


Abstract
We have developed a simple method for the quantitative detection of specific DNA or RNA molecules based on the finding that BODIPY® FL fluorescence was quenched by its interaction with a uniquely positioned guanine. This approach makes use of an oligonucleotide probe or primer containing a BODIPY® FL-modified cytosine at its 5′-end. When such a probe was hybridized with a target DNA, its fluorescence was quenched by the guanine in the target, complementary to the modified cytosine, and the quench rate was proportional to the amount of target DNA. This widely applicable technique will be used directly with larger samples or in conjunction with the polymerase chain reaction to quantify small DNA samples.
INTRODUCTION
Oligonucleotide probes modified with fluorescent dyes are widely used to detect specific DNA or RNA molecules. Moreover, if the intensity of the fluorescent emission of the probe were to vary in proportion to the amount of target as a result of hybridization, one would be able to quantify the target DNA or RNA in the samples. Hence, several hybridization assays for quantifying a specific DNA or RNA molecule in homogeneous solution have been proposed (1–8). In each case, oligonucleotide probes are designed to make use of fluorescence resonance energy transfer (FRET), a process whereby energy from a fluorescent donor is transferred to an acceptor, quenching the donor but sensitizing the emission of the acceptor. These methods require a careful design of the probes, because the two dyes must be properly positioned with respect to one another in order to accomplish the energy transfer. Unfortunately, probe design must usually be done by trial and error, making it a very time-consuming step. On the other hand, if the energy transfer were between a dye attached to the probe and a nucleic base at a specific position in the target, probe design would be much simpler.
Horn et al. (9) previously reported that the fluorescent emission from a probe modified with 4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-propionic acid (BODIPY® FL) was diminished after hybridization. In the present study, we show that this quenching was caused by the interaction between BODIPY® FL and a guanine (G) base at a particular position, and have made use of this phenomenon to develop a new method for quantitatively detecting specific DNA and RNA molecules.
MATERIALS AND METHODS
Probes and primers
BODIPY® FL (D-6140, Molecular Probes, Eugene, OR)-modified probes and primers were supplied by Takara Shuzo (Kyoto, Japan). All BODIPY® FL was attached to the 5′-end via an aminohexylphosphate linker having a six carbon spacer.
Preparation of genomic DNA samples
Human genomic DNA samples were obtained from Takara Shuzo. The samples were quantified using a Pico Green dsDNA Quantitation Kit (Molecular Probes) according to the manufacturer’s instructions and were diluted to 15 ng/µl. This solution was either used directly or after further dilution. Since one human cell contains ∼6 pg of genomic DNA and two copies of the β-globin gene, 15 ng/µl solution was calculated to contain 5000 copies/µl of human β-globin gene. The genomic DNA of Escherichia coli was prepared by culturing the bacteria overnight at 30°C in a test tube containing 5 ml of 0.8% (w/v) nutrient broth (Difco, Detroit, MI) and then extracting the genomic DNA from the cultured cells using a DNeasy™ Tissue Kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer’s instructions. The extracted DNA was quantified using a Pico Green dsDNA Quantitation Kit.
Fluorescence measurement
In the experiments for analyses of quenching phenomena, the fluorescence was measured with an LS50B luminescence spectrometer (Perkin-Elmer, Norwalk, CT); excitation and emission wavelengths were 499 and 522 nm, respectively; the slit width for each was 4 nm. The reaction mixture contained 40 nM probe, 320 nM target DNA, 50 mM NaCl, 1.0 mM MgCl2 and 50 mM Tris–HCl buffer pH 7.2. Initially, 1.936 ml of the reaction mixture, without the target DNA, was placed in a quartz cell and the fluorescence measured. Thereafter, 0.064 ml of target DNA in sterilized distilled water was added and the fluorescence was measured repeatedly until the intensity reached a plateau. All measurements were carried out at 35°C. In the experiments for quantitative detection of target DNA using BODIPY® FL-modified probe, the fluorescence intensity was monitored using a LightCycler System (Roche Diagnostics, Mannheim, Germany). The reaction mixtures (total volume: 20 µl) contained 40 nM probe, 0–160 nM target DNA, 160 nM non-target DNA, 50 mM NaCl and 50 mM Tris–HCl buffer pH 7.2. The initial temperature of the reaction mixtures was 65°C, but it was then quickly cooled to 50°C. The sequence of the probe was 5′-CCTTCCCACATCGTTT-3′, with BODIPY® FL coupled to the 5′-end. The sequence of the target DNA (positions 1027–1076 of the E.coli 23S rDNA) was 5′-AAACGATGTGGGAAGGCCCAGACAGCCAGGATGTTGGCTTAGAAGCAGCC-3′, where the underlined sequence is complementary to the probe. The sequence of the non-target DNA was the same as that of the target except the eighth base from the 5′-end was T instead of G.
Real-time quantitative PCR
PCR was carried out using a LightCycler System (Roche Diagnostics) with an initial denaturation at 95°C for 50 s followed by a cycle of denaturation at 95°C for 10 s, primer annealing at 62°C for 5 s and extension at 72°C for 17 s. The fluorescence was measured after denaturation and extension in each cycle and the PCR was stopped after 70 cycles. The sequences of primers KM29 and KM38 were 5′-GGTTGGCCAATCTACTCCCAGG-3′ and 5′-TGGTCTCCTTAAACCTGTCTTG-3′, respectively (10). C was added to the 5′-end of primer KM38 and then modified with BODIPY® FL. Primer KM29 and the modified primer KM38 were used in the experiments. The reaction mixture (20 µl) contained 2 µl of human genomic DNA solution, including 0–10 000 copies of the β-globin gene serving as the target DNA, 0–30 ng of E.coli genomic DNA serving as the non-target DNA, 0.1 µM of each primer, 2 mM MgCl2, 0.25 mg/ml of bovine serum albumin, 2 µl of reaction buffer (10× Ex Taq buffer; Takara Shuzo), 1.6 µl of dNTP mixture (2.5 mM each), 0.5 U of DNA polymerase (Takara Ex Taq; Takara Shuzo) and 0.32 µl of TaqStart antibody (Clontech Labs, Palo Alto, CA). The E.coli genomic DNA was added to equalize the amount of DNA template (30 ng) in each reaction tube.
Calculation of the fluorescence quench rate (Rn) in each cycle
To evaluate the decline in fluorescence due to the hybridization, the fluorescence intensities before and after hybridization were compared at each cycle. In this experiment, the fluorescence measured at 72°C (F72) represented the intensity after hybridization, whereas that at 95°C (F95) represented the intensity before hybridization. F72 was then divided by F95 to obtain the quench rate. Because the F72/F95 ratios varied from tube to tube, even in the early cycles when there was no detectable quenching, the value of F72/F95 for each cycle was normalized by dividing it by the value of F72/F95 obtained at the cycle occurring immediately before a significant decrease in fluorescence intensity was observed. In our experiment, the fluorescence at cycle 25 was used for normalization and the fluorescence reduction rate was calculated using the following formula:
Rn = [1 – (F72,n/F95,n)/(F72,25/F95,25)] × 100
where Rn (%) is the quench rate at cycle n, F72,n is the fluorescence intensity at 72°C at cycle n, and F95,n is the fluorescence intensity at 95°C at cycle n.
Determination of DNA sequences
PCR products were purified using MicroSpin™ S-400 HR columns (Amersham Pharmacia, Piscataway, NJ), and the DNA sequences were determined with an automatic sequence analyzer (ABI PRISM™ 377; PE Applied Biosystems, Foster City, CA) using a dye terminator cycle sequencing kit (PE Applied Biosystems).
RESULTS AND DISCUSSION
Fluorescence quenching
To identify the composition of bases that causes BODIPY® FL fluorescence to be quenched, we designed four model probes modified with BODIPY® FL at the 5′-end (Table 1) and measured the fluorescence intensities of the probe solutions before and after adding a target DNA. Of the four sets of probes and targets, a significant decrease in fluorescence intensity was observed only when the probe C9T6 was hybridized with the target A6G12 (Table 1). Moreover, probe G9A6 was much less fluorescent than the others, even before hybridization. These results suggest that a G positioned in very close proximity to BODIPY® FL is crucial for quenching the fluorescence.
Table 1. Identification of the base composition that caused the quenching of BODIPY® FL fluorescence.
| |
F0 |
F |
(F0 – F)/F0 |
| 5′-AAAAAAAAAGGGGGG-3′a3′-TTTTTTTTTTTTCCCCCC-5′b |
330 |
380 |
–0.15 |
| 5′-TTTTTTTTTCCCCCC-3′a3′-AAAAAAAAAAAAGGGGGG-5′b |
440 |
430 |
0.02 |
| 5′-GGGGGGGGGAAAAAA-3′a3′-CCCCCCCCCCCCTTTTTT-5′b |
40 |
50 |
–0.25 |
| 5′-CCCCCCCCCTTTTTT-3′a3′-GGGGGGGGGGGGAAAAAA-5′b | 360 | 30 | 0.92 |
The probes were modified with BODIPY® FL at the 5′-end. F0, fluorescence intensity (arbitrary units) in the absence of target DNA; F, fluorescence intensity after the addition of the target DNA.
aProbe sequence.
bTarget sequence.
We then attempted to determine the position of a G in the target DNA that would quench the probe upon hybridization. A series of 30 base oligonucleotides with the composition (TA)4TmGTn, where m = 2–21 and m + n = 21, served as target DNA and were hybridized with complementary 20 nt fluorescent probes having the composition A12(TA)4 or AmCAn(TA)4, where m = 0–9 and m + n = 11. Of the 20 pairs tested, a decrease in fluorescence intensity after hybridization was observed in only the four pairs listed in Table 2; no decrease was observed in the remaining sixteen pairs. By far the largest degree of quenching (72% of intensity) occurred when a C was situated at the 5′-end of the probe. In contrast, when the C was at the second position from the 5′-end, there was almost no quenching (2% of intensity) (Table 2). This strongly suggests that, by monitoring the fluorescence intensity before and after hybridization, it should be possible to quantify a target DNA or RNA using probes containing a BODIPY® FL-modified C at their 5′-ends.
Table 2. The position of G nucleotide in the targets when the reduction of fluorescence after hybridization was observed.
| |
|
F0 |
F |
(F0 – F)/F0 |
| 5′-ACAAAAAAAAAATATATATA-3′a 3′-TTTTTTTTTTTGTTTTTTTTTTATATATAT-5′b | 573.3 | 564.5 | 0.02 | |
| 5′-CAAAAAAAAAAATATATATA-3′a 3′-TTTTTTTTTTGTTTTTTTTTTTATATATAT-5′b | 521.3 | 146.2 | 0.72 | |
| 5′-AAAAAAAAAAAATATATATA-3′a 3′-TTTTTTTTTGTTTTTTTTTTTTATATATAT-5′b | 489.6 | 253.4 | 0.48 | |
| 5′-AAAAAAAAAAAATATATATA-3′a 3′-TTTTTTTTGTTTTTTTTTTTTTATATATAT-5′b | 479.7 | 406.2 | 0.15 |
F0, fluorescence intensity (arbitrary units) without the target DNA; F, fluorescence intensity after the addition of the target DNA.
aProbe sequence.
bTarget sequence.
We also designed various probes with Gs placed at different positions throughout to estimate their effect on the BODIPY® FL fluorescence and found that significant quenching caused by intramolecular interaction between G and BODIPY® FL occurred only when the G was at the 5′-end of the probe (data not shown). The influence of G was negligible within probes containing a C at their 5′-end.
Quantitative detection of target DNA
A BODIPY® FL-modified oligonucleotide probe was then used for the quantitative detection of a specific target DNA. The fluorescent probe (concentration: 40 nM) was mixed in a solution containing the target DNA (0–160 nM) as well as a non-target DNA molecule (160 nM) with a single mismatch, and the change in the fluorescence intensity of the mixture was measured before (65°C) and after (50°C) hybridization (Fig. 1). Expressed in arbitrary units, hybridization with the target DNA caused the probe fluorescence to decrease by –0.12, 4.15, 7.82, 13.55, 19.22, 22.71 and 35.87 when the concentrations of target were 0, 5, 10, 20, 30, 40 and 160 nM, respectively. Furthermore, the amount of quenching varied linearly with the concentration of the target DNA at concentrations <30 nM (r = 0.993). Similar results were obtained in the absence of the non-target DNA, indicating that coexistence with non-target DNA having even a single mismatch does not affect measurement using this method.
Figure 1.
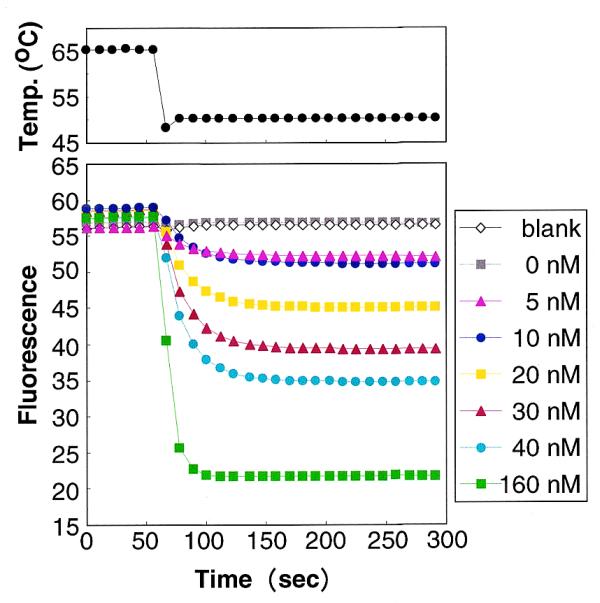
Quantitative detection of a specific DNA using a BODIPY® FL-modified probe. The concentrations of target DNA in the reaction mixtures were 0–160 nM. The blank contained the probe, NaCl and the buffer. The temperature of the reaction mixtures was initially 65°C and was then quickly cooled to 50°C.
This simple method should be widely applicable for quantifying specific DNA or RNA molecules of interest, for example rRNAs from specific microorganisms in environmental samples. By varying the concentration of the BODIPY® FL-modified probe, a wide range of concentrations of target DNA/RNA in the samples should be measurable. One drawback to this approach, however, is that relatively large amounts of target DNA/RNA are required to stimulate detectable declines in fluorescence, hence direct application of this technique may be somewhat limited.
Real-time quantitative PCR
To make this method more widely applicable, we developed a real-time quantitative PCR based on the fluorescent quenching phenomenon using a BODIPY® FL-modified primer, an approach enabling measurement of even small quantities of target DNA. To test this procedure, we quantified the human β-globin gene using the well-characterized primers, KM29 and KM38 (10). As neither of these primers have a C at their 5′-ends, a C was added to KM38 and then modified with BODIPY® FL. Primer KM29 was not modified. PCR was then carried out, and the fluorescence was measured at each PCR cycle (Fig. 2). Although the human β-globin gene does not possess a G to hybridize with the C newly added to the 5′-end of primer KM38, decreases in fluorescence intensity were observed after PCR amplification, indicating that the PCR-amplified product had acquired the additional G needed to quench the fluorescence of the probe.
Figure 2.
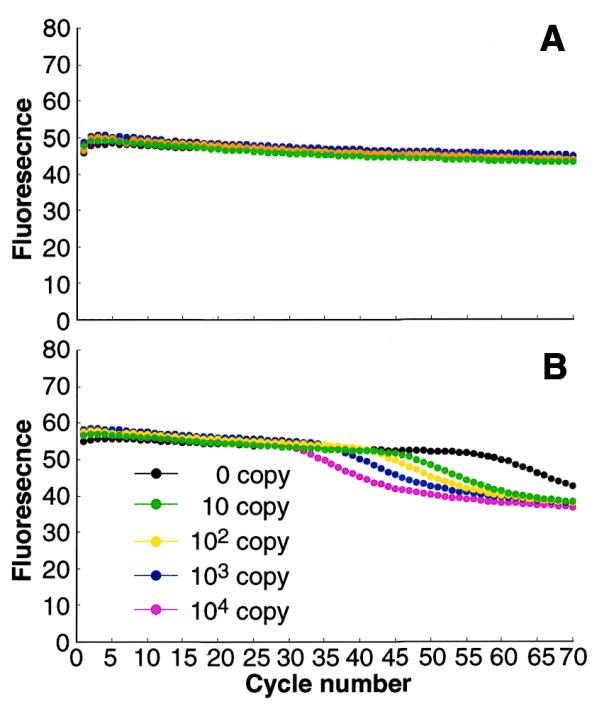
Real-time monitoring of PCR. Fluorescence intensity was measured (A) after denaturation at 95°C and (B) after extension at 72°C in each PCR cycle.
To determine the initial amount of DNA template in a sample, the fluorescence quench rate (Rn) at each cycle was calculated as described in Materials and Methods (Fig. 3), and it was found that the cycle number at which log Rn = 0.1, 0.3, 0.7, 0.9 or 1.2 was linearly related to the initial copy number of the template DNA (r > 0.99) (Fig. 4). The calibration curve showing the strongest correlation between initial copy number and cycle number (r = 0.9993) was obtained when log Rn = 0.5.
Figure 3.
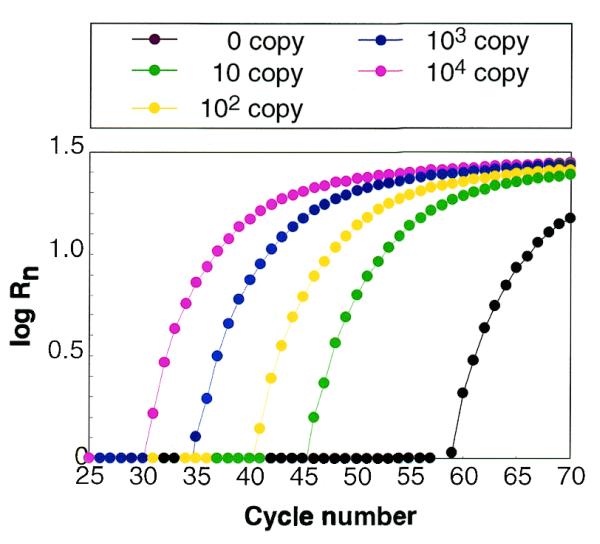
Fluorescence quench rate (Rn) at each PCR cycle. The log of Rn is expressed as function of cycle number.
Figure 4.

Calibration curves for the human β-globin gene. The correlation coefficients were 0.9980, 0.9990, 0.9993, 0.9985, 0.9989 and 0.9988, when log Rn was 0.1, 0.3, 0.5, 0.7, 0.9 and 1.2, respectively.
The PCR experiment was carried out four times, and the linear relationship between cycle number and initial template copy number was very reproducible; the correlation coefficient was >0.99 in three of the four experiments and was 0.981 in the remaining one. We also noted that only one target molecule was detectable using this method (data not shown). Our results thus strongly suggest that determination of the initial template copy number is possible using a primer containing a BODIPY® FL-modified C at the 5′-end. Furthermore, even when preliminarily designed primers do not have a C at the 5′-end, its addition enables use of this technique.
Fluorescence also declined in the sample containing no template (Figs 2 and 3), but this decline occurred at very high cycle numbers, hence this false reaction was easily distinguished from the quenching that resulted from amplification of the target DNA. Electrophoresis of the PCR product from this sample yielded one band of ∼300 bp, which was found to be a sequence derived from the E.coli genome added as the non-target DNA. In contrast, sequence analysis of the PCR products from samples containing 10–10 000 initial copies of the target DNA were found to be identical to the target.
The established methods for real-time quantitative PCR mainly use sequence-specific fluorescent probes/primers or double-stranded DNA-specific dyes to measure PCR products. Probe/primer-based methods (4–8) rely on FRET to achieve the necessary sensitivity for detection, but the design of suitable probes is usually a time-consuming step. Dye-based methods (11–15) are comparatively simple but are not as sensitive or reliable, since the fluorescence depends solely on the amount of double-stranded DNA, which includes not only specific products but also non-specific ones. Compared with these approaches, the new PCR method described here is simpler and more sensitive. Thus, we have developed simple and widely applicable methods for the detection of specific DNA/RNA molecules of interest. By using oligonucleotide probes/primers modified with BODIPY® FL, targeted DNA/RNA molecules can be quantitatively detected in samples by monitoring the fluorescence intensity of the probe before and after hybridization.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr K.Nakamura of the National Institute of Bioscience and Human-Technology for his valuable advice. This work was supported by the Industrial Science and Technology Frontier Program of the New Energy and Industrial Technology Development Organization (NEDO), Japan.
References
- 1.Cardullo R.A., Agrawal,S., Flores,C., Zamecnik,P.C. and Wolf,D.E. (1988) Detection of nucleic acid hybridization by nonradiative fluorescence resonance energy transfer. Proc. Natl Acad. Sci. USA, 85, 8790–8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morrison L.E., Halder,T.C. and Stols,L.M. (1989) Solution-phase detection of polynucleotides using interacting fluorescent labels and competitive hybridization. Anal. Biochem., 183, 231–244. [DOI] [PubMed] [Google Scholar]
- 3.Parkhurst K.M. and Parkhurst,L.J. (1995) Kinetic studies by fluorescence resonance energy transfer employing a double-labeled oligonucleotide: hybridization to the oligonucleotide complement and to single-stranded DNA. Biochemistry, 34, 285–292. [DOI] [PubMed] [Google Scholar]
- 4.Tyagi S. and Kramer,F.R. (1996) Molecular beacons: probes that fluoresce upon hybridization. Nat. Biotechnol., 14, 303–308. [DOI] [PubMed] [Google Scholar]
- 5.Heid C.A., Stevens,J., Livak,K.J. and Williams,P.M. (1996) Real time quantitative PCR. Genome Res., 6, 986–994. [DOI] [PubMed] [Google Scholar]
- 6.Nazarenko I.A., Bhatnagar,S.K. and Hohman,R.J. (1997) A closed tube format for amplification and detection of DNA based on energy transfer. Nucleic Acids Res., 25, 2516–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whitcombe D., Theaker,J., Guy,S.P., Brown,T. and Little,S. (1999) Detection of PCR products using self-probing amplicons and fluorescence. Nat. Biotechnol., 17, 804–807. [DOI] [PubMed] [Google Scholar]
- 8.Wittwer C.T., Herrmann,M.G., Moss,A.A. and Rasmussen,R.P. (1997) Continuous fluorescence monitoring of rapid cycle DNA amplification. Biotechniques, 22, 130–131, 134–138. [DOI] [PubMed] [Google Scholar]
- 9.Horn T., Chang,C.A. and Urdea,M.S. (1997) Chemical synthesis and characterization of branched oligodeoxyribonucleotides (bDNA) for use as signal amplifiers in nucleic acid quantification assays. Nucleic Acids Res., 25, 4842–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saiki R.K., Gelfand,D.H., Stoffel,S., Scharf,S.J., Higuchi,R., Horn,G.T., Mullis,K.B. and Erlich,H.A. (1988) Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science, 239, 487–491. [DOI] [PubMed] [Google Scholar]
- 11.Higuchi R., Fockler,C., Dollinger,G. and Watson,R. (1993) Kinetic PCR analysis: real-time monitoring of DNA amplification reactions. Biotechnology, 11, 1026–1030. [DOI] [PubMed] [Google Scholar]
- 12.Ishiguro T., Saitoh,J., Yawata,H., Yamagishi,H., Iwasaki,S. and Mitoma,Y. (1995) Homogeneous quantitative assay of hepatitis C virus RNA by polymerase chain reaction in the presence of a fluorescent intercalater. Anal. Biochem., 229, 207–213. [DOI] [PubMed] [Google Scholar]
- 13.Ririe K.M., Rasmussen,R.P. and Wittwer,C.T. (1997) Product differentiation by analysis of DNA melting curves during the polymerase chain reaction. Anal. Biochem., 245, 154–160. [DOI] [PubMed] [Google Scholar]
- 14.Woo T.H., Patel,B.K., Cinco,M., Smythe,L.D., Symonds,M.L., Norris,M.A. and Dohnt,M.F. (1998) Real-time homogeneous assay of rapid cycle polymerase chain reaction product for identification of Leptonema illini. Anal. Biochem., 259, 112–117. [DOI] [PubMed] [Google Scholar]
- 15.Morrison T.B., Weis,J.J. and Wittwer,C.T. (1998) Quantification of low-copy transcripts by continuous SYBR Green I monitoring during amplification. Biotechniques, 24, 954–958, 960, 962. [PubMed] [Google Scholar]