John Boothroyd (Stanford University), and GFP+ parasites were from Gustavo Arrizabalaga (University of Idaho). All parasites and host cells were routinely tested for mycoplasma contamination by using the MycoAlert assay kit (Lonza, Basal, Switzerland) and were found to be negative.
Synchronized invasion, attachment, and gliding motility assays.
For all assays, GFP+ parasites were harvested from infected, but nonlysed HFFs. Parasites were released from their host cells by passing them three times through a 27-gauge syringe needle. High potassium-based Toxoplasma synchronous invasion assays were performed essentially as described previously (16). Briefly, freshly egressed parasites were added to host cells in high K+ Endo buffer at a multiplicity of infection (MOI) of 10:1 (parasite/host cell), followed by incubation for 20 min at 37°C to allow parasites to adhere to the host cells. The buffer was then removed and gently replaced with prewarmed invasion medium (DMEM plus 10% FBS). Temperature shift-based synchronized invasion assays were performed by adding parasites to host cells at room temperature and then immediately centrifuging the cells at 250 × g for 3 min, followed by incubation at room temperature for an additional 5 min. The plates were then placed on a 37°C heat block. At each time point, the cells were fixed with 0.1% glutaraldehyde–4% formaldehyde for 15 min and then stained with rabbit anti-SAG1 (from John Boothroyd) and Alexa Fluor 594-conjugated goat anti-rabbit IgG (Invitrogen). Coverslips were mounted on glass slides in DAPI (4′,6′-diamidino-2-phenylindole)-containing Vectashield (Vector Laboratories, Burlingame, CA). A minimum of 10 randomly selected fields (i.e., a minimum of 200 parasites) were counted for each sample to determine whether they were extracellular (GFP+/SAG1+) or intracellular (GFP+/SAG1−). Invasion was calculated by dividing the number of intracellular parasites by the total number of parasites counted.
Nocodazole, demecolcine, and taxol were purchased from Sigma (St. Louis, MO) and were added at a concentration of 1 μM to host cells 3 h before infection, which was the minimum time and concentration necessary for complete microtubule disruption without having dramatic effects on host cell morphology (see Fig. S1 in the supplemental material and data not shown). To assess whether the drugs had unexpected irreversible effects on the parasite, freshly egressed parasites were pretreated with the drug for 1 h at 37°C. Parasites were then rinsed two times in medium and resuspended in high-K+ buffer before being added to host cells.
Assays to measure parasite contact with and intimate attachment to host cells were performed essentially as previously described but with minor modifications (17). Briefly, GFP+ parasites were added to confluent HFF monolayers (MOI of 10:1) in high-potassium buffer and then incubated for 20 min at 37°C to allow parasites to settle onto the monolayer. The low-potassium buffer was then added, and the cells were fixed by adding 0.1% glutaraldehyde–4% formaldehyde directly to cells without rinsing so as to not disturb the parasites. A similar procedure was followed to assess intimate attachment except that, prior to fixation, the parasites and cells were rinsed with ice-cold phosphate-buffered saline (PBS) to remove parasites loosely associated with host cells as performed by Kafsack et al. (17). Parasite contact and attachment were quantified by counting the total numbers of remaining GFP+ parasites in 10 random fields (a minimum of 200 parasites).
Gliding motility assays were performed on mock- or nocodazole-treated HFFs plated on 12-mm2 coverslips that were fixed with 0.1% glutaraldehyde–4% formaldehyde for 15 min at room temperature and extensively washed with PBS. GFP+ parasites (107) were added, followed by incubation for 1 h at 37°C. Parasites and cells were fixed and then blocked with 3% bovine serum albumin (BSA), and parasite plasma membrane deposits left behind by the gliding parasite were detected with rabbit anti-SAG1 as previously described (10).
Confocal microscopy.
GFP-tubulin cells were infected with tRFP+ parasites using the potassium-based synchronized invasion assay. At the indicated time points, cells were fixed with glutaraldehyde-formaldehyde and blocked with 3% BSA in PBS. Nonpermeabilized cells were stained with rabbit anti-SAG1 and Alexa Fluor-647 goat anti-rabbit (Invitrogen). Coverslips were mounted on glass slides and serial z-stack images (0.25-μm steps/section) were collected with a Leica SP2 MP confocal microscope. Images were deconvoluted and three-dimensional (3D) reconstructions developed using the Volocity 3D imaging software (Perkin-Elmer Waltham, MA). To properly assign colocalization with the moving junction, 3D reconstructions were necessary for all image analysis except for RON4, as discussed below. f-actin was detected in 3% paraformaldehyde-fixed HFFs with Alexa Fluor 568-conjugated phalloidin (Invitrogen). Microtubules were scored as localized to the parasite moving junction if one or more microtubules spanned across the width of the moving junction. If microtubules were not associated with the moving junction but spanned across the width (but not the length) of an invading parasite a score of “distal to moving junction” was assigned. Images were independently analyzed by two individuals in a nonblinded fashion, and their counting was found to significantly correlate with each other and thus was used to generate averages and standard deviations. Costaining f-actin and microtubules was not possible because glutaraldehyde damages actin filaments (21), and microtubules are poorly preserved in cells fixed only with formaldehyde. To detect HA-tagged CD59, cells were formaldehyde fixed, stained with rabbit anti-SAG1 Alexa Fluor-594 chicken anti-rabbit (Invitrogen), permeabilized with 0.1% Triton X-100, and then stained with rat anti-HA (Roche Indianapolis, IN), and Alexa Fluor-488 goat anti-rat (Invitrogen). Images were analyzed by confocal microscopy as described above.
To detect RON4, cells were fixed with ice-cold methanol for 5 min and stained with rabbit anti-RON4 (from John Boothroyd) and Pacific Orange-conjugated goat anti-rabbit IgG (Invitrogen). SAG1 was detected in the RON4-stained cells with mouse anti-SAG1 (Abcam, Cambridge, MA) and Alexa Fluor 647-conjugated chicken anti-mouse IgG (Invitrogen). Because the cells were fixed with methanol and lost significant volumetric detail, 3D projections were not informative. Thus, the RON4 images were analyzed as a 2D maximal projection.
Video microscopy.
A 35-mm2 glass-bottom dish (World Precision Instruments, Sarasota, FL) containing either mock- or nocodazole-treated GFP+-tubulin cells was placed on a prewarmed heated microscope stage. tRFP+ parasites in high K+ buffer were then added for 20 min. The medium was removed, prewarmed invasion medium was added, the stage was raised, and then a field for imaging was selected. It was determined that this process took ∼45 s to complete. Imaging then proceeded for 15 min, with two z-stack images (1 μm between sections) collected at each time point using a ×63 0.9 NA water-immersion lens on a Leica SP2 MP confocal microscope (Leica). Time-lapse images were then analyzed with the Volocity software.
RESULTS
Disruption of the host microtubule cytoskeleton transiently decreases Toxoplasma invasion.
Toxoplasma gondii invasion is a multistep, complex process in which parasites adhere to and then penetrate into cells (5). These two steps can be dissociated by adding parasites to cells in a high-potassium-containing buffer, which permits attachment but blocks parasite penetration (16). Penetration can then be stimulated by replacing the medium with a low-potassium-containing buffer (e.g., DMEM) that is permissive for invasion. By allowing parasites to adhere to host cells in the high-potassium buffer and then switching to low-potassium buffer, parasite invasion can be synchronized. Using this synchronized invasion assay, we measured Toxoplasma invasion following detailed time points. Thus, freshly egressed GFP-expressing tachyzoites were added to HFFs in high potassium buffer and after 20 min parasite invasion was stimulated by adding invasion buffer. At each time point, the cells were fixed and stained with antisera to the Toxoplasma surface protein SAG1 without permeabilization to discriminate between intracellular and extracellular parasites. The data indicated that parasite invasion began within 1 min of adding invasion medium and rapidly increased during the first 4 min, at which time 70% of the total number of parasites that would invade in the first hour had invaded. Over the next 56 min, parasite invasion continued but at a significantly reduced rate (Fig. 1A).
Fig. 1.
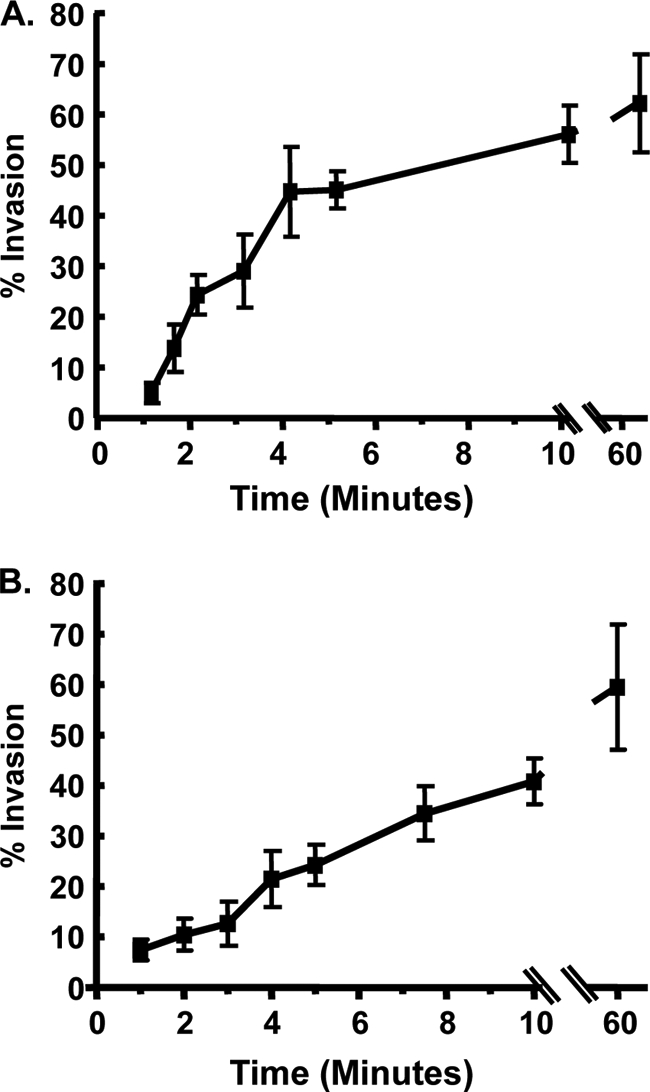
Invasion kinetics. (A) GFP+ parasites were added to HFFs in high-K+ buffer, and 20 min later invasion buffer was added to stimulate invasion. At the indicated times, the cells were fixed, and nonpermeabilized cells were stained with anti-SAG1 antibody to discriminate between extracellular and intracellular parasites. The averages and standard deviations of three independent experiments counting a minimum of 200 randomly selected parasites for each sample are shown. (B) GFP+ parasites were added to HFFs at room temperature and centrifuged for 3 min at 250 × g. The cells were then transferred to a 37°C heat block to stimulate invasion. At the indicated times, cells were fixed and then stained with anti-SAG1 to discriminate between extracellular and intracellular parasites.The averages and standard deviations of three independent experiments counting a minimum of 200 randomly selected parasites for each sample are shown.
We next used a temperature-based synchronized parasite invasion as a second method to examine invasion kinetics. Parasites were added to host cells at room temperature, which is a temperature nonpermissive for invasion, and then immediately centrifuged at 250 × g for 3 min at room temperature. Invasion was initiated by transferring the cells to a 37°C heat block, after which the cells were fixed at specific times, and invasion was quantified by differential SAG1 staining. Relative to potassium-synchronized parasites, temperature-synchronized parasites invaded more slowly requiring 10 min before 70% of the parasites invaded (Fig. 1B).
We next tested how loss of host microtubules impacted Toxoplasma invasion by comparing invasion in the absence or presence of the microtubule depolymerizing agent, nocodazole. Mock- and nocodazole-treated host cells were infected with parasites using the potassium synchronization assay. The cells were fixed 1, 2, 5, and 60 min later, and invasion was quantified by differential SAG1 staining. The data indicated that in nocodazole-treated host cells, parasite invasion was reduced by more than 70% 1 min after adding invasion buffer and by ca. 50% after 2 min (Fig. 2A). By 5 min, no significant differences between mock- and drug-treated cells were observed (P = 0.58). Similarly, early but not late parasite invasion was significantly reduced when host cells were treated with demecolcine, which also destabilizes microtubules through a mechanism distinct from nocodazole, or taxol, which prevents microtubule depolymerization (Fig. 2B). The effect of these drugs on parasite invasion was not cell type specific since early, but not late, invasion of Vero and CHO cells was also reduced by nocodazole (see Fig. S2 in the supplemental material). Finally, nocodazole had a comparable effect on parasite invasion when invasion was synchronized using the temperature-shift assay (Fig. 2C).
Fig. 2.

Host microtubules are important for Toxoplasma Invasion. (A) HFFs were treated with dimethyl sulfoxide (DMSO) or 1 μM nocodazole for 3 h at 37°C. GFP+ parasites were then added to the cells in vehicle- or drug-containing high-K+ buffer and incubated for 20 min. K+ buffer was removed and replaced with DMSO- or nocodazole-containing invasion buffer. At the indicated times, cells were fixed, and the numbers of intracellular and extracellular parasites were determined by differential SAG1 staining. The averages and standard deviations of three independent experiments counting a minimum of 200 randomly selected parasites for each sample are shown. *, P < 0.05; **, P < 0.005 (Student t test). (B) HFFs were mock treated or treated with a 1 μM concentration of the indicated drug for 3 h at 37°C and then infected with GFP+ parasites using the potassium synchronization method. At the indicated times, the cells were fixed and stained with anti-SAG1 to discriminate between extracellular and intracellular parasites. The averages and standard deviations of three independent experiments counting a minimum of 200 randomly selected parasites for each sample are shown (*, P < 0.05; **, P < 0.005 [Student t test]). (C) DMSO- or nocodazole-treated HFFs were infected with GFP+ parasites using the temperature-shift synchronization method. At the indicated times, cells were fixed and stained with anti-SAG1 to discriminate between extracellular and intracellular parasites. The averages and standard deviations of three independent experiments counting a minimum of 200 randomly selected parasites for each sample are shown (**, P < 0.005 [Student t test]). (D) “Treated HFFs” are host cells pretreated with 1 μM nocodazole or DMSO for 3 h at 37°C and then infected with GFP+ parasites. “Treated parasites” are extracellular GFP+ parasites treated with 1 μM nocodazole or DMSO in DMEM for 1 h at 37°C and then washed. The washed parasites were added to host cells in high-K+ buffer, and then invasion buffer was added. Two minutes later, the cells were fixed, and the numbers of invaded parasites were determined by SAG1 differential staining. The averages and standard deviations of three independent experiments counting a minimum of 200 randomly selected parasites for each sample are shown (*, P < 0.05 [Student t test]).
Having established that host cell invasion was similarly affected by nocodazole with both synchronization methods, we chose to perform the rest of our assays with the potassium shift method since it more efficiently synchronizes invasion, which is a conclusion reached by others (16). This is most likely because parasites reach equilibrium more quickly and uniformly after the buffer switch than by shifting a room temperature plate of cells to a 37°C heat block. In addition, after parasites are pelleted onto host cells at room temperature they must first become intimately associated with the host cell since microneme secretion is negligible at room temperature (6).
Nocodazole is unlikely to target the parasite's microtubule cytoskeleton since Toxoplasma tubulin is only sensitive to nocodazole at concentrations 1,000-fold higher than those used here (26). In addition, the parasite's subpellicular microtubules are not dynamic during invasion and only disassemble as parasites progress through the cell cycle and replicate (32). For this reason, Toxoplasma invasion is unaffected even when extracellular tachyzoites are incubated with microtubule inhibitors that can specifically target the parasite's, but not the host's, microtubule cytoskeleton (32). To further ensure that nocodazole was not impacting the parasites in an unforeseen and irreversible manner, extracellular parasites were pretreated with nocodazole, washed, and then evaluated in the invasion assay using untreated host cells. As expected, pretreating parasites with nocodazole had no detectable effect on invasion (Fig. 2D). Although, we cannot rule out nocodazole having a reversible affect on the parasite, our data together with the above-mentioned studies indicate that the effects of nocodazole and the other microtubule inhibitors on parasite invasion are a consequence of the drugs affecting the host microtubule cytoskeleton.
Host cell microtubules localize to the moving junction of early invading parasites.
We next assessed host microtubule localization and organization during parasite invasion by infecting GFP-tubulin expressing host cells with tRFP+ parasites using the synchronized invasion assay. At 2 min after the addition of invasion buffer, the cells were fixed and stained with anti-SAG1 antisera without permeabilizing the cells. Confocal z-stacks underwent 3D reconstruction to accurately determine the location and orientation of host microtubules relative to an invading parasite, which was identified as a tRFP+ parasite that was partially SAG1+. Host microtubules were found to fully span across the width of ∼70% of those parasites in the process of invasion (see Fig. 3A and B, Fig. 4, and see Fig. S3 and Movies S1 to S4 in the supplemental material). The majority (>90%) of these parasites had microtubules localized to the moving junction, which is defined as a visible constriction on an invading parasite, as well as the position demarcating SAG1 staining (1). However, host microtubule association with the moving junction was not required for the formation of a visible constriction on an invading parasite since we never observed a partially SAG1+ parasite that lacked a constriction (Fig. 3C and D and see Movies S5 and S6 in the supplemental material). We noted from the 3D reconstructions that host microtubules were asymmetrically concentrated on one side of the moving junction, although the significance of this observation is unclear. Finally, we confirmed that microtubules were indeed localized to the moving junction by showing that microtubule staining at the constriction of the invading parasite colocalized with RON4 (Fig. 5), which is one component of the moving junction (1, 20).
Fig. 3.

Host microtubules localize to the moving junction during Toxoplasma invasion. GFP-tubulin cells were infected with tRFP+ parasites using the synchronized invasion assay. Two minutes later, cells were fixed and processed for confocal microscopy. The left panel of each row is a low-magnification image, and the white box indicates the zoomed-in areas shown in the other panels. The rightmost panel shows the same area rotated around the x axis to show the underside of the invading parasite. (A and B) Representative 3D reconstructions of invading parasites with host microtubules associated with the moving junction (arrows). (C and D) Representative images of invading parasites without host microtubule association. Scale bars, 3 μm.
Fig. 4.

Host microtubules associate with the moving junction of early, but not late, invading parasites. GFP-tubulin cells were infected with tRFP+ parasites for 2 or 4 min and then fixed and analyzed by confocal microscopy. 3D reconstructions were used to determine how often and where (relative to the moving junction) host microtubules were associated with invading parasites. Means and standard deviations from three independent experiments counting 15 randomly selected invading parasites for each sample are shown (*, P < 0.005 [Student t test]).
Fig. 5.

Host microtubules and RON4 colocalization at the moving junction. GFP-tubulin cells were infected with tRFP+ parasites for 2 min, methanol fixed, and then stained to detect RON4 and SAG1. The white arrows are pointing to the moving junction identified as a ring of RON4 protein at the parasite constriction (scale bar, 5 μm). The RON4 staining in front of the moving junction is apical nonexocytosed protein (1).
The effect that nocodazole had on early Toxoplasma invasion suggested that host microtubules would be associated with parasites invading soon after stimulating invasion but not with those invading at later time points. This hypothesis was tested by comparing the frequency that host microtubules were associated with the moving junction of parasites fixed 4 min (late invaders) after adding invasion medium to those fixed after 2 min (early invaders). As discussed above, host microtubules were associated with a large majority of parasites fixed 2 min after the addition of invasion medium (Fig. 4). In contrast, host microtubules were associated with <30% of the moving junction of those parasites that were fixed 4 min after stimulating parasite invasion (Fig. 4 and see Fig. S4 in the supplemental material). Regardless of the fixation time point (see Fig. S4 in the supplemental material) or whether parasites were invading mock or nocodazole treated (data not shown), a constriction was observed in all of the partially SAG1+ parasites.
Association of host microtubules specifically with those parasites invading early, but not later, after stimulating invasion suggested that microtubule localization at the moving junction was not a general consequence of the invading parasites displacing host cytoplasmic structures. To further address this issue, we examined host f-actin localization during parasite invasion. In contrast to microtubules, we could only detect f-actin at the moving junction of <20% of invading parasites (see Fig. S5 in the supplemental material). Taken together, the time-dependent host microtubule localization to the moving junction, asymmetric microtubule organization at the moving junction, and the apparent lack of f-actin staining at the moving junction strongly suggests that host microtubule localization at the moving junction is specifically associated with early invading parasites.
Host microtubules hasten parasite penetration.
Parasite invasion consists of at least four stages: host cell contact, intimate attachment, gliding motility, and penetration (5). To assess whether disruption of host microtubules affected the ability for parasites to engage in loose, low-affinity contact with the host cell, parasites were added to mock- or nocodazole-treated cells in high potassium buffer and then switched to invasion buffer for 2 or 60 min, at which time the cells were fixed without washing so as to not remove the loosely attached parasites. No differences were observed in the numbers of parasites remaining associated with the mock- or nocodazole-treated host cells (Fig. 6A). To next test whether nocodazole affects intimate attachment, the assay described above was repeated, but loosely associated parasites were removed by rigorous washing before the host cells were fixed. Nocodazole had no significant impact at either time point on the parasites remaining under these conditions (Fig. 6B). Numbers of intimately attached parasites remaining after 2 min increased at the 60-min time point, which is consistent with the finding that only a certain percentage of contact stage parasites will rapidly release their micronemes, while others will take longer to become intimately attached (17). Taken together with the finding that nocodazole reduced early but not late invasion, these data demonstrate that nocodazole has no detectable impact on parasite contact or intimate attachment. Coupled with the fact that nocodazole did not appear to have severe effects on host cell morphology under these conditions (see Fig. S1 in the supplemental material), these data also indicate that differences in parasite invasion are not due to the drug altering host plasma membrane surface area.
Fig. 6.

Disruption of host microtubules has no apparent affect on parasite contact, intimate attachment, or gliding motility. (A) GFP+ parasites in high-K+ buffer were added to mock-treated (▪) or nocodazole-treated (□) HFFs and switched to invasion medium for the indicated times. The medium was then removed, and the cells were fixed without washing. The numbers of cell-associated GFP+ parasites and host nuclei were counted. Means and standard deviations from three independent assays counting 10 random fields for each sample are shown. (B) GFP+ parasites in high-K+ buffer were added to mock-treated (▪) or nocodazole-treated (□) HFFs. The medium was removed, after which the cells were washed three times with ice-cold PBS and then fixed. The numbers of cell-associated GFP+ parasites and host nuclei were counted. The means and standard deviations from three independent experiments counting 10 random fields in each sample are shown. (C) GFP+ parasites were added to formaldehyde-fixed and washed mock- or nocodazole-treated cells for 60 min at 37°C. The cells were fixed and stained with anti-SAG1. Representative images from three independent experiments are shown.
Gliding motility is a specialized mode of parasite motility on the host cell surface that precedes host cell penetration. A common readout for gliding motility is detection of parasite-derived surface depositions that appear as long, circular trails on serum-coated glass coverslips (10, 13). To examine whether disrupting host microtubules affected gliding motility, GFP+ parasites were incubated at 37°C with formaldehyde-fixed host cells that were either mock or nocodazole treated. After 60 min, the cells were fixed and stained to detect SAG1 deposited by motile parasites. No dramatic differences in SAG1 deposition were observed (Fig. 6C). It should be noted, however, that SAG1 deposition on host cells was not as widespread as on serum-coated coverslips. In addition, these depositions appeared as periodic deposits rather than the long continuous ones left on glass surfaces. The basis for these differences is unclear, but their irregular shape and size precluded us from being able to develop quantifiable criteria (e.g., trail length, intensity, etc.).
As Toxoplasma penetrates into its host cell, the parasite selectively incorporates host plasma membrane GPI-linked proteins into the developing PV, while excluding single-pass transmembrane proteins (7, 24). To test whether disrupting host microtubules affected plasma membrane partitioning during penetration, cells stably expressing GPI or transmembrane versions of HA-tagged CD59, which is a naturally occurring GPI-linked protein (28, 31, 34), were mock or nocodazole treated and then infected with tRFP+ parasites for 2 min using the synchronized invasion assay. PV association of either version of epitope-tagged CD59 was defined by using the criteria developed by Mordue et al. (24): a rim of staining enveloping the internalized region of the parasite. The incorporation of HA-CD59GPI into the developing PV was not significantly altered in nocodazole-treated host cells (Fig. 7A; CD59GPI). Using HA-CD59, we did note a slight but not statistically significant increase in HA-CD59 associated with parasites invading nocodazole-treated cells (P = 0.078 [Student t test]). Thus, nocodazole did not significantly impact exclusion of HA-CD59 from the forming PV (Fig. 7B; CD59).
Fig. 7.

Host microtubules are dispensable for membrane sieving during parasitophorous vacuole formation. HA-CD59GPI (A) and HA-CD59 (B) were infected with tRFP+ parasites by using the synchronized infection assay. At 2 min after the addition of invasion buffer, the cells were fixed and stained to detect extracellular SAG1 (magenta). The cells were then permeabilized and stained with anti-HA (green) to detect HA-CD59. Representative 3D projections are shown. Scale bar, 7 μm. The graphs represent quantifications of the frequency of CD59 incorporation into the PV from a minimum of 30 parasites that either partially or completely invaded the host cell.
Confocal fluorescence video microscopy imaging of tRFP+ parasite invasion of GFP-tubulin cells was next used to measure rates of parasite invasion. To ensure that rates were properly determined, only parasites for which we could definitively identify the moving junction (defined as a constriction) starting at the anterior end and ending at the posterior end were included in the analysis. In mock-treated host cells, parasite invasion was completed within 23.1 ± 6.0 s (n = 20) (Fig. 8A). Similarly, penetration into nocodazole-treated cells was completed in 24.7 ± 10.3 s (n = 29), which was not significantly different than mock-treated cells (P > 0.79 Student t test). These data are also consistent with previous studies reporting that tachyzoites require 25 to 30 s to fully penetrate into the host cell (25, 35). Finally, these data further support our conclusion regarding gliding motility from the SAG1 deposition assays (Fig. 6C) since the parasite's acto-myosin machinery power both gliding motility and host cell penetration (5, 13).
Fig. 8.
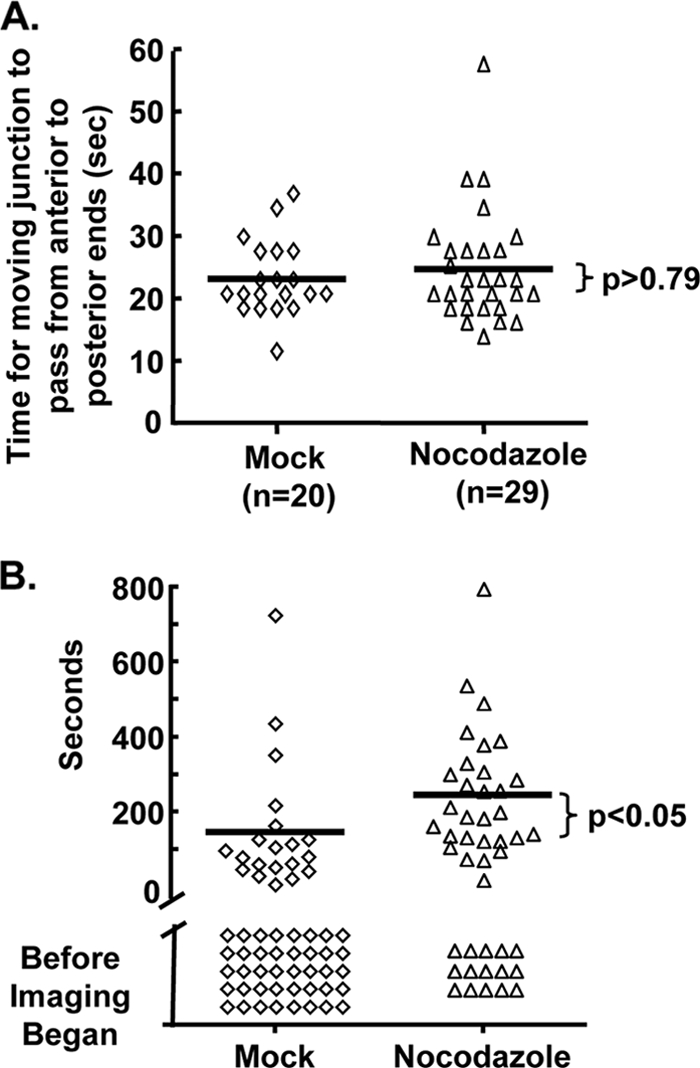
Nocodazole increases the time needed for Toxoplasma to initiate host cell penetration. (A) tRFP+ parasites were incubated with mock- or nocodazole-treated GFP-tubulin host cells for 20 min at 37°C. Invasion medium was then added, and confocal images were captured (two z-stacks separated by 1 μm). Images were compiled into movies by using Volocity imaging software and analyzed to identify parasites whose moving junctions were first detected at the anterior end of the parasite and progressed to the posterior end. Shown are the observed times necessary for the moving junction to pass from the anterior to the posterior ends of the parasite from four independent assays. Bars represent the mean times, which are not statistically different (Student t test, P = 0.79). (B) Real-time confocal video microscopy was used to determine the times after the addition of invasion media that the moving junction was first detected at the anterior end of the parasite. Parasites that invaded before imaging began are denoted as “Before Imaging Began.” Bars represent the means of the times for only parasites where we could detect formation of the moving junction and are statistically different between mock- and nocodazole-treated cells (P < 0.005, Mann-Whitney t test).
Collectively, our data revealed that parasite attachment, motility, and penetration rate were independent of host microtubules. These data led us to hypothesize that host microtubules shortened the time before parasites initiated invasion. To test this hypothesis, we used fluorescence video microscopy to determine how long after adding invasion media the moving junction could first be detected at the anterior end of the parasite. Since it took approximately 45 s after the addition of invasion buffer to select a field and initiate imaging, we expected that some parasites would have already completely invaded its host cell. We identified invaded parasites by two criteria: (i) the parasites remained stationary, and (ii) the parasites displaced host microtubules but were not surrounded by them, which is consistent with other work showing that host microtubules associate with the PV at least 60 min postinvasion (37).
In the absence of nocodazole, ca. 65% of parasites completed invasion before imaging could be started (Fig. 8B). Parasites that invaded after initiating invasion did so after ∼147 s. In contrast, significantly fewer parasites penetrated nocodazole-treated cells before imaging began and those parasites that had invaded began after ∼245 s, which was significantly later than mock-treated cells. Together, these data demonstrate that host microtubules act to shorten the time before parasites initiate host cell invasion.
DISCUSSION
Host cell invasion, a critical step in Toxoplasma's lytic life cycle, is composed of a series of finely orchestrated events involving attachment, gliding motility, moving junction formation, and penetration. The host cell factors involved in each of these stages are unknown. Here, we demonstrated that Toxoplasma invasion is temporally dependent on host microtubules. In contrast to the well-established function of microtubules during endocytosis, as well as during bacterial or viral host cell invasion (8, 15, 27, 29, 40), host microtubules affected Toxoplasma invasion by hastening the time that parasites initiated host cell invasion. These data are significant in that they define a host cell structure that functions during Toxoplasma invasion. In addition, they are, to our knowledge, the first demonstration that host microtubules define when an intracellular pathogen initiates host cell penetration.
Several critical cellular processes are dependent on the microtubule cytoskeleton, including cytokinesis, cell shape, and membrane trafficking. Since our assays were done both with quiescent fibroblasts and replicating cells (Vero, CHO, and LLCPK-1), it is unlikely that cell cycle regulation is associated with how host microtubules promote early parasite infection. In addition, dramatic changes in cell shape, which could affect the available surface area for parasite attachment, do not appear to underlie the effect of nocodazole on parasite invasion for two reasons. First, under the conditions in which we performed our assays, we did not note significant changes in cell shape or morphology. Second, the drug had no significant effect on numbers of parasites in contact with host cells. Moreover, we could not detect differences in numbers of intimately attached parasites, indicating that host microtubules have no apparent effect on micronemal adhesin secretion or for host plasma membrane expression of their receptors. Finally, the lack of an effect of nocodazole on the rate of parasite penetration indicates that host microtubule-based trafficking does not help the parasites to propel themselves into the host cell.
Host microtubule hastening of tachyzoite penetration could be a result of three different but not necessarily mutually exclusive mechanisms. First, host microtubules may organize a host plasma membrane-based protrusion that acts as a fulcrum for gliding parasites to use as a pivot to begin to penetrate into the host cell. The concentration of host microtubules on one side of the moving junction suggests that once a parasite encounters such a structure the parasite would begin to invade while the structure remained stable at the site of parasite invasion. Second, host microtubules may provide support to help withstand the compressive force induced by a parasite in contact with the host cell. Such a hypothesis is consistent with the tensegrity model of cellular responses to mechanical force. In this model, the cells are prestressed structures that use microtubules to provide resistance to compression (14). As examples, microtubules are required to help cells respond to mechanical stresses such as artificial stimuli (e.g., glass pipette-mediated plasma membrane deformation) or physiological stimuli (e.g., cardiomyocyte contractility or endothelial cell responses to thrombin) (3, 14, 38).
A third possibility is that host microtubules may aid in promoting the efficient formation of a functional moving junction. The moving junction complex is composed of the micronemal protein AMA1 and the preformed rhoptry-derived RON complex that only bind one another after they are secreted (1, 20). The RON complex is composed of several proteins that include RON2, which is associated with the host plasma membrane most likely as a multipass transmembrane protein, and RON4, RON5, and RON8 that are exposed to the host cytosol (2, 33). One current model of rhoptry secretion is that rhoptry proteins are released into the host cell within membranous whorled-structures (12). While trafficking of some rhoptry proteins to the interior of the cell is host microtubule dependent (12), it is unknown what would direct the RON complex to the host plasma membrane. Thus, we propose a model where the RON complex is secreted into the membranous whorls, associates with host microtubules either directly via one of its host cytoplasm-exposed proteins or indirectly by binding a microtubule-binding protein and uses these host microtubules to traffic to the host plasma membrane where it can bind AMA1 and form the moving junction. When host microtubules are disrupted, RON complex delivery to the plasma membrane would be less efficient and thus cause a lag in the time before parasites could begin to invade.
Association of host microtubules with the moving junction was defined as a specific process since host microtubules were associated significantly more often with early invading parasites than late invading ones. In addition, we and others (9) failed to detect host f-actin at the moving junction of a majority of invading parasites. Finally, host microtubules were asymmetrically organized at the moving junction instead of evenly dispersed around it. Thus, we do not believe that microtubule association with the moving junction was simply due to the enlarging PV pushing host cytoplasmic contents aside. Whether asymmetric organization is functionally significant is unclear, but this observation is consistent with all three of our hypotheses. For example, the microtubule-based plasma membrane protrusion used by the parasite as a pivot may remain intact as a parasite continues to invade. Likewise, the microtubules may need to remain intact to help the cell withstand the compressive forces induced as a parasite continues to invade. Finally, host microtubules may not disperse after aiding in helping to sort rhoptry proteins.
Our data showed that nocodazole had a dramatic affect on early parasite invasion, which was overcome at later time points. However, it is unclear whether these data indicate that Toxoplasma gains entry to its host cell using distinct sets of host factors/pathways (at least one of which is rapid and microtubule dependent). Other pathogens with wide host cell tropisms also utilize multiple host cell pathways to gain entry. For example, Trypanosoma cruzi can infect host cells by either recruiting host lysosomes or by activating host phosphoinositide 3-kinase (36, 39). It is also possible that host microtubules promote early Toxoplasma invasion by increasing the probability that a given parasite will successfully be able to initiate invasion. Our future studies will be aimed at discriminating between these two possibilities.
Previously, Gonzalez et al. reported that host f-actin was enriched at the moving junction in over 80% of invading parasites (11). In our experiments, we detected f-actin significantly less frequently at the moving junction. The basis for this difference is unclear but could be due to differences in methodology and/or host cells. However, it is clear that both cytoskeletal networks interact in diverse biological processes including vesicular transport, growth cone formation, tensegrity, and cytokinesis (4, 14, 18, 22). Thus, it is possible that coordination between both host actin and microtubule cytoskeletons regulate Toxoplasma invasion and future experiments will address this.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jim Henthorn for technical assistance and Peter Bradley, Vern Carruthers, Bjorn Kafsack, and Jess Tyler for critical reading of the manuscript and helpful discussions.
This study was funded by grants from the American Cancer Society (MBC-114461) and NIH/NIAID (AI069986).
Footnotes
Supplemental material for this article may be found at http://ec.asm.org/.
Published ahead of print on 30 April 2010.
REFERENCES
- 1.Alexander D. L., Mital J., Ward G. E., Bradley P., Boothroyd J. C. 2005. Identification of the moving junction complex of Toxoplasma gondii: a collaboration between distinct secretory organelles. PLoS Pathog. 1:e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Besteiro S., Michelin A., Poncet J., Dubremetz J. F., Lebrun M. 2009. Export of a Toxoplasma gondii rhoptry neck protein complex at the host cell membrane to form the moving junction during invasion. PLoS Pathog. 5:e1000309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brangwynne C. P., MacKintosh F. C., Kumar S., Geisse N. A., Talbot J., Mahadevan L., Parker K. K., Ingber D. E., Weitz D. A. 2006. Microtubules can bear enhanced compressive loads in living cells because of lateral reinforcement. J. Cell Biol. 173:733–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campellone K. G., Webb N. J., Znameroski E. A., Welch M. D. 2008. WHAMM is an Arp2/3 complex activator that binds microtubules and functions in ER to Golgi transport. Cell 134:148–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carruthers V., Boothroyd J. C. 2007. Pulling together: an integrated model of Toxoplasma cell invasion. Curr. Opin. Microbiol. 10:83–89 [DOI] [PubMed] [Google Scholar]
- 6.Carruthers V. B., Giddings O. K., Sibley L. D. 1999. Secretion of micronemal proteins is associated with toxoplasma invasion of host cells. Cell. Microbiol. 1:225–235 [DOI] [PubMed] [Google Scholar]
- 7.Charron A. J., Sibley L. D. 2004. Molecular partitioning during host cell penetration by Toxoplasma gondii. Traffic 5:855–867 [DOI] [PubMed] [Google Scholar]
- 8.Damm E. M., Pelkmans L., Kartenbeck J., Mezzacasa A., Kurzchalia T., Helenius A. 2005. Clathrin- and caveolin-1-independent endocytosis: entry of simian virus 40 into cells devoid of caveolae. J. Cell Biol. 168:477–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.da Silva C. V., da Silva E. A., Cruz M. C., Chavrier P., Isberg R., Mortara R. A. 2008. ARF6, PI3-kinase and host cell actin cytoskeleton in Toxoplasma gondii cell invasion. Biochem. Biophys. Res. Commun. 378:656–661 [DOI] [PubMed] [Google Scholar]
- 10.Dobrowolski J. M., Sibley L. D. 1996. Toxoplasma invasion of mammalian cells is powered by the actin cytoskeleton of the parasite. Cell 84:933–939 [DOI] [PubMed] [Google Scholar]
- 11.Gonzalez V., Combe A., David V., Malmquist N. A., Delorme V., Leroy C., Blazquez S., Menard R., Tardieux I. 2009. Host cell entry by apicomplexa parasites requires actin polymerization in the host cell. Cell Host Microbe 5:259–272 [DOI] [PubMed] [Google Scholar]
- 12.Hakansson S., Charron A. J., Sibley L. D. 2001. Toxoplasma evacuoles: a two-step process of secretion and fusion forms the parasitophorous vacuole. EMBO J. 20:3132–3144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hakansson S., Morisaki H., Heuser J., Sibley L. D. 1999. Time-lapse video microscopy of gliding motility in Toxoplasma gondii reveals a novel; biphasic mechanism of cell locomotion. Mol. Biol. Cell 10:3539–3547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ingber D. E. 2003. Tensegrity I: cell structure and hierarchical systems biology. J. Cell Sci. 116:1157–1173 [DOI] [PubMed] [Google Scholar]
- 15.Jin M., Snider M. D. 1993. Role of microtubules in transferrin receptor transport from the cell surface to endosomes and the Golgi complex. J. Biol. Chem. 268:18390–18397 [PubMed] [Google Scholar]
- 16.Kafsack B. F., Beckers C., Carruthers V. B. 2004. Synchronous invasion of host cells by Toxoplasma gondii. Mol. Biochem. Parasitol. 136:309–311 [DOI] [PubMed] [Google Scholar]
- 17.Kafsack B. F., Carruthers V. B., Pineda F. J. 2007. Kinetic modeling of Toxoplasma gondii invasion. J. Theor. Biol. 249:817–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kunda P., Baum B. 2009. The actin cytoskeleton in spindle assembly and positioning. Trends Cell Biol. 19:174–179 [DOI] [PubMed] [Google Scholar]
- 19.LaChapelle S., Tweten R. K., Hotze E. M. 2009. Intermedilysin-receptor interactions during assembly of the pore complex: assembly intermediates increase host cell susceptibility to complement-mediated lysis. J. Biol. Chem. 284:12719–12726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lebrun M., Michelin A., El Hajj H., Poncet J., Bradley P. J., Vial H., Dubremetz J. F. 2005. The rhoptry neck protein RON4 re-localizes at the moving junction during Toxoplasma gondii invasion. Cell Microbiol. 7:1823–1833 [DOI] [PubMed] [Google Scholar]
- 21.Lehrer S. 1981. Damage to actin filaments by glutaraldehyde: protection by tropomyosin. J. Cell Biol. 90:459–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lowery L. A., Van Vactor D. 2009. The trip of the tip: understanding the growth cone machinery. Nat. Rev. Mol. Cell. Biol. 10:332–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Montoya J. G., Liesenfeld O. 2004. Toxoplasmosis. Lancet 363:1965–1976 [DOI] [PubMed] [Google Scholar]
- 24.Mordue D. G., Desai N., Dustin M., Sibley L. D. 1999. Invasion by Toxoplasma gondii establishes a moving junction that selectively excludes host cell plasma membrane proteins on the basis of their membrane anchoring. J. Exp. Med. 190:1783–1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morisaki J., Heuser J., Sibley L. 1995. Invasion of Toxoplasma gondii occurs by active penetration of the host cell. J. Cell Sci. 108:2457–2464 [DOI] [PubMed] [Google Scholar]
- 26.Morrissette N. S., Sibley L. D. 2002. Disruption of microtubules uncouples budding and nuclear division in Toxoplasma gondii. J. Cell Sci. 115:1017–1025 [DOI] [PubMed] [Google Scholar]
- 27.Parton R. G., Joggerst B., Simons K. 1994. Regulated internalization of caveolae. J. Cell Biol. 127:1199–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rollins S. A., Sims P. J. 1990. The complement-inhibitory activity of CD59 resides in its capacity to block incorporation of C9 into membrane C5b-9. J. Immunol. 144:3478–3483 [PubMed] [Google Scholar]
- 29.Sodeik B., Ebersold M. W., Helenius A. 1997. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 136:1007–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spear W., Chan D., Coppens I., Johnson R. S., Giaccia A., Blader I. J. 2006. The host cell transcription factor hypoxia-inducible factor 1 is required for Toxoplasma gondii growth and survival at physiological oxygen levels. Cell. Microbiol. 8:339–352 [DOI] [PubMed] [Google Scholar]
- 31.Stefanova I., Hilgert I., Kristofova H., Brown R., Low M. G., Horejsi V. 1989. Characterization of a broadly expressed human leucocyte surface antigen MEM-43 anchored in membrane through phosphatidylinositol. Mol. Immunol. 26:153–161 [DOI] [PubMed] [Google Scholar]
- 32.Stokkermans T. J., Schwartzman J. D., Keenan K., Morrissette N. S., Tilney L. G., Roos D. S. 1996. Inhibition of Toxoplasma gondii replication by dinitroaniline herbicides. Exp. Parasitol. 84:355–370 [DOI] [PubMed] [Google Scholar]
- 33.Straub K., Cheng S., Sohn C., Bradley P. 2009. Novel components of the Apicomplexan moving junction reveal conserved and coccidia-restricted elements. Cell. Microbiol. 11:590–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sugita Y., Tobe T., Oda E., Tomita M., Yasukawa K., Yamaji N., Takemoto T., Furuichi K., Takayama M., Yano S. 1989. Molecular cloning and characterization of MACIF, an inhibitor of membrane channel formation of complement. J. Biochem. 106:555–557 [DOI] [PubMed] [Google Scholar]
- 35.Suss-Toby E., Zimmerberg J., Ward G. E. 1996. Toxoplasma invasion: the parasitophorous vacuole is formed from host cell plasma membrane and pinches off via a fission pore. Proc. Natl. Acad. Sci. U. S. A. 93:8413–8418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tardieux I., Webster P., Ravesloot J., Boron W., Lunn J. A., Heuser J. E., Andrews N. W. 1992. Lysosome recruitment and fusion are early events required for trypanosome invasion of mammalian cells. Cell 71:1117–1130 [DOI] [PubMed] [Google Scholar]
- 37.Walker M. E., Hjort E. E., Smith S. S., Tripathi A., Hornick J. E., Hinchcliffe E. H., Archer W., Hager K. M. 2008. Toxoplasma gondii actively remodels the microtubule network in host cells. Microbes Infect. 10:1440–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang N., Naruse K., Stamenovic D., Fredberg J. J., Mijailovich S. M., Tolic-Norrelykke I. M., Polte T., Mannix R., Ingber D. E. 2001. Mechanical behavior in living cells consistent with the tensegrity model. Proc. Natl. Acad. Sci. U. S. A. 98:7765–7770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woolsey A. M., Sunwoo L., Petersen C. A., Brachmann S. M., Cantley L. C., Burleigh B. A. 2003. Novel PI 3-kinase-dependent mechanisms of trypanosome invasion and vacuole maturation. J. Cell Sci. 116:3611–3622 [DOI] [PubMed] [Google Scholar]
- 40.Yoshida S., Sasakawa C. 2003. Exploiting host microtubule dynamics: a new aspect of bacterial invasion. Trends Microbiol. 11:139–143 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.