

Abstract
T-cell immunity has been claimed as the main immunoprotective mechanism against Paracoccidioides brasiliensis infection, the most important fungal infection in Latin America. As the initial events that control T-cell activation in paracoccidioidomycosis (PCM) are not well established, we decided to investigate the role of CD28, an important costimulatory molecule for the activation of effector and regulatory T cells, in the immunity against this pulmonary pathogen. Using CD28-deficient (CD28−/−) and normal wild-type (WT) C57BL/6 mice, we were able to demonstrate that CD28 costimulation determines in pulmonary paracoccidioidomycosis an early immunoprotection but a late deleterious effect associated with impaired immunity and uncontrolled fungal growth. Up to week 10 postinfection, CD28−/− mice presented increased pulmonary and hepatic fungal loads allied with diminished production of antibodies and pro- and anti-inflammatory cytokines besides impaired activation and migration of effector and regulatory T (Treg) cells to the lungs. Unexpectedly, CD28-sufficient mice progressively lost the control of fungal growth, resulting in an increased mortality associated with persistent presence of Treg cells, deactivation of inflammatory macrophages and T cells, prevalent presence of anti-inflammatory cytokines, elevated fungal burdens, and extensive hepatic lesions. As a whole, our findings suggest that CD28 is required for the early protective T-cell responses to P. brasiliensis infection, but it also induces the expansion of regulatory circuits that lately impair adaptive immunity, allowing uncontrolled fungal growth and overwhelming infection, which leads to precocious mortality of mice.
It has long been appreciated that cellular immunity is the most important resistance mechanism against fungal infections (14, 36, 64). CD4+ and CD8+ T-cell subpopulations have been described to have a fundamental role in the control of fungal growth, and disease severity is also controlled by regulatory T (Treg) cells, which prevent tissue pathology by controlling excessive inflammatory reactions (25, 45, 46, 65). Similar to other deep mycoses, the severity of paracoccidioidomycosis (PCM), the most severe pulmonary mycosis in Latin America, is controlled by cellular immunity and cytokine-activated phagocytes that are able to kill Paracoccidioides brasiliensis, the etiological agent of this infection (10, 20, 30, 60, 61). In humans and in murine models of PCM, resistance to the disease is associated with the secretion of gamma interferon (IFN-γ) and other type 1 cytokines, whereas impaired Th1 immunity and the prevalent secretion of Th2 cytokines correlate with a systemic and progressive disease (2, 6, 39, 59, 76). Studies with CD4+ and CD8+ T-cell-deficient mice revealed that both T-cell subsets are involved in the protective immunity against P. brasiliensis infection and indicated the prominent role of CD8+ T cells (3, 21, 25). Besides the prevalent Th2 immunity, recent investigations have described alternative mechanisms underlying T-cell dysfunction in humans and experimental PCM. Increased apoptosis and overexpression of Fas and FasL in T cells suggest that activation-induced cell death (AICD) is a mechanism that controls T-cell expansion during the active disease (13, 19). In addition, the increased expression of CTLA-4 and the expansion of Treg cells were associated with severe patterns of the disease (24, 45, 46, 56). Thus, in addition to cytokine imbalance, other regulatory mechanisms can actively participate in the unresponsiveness of T cells in P. brasiliensis-infected hosts.
Optimal activation, proliferation, and cytokine production by antigen-specific T cells require two distinct signals from dendritic cells or other antigen-presenting cells (APCs). After T-cell receptor (TCR) occupancy by the antigen epitope/major histocompatibility (MHC) complex (first signal), a second signal is mediated by costimulatory molecules (43, 63), such as CD28 on T cells and their counter-receptors CD80 (B7-1) and CD86 (B7-2) expressed by APCs (1, 34). Soluble molecules, such as cytokines and chemokines, also participate in the activation process, which drives and controls T-cell numbers and fates (1). CD28 enhances the TCR-triggered activation of naïve T cells, promotes interleukin-2 (IL-2) secretion and prevents T-cell anergy (1, 37). Alternatively, CD28-independent T-cell activation can occur if a strong and sustained antigen-specific signal is available (40, 81). Like CD28, two other molecules, cytotoxic T-lymphocyte antigen-4 (CTLA-4) and mouse inducible costimulatory molecule (ICOS), are selectively expressed by T cells, but the expression of these molecules depends on previous cell activation (50, 71). More recently, evidence has emerged that CD28 family members are also crucial regulators of natural and induced regulatory (CD4+CD25+Foxp3+) T cells (9). These cells are induced in the thymus and in the periphery, respectively, and control self-tolerance and the activation of several components of innate and adaptive immunity (68). Treg cells can suppress immune responses through the production of immunosuppressive cytokines (mainly IL-10 and transforming growth factor β [TGF-β]), through the induction of the apoptosis of effector T cells and through the modification of the functional properties of antigen-presenting cells (70, 78).
Immunoprotection against microorganisms has been shown to be either CD28 dependent or independent. CD28-deficient (CD28−/−) mice are highly susceptible to infection with Salmonella enterica serovar Typhimurium due to the poor ability of these mice to secrete IFN-γ (51). During some viral and parasitic infections, CD28 was shown to be required to mediate CD8+ T-cell immunoprotection (8, 53). In contrast, CD28−/− mice infected with Mycobacterium bovis or Listeria monocytogenes control the bacterial burden and develop cell-mediated immunity (35, 52). In primary and opportunistic fungal infections, CD28 costimulation controls protective immunity, the expansion and function of regulatory T cells, and the intensity of inflammatory reactions (5, 54, 55, 66, 84).
Because CD28 is critical for T-cell activation in fungal infections, we investigated its role in a murine model of P. brasiliensis infection. We show that CD28 costimulation exerts contrasting roles in pulmonary PCM. Early in infection, CD28 expression results in efficient adaptive immunity that is able to control fungal growth. Late in infection, however, this costimulatory molecule induces significant expansion of regulatory T cells, diminished immunity, and uncontrolled fungal growth that eventually leads to the death of the mice. In contrast, the absence of CD28 costimulation results in impaired T-cell immunity, which appeared to be compensated by the absence of Treg cell expansion. This weak but persistent immunity was able to partially control fungal growth, organize granulomatous lesions, and guarantee the enhanced survival of the mice, suggesting the relative protection conferred by CD28-independent mechanisms.
MATERIALS AND METHODS
Mice.
Breeding pairs of homozygous CD28-deficient (CD28−/−) and wild-type (WT) control C57BL/6 mice (intermediate susceptibility to P. brasiliensis) were bred at the University of São Paulo animal facilities under specific-pathogen-free (SPF) conditions in closed-top cages. Clean food and water were given ad libitum. Mice were 8 to 11 weeks of age at the time of infection, and procedures involving animals and their care were approved by the Ethics Committee on Animal Experiments from Instituto de Ciências Biomédicas, Universidade de São Paulo.
Fungus and mice infection.
P. brasiliensis 18 isolate (Pb18), which is highly virulent, was used throughout the study. To ensure the maintenance of its virulence, the isolate was used after three serial animal passages (38). Pb18 yeast cells were then maintained by weekly subcultivation in semisolid Fava Netto culture medium (29) at 35°C and used on the seventh day of culture. The fungal cells were washed in phosphate-buffered saline (PBS; pH 7.2) and counted in a hemocytometer, and the suspension was adjusted to 20 × 106 fungal cells/ml. The viability of fungal suspension, determined by Janus Green B vital dye (Merk, Darmstadt, Germany), was always higher than 80%. Mice were anesthetized and submitted to intratracheal (i.t.) P. brasiliensis infection as previously described (20). Briefly after intraperitoneal anesthesia, the animals were infected with 1 × 106 Pb18 yeast cells, contained in 50 μl of PBS, by surgical i.t. inoculation, which allowed dispensing of the fungal cells directly into the lungs. The skins of the animals were then sutured, and the mice were allowed to recover under a heat lamp. Mice were studied during an early period (2 and 10 weeks after infection) and a late period (16 and 26 weeks). Two or three experiments were performed separately.
Assay for organ CFU.
The number of viable microorganisms in infected organs (lung, liver, and spleen) from experimental and control mice were determined by counting the number of CFU. Animals (n = 6 to 8) from each group were sacrificed, and the enumeration of viable organisms was done as previously described (74). Briefly, aliquots (100 μl) of the cellular suspensions and serial dilutions were plated on brain heart infusion agar (Difco, Detroit, MI) supplemented with 4% (vol/vol) horse serum (Instituto Butantan, São Paulo, Brazil), and 5% P. brasiliensis 192 culture filtrate, the last constituting a source of growth-promoting factor. The plates were incubated at 35°C, and colonies were counted daily until no increase in counts was observed. The number (log10) of viable P. brasiliensis colonies per gram of tissue were expressed as means ± standard errors (SE).
Mortality rates.
Mortality studies were performed with CD28−/− and control WT mice inoculated i.t. with 1 × 106 yeast cells or PBS (n = 9 to 11). Deaths were registered daily for a 400-day period, and experiments were repeated twice.
NO production.
Nitric oxide (NO) production was quantified by the accumulation of nitrite in the tissue homogenates by a standard Griess reaction. Briefly, 50 μl of supernatants was removed from 24-well plates and incubated with an equal volume of Griess reagent (1% sulfanilamide, 0,1% naphthylene diamine dihydrochloride, 2.5% H3PO4) at room temperature for 10 min. The absorbance at 550 nm was determined with a microplate reader. The conversion of absorbance to micromolar NO was deduced from a standard curve by using a known concentration of NaNO2 diluted in RPMI medium. All determinations were performed in duplicate and expressed as micromolar NO.
Measurement of serum P. brasiliensis-specific isotypes.
Specific isotype levels (total IgG, IgM, IgA, IgG1, IgG2a, IgG2b, and IgG3) were measured by a previously described enzyme-linked immunosorbent assay (ELISA) (20) employing a cell-free antigen (18) prepared by using a pool of different P. brasiliensis isolates (Pb339, Pb265, and Pb18). The average of the optical densities obtained with sera from control mice (PBS inoculated) diluted 1:20 was considered the cutoff for each respective isotype. Optical densities for each dilution of experimental sera were compared to the control values. The titer for each sample was expressed as the reciprocal of the highest dilution that presented an absorbance higher than the cutoff.
Measurement of cytokines.
Mice were infected i.t. with P. brasiliensis (n = 6 to 8), and their right lungs were aseptically removed and individually disrupted in 5.0 ml of PBS. Supernatants were separated from cell debris by centrifugation at 2,000 × g for 15 min, passed through 0.22-μm-pore-size filters (Millipore, Bedford, MA), and stored at −70°C. The levels of IL-2, IL-12, IFN-γ, tumor necrosis factor alpha (TNF-α), IL-4, IL-5, IL-10, granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-6, IL-23, IL-17, and TGF-β were measured by capture ELISA with antibody pairs purchased from Pharmingen. The ELISA procedure was performed according to the manufacturer's protocol. The concentrations of cytokines were determined with reference to a standard curve for several 2-fold dilutions of murine recombinant cytokines. As an additional control, lung homogenates were added to recombinant cytokines used to obtain standard curves; no interference was detected, indicating the absence of inhibitory substances (e.g., soluble cytokine receptors).
Lung and liver leukocyte isolation.
Lungs from each mouse were excised, washed in PBS, minced, and digested enzymatically for 1 h in 15 ml/lung of digestion buffer (RPMI, 5% fetal calf serum, 1 mg/ml collagenase [Sigma Aldrich Inc.], and 30 μg/ml DNase). Livers from individual mice were obtained and submitted to organ perfusion using 10.0 ml of warm PBS via the portal vein, and organ fragments were pressed through a 70-μm cell strainer (Becton Dickson). After erythrocyte lysis using NH4Cl buffer, cells were washed, resuspended in complete media, and centrifuged for 30 min at 1,200 × g in the presence of 20% Percoll (Sigma) to separate leukocytes from cell debris and epithelial cells. Total leukocyte numbers were assessed in the presence of trypan blue using a hemocytometer; viability was always higher than 85%. The absolute number of a leukocyte subset was equal to the percentage of that cell subset multiplied by the total number of leukocytes recovered from the digested organ divided by 100.
Flow cytometry analysis.
For surface staining alone, leukocytes were washed and resuspended at a concentration of 1 × 106 cells/ml in staining buffer (1× PBS, 2% serum calf bovine, and 0.5% NaN3). Fc receptors were blocked by the addition of unlabeled anti-CD16/32 (Fc block; BD Pharmingen, San Diego, CA). The leukocytes were then stained for 20 min at 4°C with the optimal dilution of each antibody or fluorescein isothiocyanate (FITC) or phycoerythrin (PE) conjugated (BD Pharmingen). The following antibodies were used: CD11b+, CD11c+, Iab+, CD80+, CD86+, CD40+, CD4+, CD8+, CD44+, CD25+, CD69+, CTLA-4+, ICOS+, and FasL+. Cells were washed twice with staining buffer resuspended in 100 μl, and an equal volume of 2% formalin was added to fix the cells. The stained cells were analyzed immediately on a FACSCalibur flow cytometer (BD Biosciences, CA) using the CellQuest software (BD Biosciences) gating on macrophages or lymphocytes as judged from forward and side light scatter. Ten thousand cells were counted, the data were expressed as the percentage or the absolute number of positive cells, which was calculated through the percentage obtained by a fluorescence-activated cell sorter (FACS), and the number of cells was determined in Neubauer chambers. The intracellular detection of FoxP3, the X-linked forkhead/winged helix transcription factor, in leukocytes obtained from the lung lesions was performed in fixed and permeabilized cells using Cytofix/Cytoperm (BD Biosciences). Initially, the cells were labeled with antibodies for cell surface molecules, such as FITC-conjugated anti-CD4 and PE-conjugated anti-CD25. Next, the cells were fixed, permeabilized, and stained with Cy-conjugated anti-FoxP3 for 90 min at 4°C. Cells were then washed twice with staining buffer and resuspended in 100 μl, and an equal volume of 2% formalin was added to fix the cells. A minimum of 20,000 events were acquired on the FACSCalibur flow cytometer using the CellQuest software, as described above. Surface staining of CD25 and intracellular FoxP3 expression were back-gated on the CD4 T-cell population. For flow cytometric analysis of apoptotic and necrotic lymphocytes, annexin V and propidium iodide labeling was used (82).
In vivo depletion of CD8+ T cells.
The H-35 hybridoma (rat IgG1, anti-mouse CD8 monoclonal antibody [MAb]) was grown intraperitoneally (i.p.) in pristane (Sigma Chemical Co., St. Louis, MO)-primed BALB/c nu/nu male mice. The antibodies were purified from ascites as described elsewhere (49) and assessed for purity by SDS-PAGE electrophoresis. Groups of WT and CD28−/− C57BL/6 mice were given 200 μg of the H-35 MAbs or normal rat IgG (controls) by the i.p. route, 48 and 24 h before infection with P. brasiliensis cells, and 150 μg of the H-35 MAbs or rat IgG at days 6 and 12 after infection. Mice were sacrificed 48 h later, and lungs analyzed for CFU counts and the presence of lymphocytes and macrophages.
Histopathologic and morphometrical analyses.
Groups of CD28−/− mice and their WT counterparts were killed at the second and tenth week postinfection. Lungs were collected, fixed in 10% formalin, and embedded in paraffin. Five-micrometer sections were stained by hematoxylin-eosin (H&E) for an analysis of the lesions and silver stained for fungal evaluation. Pathological changes were analyzed based on the size, morphology, and cell composition of granulomatous lesions, presence of fungi, and intensity of the inflammatory infiltrates. Morphometrical analysis was performed using a Nikon DXM 1200c digital camera (magnification of ×10) and Nikon NIS Elements AR 2.30 software. The area of lesions was measured (in μm2) in 10 microscopic fields per slide (n = 4 to 6). Results were expressed as the mean (± standard error of the mean [SEM]) total area of lesions for each animal.
Statistical analysis.
All values are means ± SEM, unless otherwise indicated. Differences between two means were evaluated by Student's t test. Differences between survival times were determined with the log rank test using GraphPad Prism 5 for Windows (GraphPad Software). P values of ≤0.05 were considered significant.
RESULTS
CD28 expression induces an early protective but a late deleterious effect in the control of fungal growth.
The evolution of the disease of i.t. infected CD28−/− mice and WT controls was monitored by counts of CFU in the lung, liver, and spleen at different times postinfection (2, 10, 16, and 26 weeks). At weeks 2 and 10 of infection, CD28-deficient mice showed higher counts of CFU in their lungs (Fig. 1 A) than WT mice. At week 2, fungal dissemination to the liver was observed with CD28−/− mice, but the difference between the control and CD28−/− mice was significant only at week 10 (Fig. 1B). Unexpectedly, late in infection (weeks 16 and 26), no difference in the pulmonary fungal burden was observed between the two mouse strains. Importantly, by week 16, a reversion in the severity of the hepatic infection was detected: fungal loads of the WT mice were significantly higher than those of the CD28−/− mice, indicating a vigorous dissemination or uncontrolled fungal growth in the WT strain. This difference was even higher at week 26 postinfection, when the livers of the WT mice showed 6.12 ± 0.10 viable yeast cells, while 4.11 ± 0.52 fungal cells were detected in CD28−/− mice. No difference in fungal burdens was detected in the spleens (data not shown).
FIG. 1.

Early in infection CD28 is protective, while at the chronic phase, CD28 signaling exerts a deleterious effect. CD28−/− and control WT C57BL/6 mice were i.t. infected with 1 × 106 fungal cells, and severity of infection was analyzed by CFU counts at four postinfection periods. (A) At weeks 2 and 10 after infection, increased fungal burdens in the lungs of CD28-deficient mice were observed. (B) By week 10, an increased dissemination to livers of CD28−/− mice was also seen. Unexpectedly, at week 16, a marked fungal growth was detected in the lungs, and the number of fungal cells in the livers of WT mice supplanted that observed with the deficient strain. At week 26 postinfection, no differences in the number of pulmonary CFU were noted, but the number of viable yeasts in the livers of WT mice remained higher than that of CD28-deficient mice. The points represent means ± SEM of the numbers of log10 CFU obtained from groups of six to eight mice. The results are representative of 3 experiments (except week 16). *, P < 0.05, and ***, P < 0.001, compared with WT controls.
CD28-deficient mice present diminished mortality rates.
To better characterize the effect of CD28 signaling in the outcome of pulmonary paracoccidioidomycosis, mortality in P. brasiliensis-infected CD28-deficient and control mice was followed over a 57-week period (Fig. 2). Unexpectedly, WT mice showed increased mortality rates compared to CD28−/− mice. Thus, late in infection, CD28-dependent mechanisms appeared to result in deleterious effects to P. brasiliensis-infected hosts.
FIG. 2.
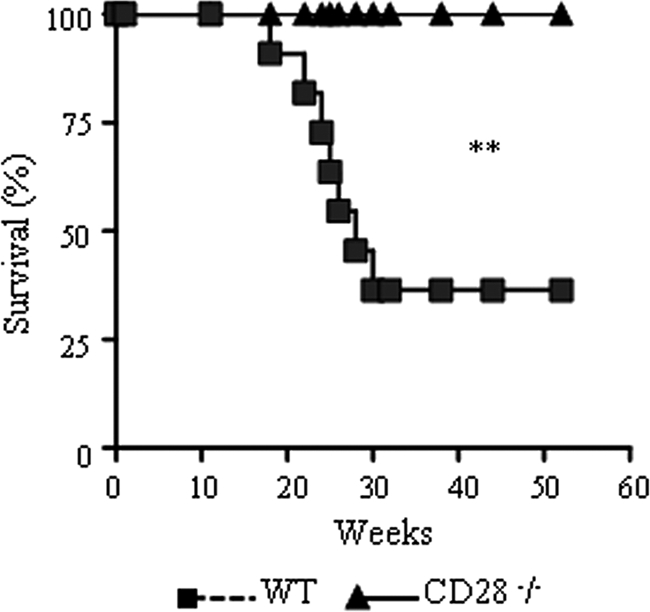
CD28 costimulation results in increased mortality rates of P. brasiliensis-infected mice. Survival times of CD28−/− and WT control mice after i.t. infection with 1 × 106 P. brasiliensis yeast cells were determined for a period of 57 weeks. Significant differences in mortality rates were detected; the results are representative of two independent experiments; (n = 6). **, P < 0.01.
Absence of CD28 signaling leads to decreased synthesis of nitric oxide (NO) and an impaired humoral immune response.
Synthesis of nitric oxide by cytokine-activated macrophages has been reported to be the most important mechanism in the control of P. brasiliensis growth in mice (11, 22). Accordingly, the better control of the fungal burden by the CD28-normal mice early in the infection was concomitant with increased levels of nitric oxide in the lungs and liver homogenates of these mice (Fig. 3 A).
FIG. 3.

CD28 deficiency determines impaired production of NO and decreased humoral immunity. Levels of nitric oxide (A) and P. brasiliensis-specific antibodies (B) were assayed, respectively, in organ homogenates and sera of CD28−/− and WT mice i.t. infected with 1 × 106 yeast cells. NO was measured by the Griess reaction, and sera were assayed for total IgG, IgM, IgA, IgG1, IgG2a, IgG2b, and IgG3 by using an isotype-specific ELISA as detailed in Materials and Methods. The bars depict means ± SE of NO levels or serum titers (6 to 8 mice per group). The results are representative of 3 experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001, compared with WT controls.
Although specific antibodies may have a protective role in some fungal infections (23), antibody production in human and experimental PCM is a marker of disease severity (14, 17, 28). In addition, almost all P. brasiliensis components are T-dependent antigens and require Th cells to induce antibody production (12). In the severe forms of PCM, there is a prevalence of Th2 isotypes (IgG2b, IgG1, and IgA), while in the benign infection, IgG2a, a Th1 isotype, is preferentially produced (14). Because CD28 deficiency was shown to impair antibody production (73) and influenced the severity of P. brasiliensis infection, we decided to characterize humoral immunity at the second and tenth weeks postinfection. At week 2, the increased fungal loads of CD28−/− mice were associated with decreased levels of IgM compared with the control group. At week 10, the more severe and disseminated disease of CD28−/− mice paralleled a prominent impairment in the synthesis of all IgG isotypes except IgG3 (Fig. 3B). As a whole, the data on nitric oxide and antibody production demonstrate that CD28 signaling is necessary for the activation of the immune response against P. brasiliensis.
CD28-deficient mice produce decreased levels of pro- and anti-inflammatory cytokines.
Similar to what has been observed with other intracellular infections, proinflammatory cytokines (IFN-γ, TNF-α, and IL-12) have been shown to be protective, while anti-inflammatory cytokines (IL-4, IL-10, and TGF-β) are prevalent in the severe forms of PCM (6, 14, 15, 47). Cytokines associated with the Th17 pattern of immunity, however, have been shown to exert both protective and deleterious effects in pulmonary PCM: they increase the number and effector activity of neutrophils but also induce increased inflammatory reaction mixtures (46). Here, the levels of type 1 (IL-12, TNF-α, IL-2, and IFN-γ,), type 2 (IL-4, IL-5, and IL-10), and Th17-associated (IL-6, TGF-β, IL-23, and IL-17) cytokines were measured in the lungs and livers of CD28−/− mice and their normal counterparts at weeks 2 and 10 after P. brasiliensis infection. We could verify that all types of cytokines were present in the lungs and livers of CD28-deficient and WT mice (Fig. 4). The liver homogenates, however, exhibited the most important differences determined by CD28 deficiency. At week 2 postinfection, IL-2, IL-4, and GM-CSF appeared at significantly lower levels in the lungs and livers of CD28−/− mice than in those of WT mice (Fig. 4A); the other cytokines assayed appeared at equivalent levels (data not shown). At week 10, diminished levels of IL-12 and TNF-α were present in the lungs, whereas lower levels of pro- and anti-inflammatory cytokines (IL-12, TNF-α, IFN-γ, IL-10 and GM-CSF) were observed in the liver homogenates of CD28-deficient mice (Fig. 4B). At this postinfection time point, no differences in IL-6, IL-23, or IL-17 were detected in organ homogenates, but TGF-β was detected at lower levels in the lung and liver homogenates of CD28−/− mice (Fig. 4B).
FIG. 4.

CD28 deficiency causes a reduced production of pro- and anti-inflammatory cytokines in the lungs and livers of P. brasiliensis-infected mice. Levels of cytokines in organ homogenates of CD28−/− and WT mice were measured after i.t. infection with 1 × 106 yeast cells. Lungs and livers were collected at weeks 2 and 10 after infection and disrupted in 5.0 ml of PBS, and supernatants were analyzed for cytokine content by capture ELISA. (A) At week 2 after infection, CD28-deficient mice presented reduced levels of IL-2, IL-4, and GM-CSF in the lungs and livers. (B) Decreased levels of IL-12, TNF-α, and TGF-β in the lungs and IL-12, TNF-α, IFN-γ, IL-10, and TGF-β in the livers of deficient mice were seen at week 10 postinfection. The bars depict means ± SEM of cytokine levels (6 to 8 animals per group). *, P < 0.05; **, P < 0.01; ***, P < 0.001, compared with WT controls.
Early in infection, CD28 costimulation leads to the increased influx of activated macrophages, T cells, and regulatory T cells to the site of infection.
To characterize the inflammatory infiltrates in the lung, the presence and activation status of monocytes/macrophages and T cells were studied at week 10 after P. brasiliensis infection. Figure 5 A shows that increased numbers of CD11b+-activated macrophages were present in the lungs of WT mice. CD11b+ cells expressing costimulatory molecules were detected in diminished numbers in the lungs of CD28-deficient mice. The expression of CD69, CD25, CD44, ICOS, CTLA-4, and FasL by T cells freshly isolated from the lungs was used to determine the activation profile of CD4+ and CD8+ T cells that had migrated to the lungs of P. brasiliensis-infected mice. The marker CD69 is a very early activation antigen (86), as is CD25, the α-chain of the interleukin-2 receptor (69), and both markers are rapidly upregulated on activated T cells. CTLA-4 is a costimulatory molecule of which the main function is to attenuate the expansion and cytokine production of recently activated T cells (67). The expression of high levels of CD44 (CD44high) has been observed with effector or memory T cells (80). Increased numbers of CD4+ T cells expressing CD44high, CD25, CTLA-4, and ICOS were observed with the lungs of CD28-sufficient mice (Fig. 5B), demonstrating that a high number of activated T cells migrated to the site of infection. Conversely, CD8+ T cells migrated in higher numbers into the lungs of CD28-deficient mice. The increased number of CD8+CD44low cells indicated, however, the prevalence of a naïve phenotype (Fig. 5C). No differences in CD69 expression were observed in CD8+ T cells, but upregulated expression of FasL was detected in WT mice. Because the expression of CD28 is necessary for the expansion of Treg cells (33, 77), the presence of CD4+CD25+Foxp3+ T cells in the lungs of both mouse strains was evaluated. Consistent with previous reports, increased expression of Foxp3 was observed in the CD4+CD25+ cells of CD28-normal mice (Fig. 5D). Because upregulated expression of FasL was detected in the lymphocytes of WT mice, we also assessed the number of apoptotic and necrotic cells in the lungs of infected mice. Increased numbers of both apoptotic and necrotic cells were detected in WT mice (Fig. 5E).
FIG. 5.

WT mice presented an increased influx of activated macrophages, T cells, and regulatory CD4+CD25+FoxP3+ T cells to the lungs, whereas CD28-deficient mice showed an increased presence of naïve CD8+ T cells. Characterization of leukocyte subsets and the activation profile of cells by flow cytometry in the lung-infiltrating leukocytes (LIL) from CD28−/− and WT mice inoculated i.t. with 1 × 106 P. brasiliensis yeast cells. At week 10 after infection, lung cell suspensions were obtained and stained as described in Materials and Methods. The acquisition and analysis gates were restricted to macrophages or lymphocytes. (A) Macrophages (møs); (B) CD4+ T cells; (C) CD8+ T cells. (D) To characterize the expansion of regulatory T cells in LIL, surface staining of CD25+ and intracellular FoxP3 expression were back-gated on the CD4+ T-cell population. (E) The number of apoptotic and necrotic leukocytes was assessed by flow cytometry using FITC-labeled annexin V and propidium iodide. The data represent the mean ± SEM of the results from 6 to 8 mice per group and are representative of two independent experiments. *, P < 0.05; **, P < 0.01; and ***, P < 0.001, compared with WT controls. Nr, number.
In the chronic phase, WT mice showed a progressive deactivation of inflammatory cells associated with the persistent presence of regulatory T cells.
Because the infection in WT mice was seen to be more severe than in CD28−/− mice from week 16 onward, we decided to characterize the inflammatory cell infiltrates of the lungs and livers at week 16 postinfection. Flow cytometric analysis of lung cells showed that almost all of the increased activation markers of macrophages and CD4+ and CD8+ T cells observed with WT mice at week 10 postinfection were lost during this later period (Fig. 6 A, B, and C). The only significant difference that was maintained was the increased expression of CD25 (a marker of activated and regulatory T cells) on the CD4+ T cells of WT mice. Indeed, the CD4+CD44high and CD8+CD44high phenotypes were observed in very low numbers in both mouse strains (Fig. 6B and C). Interestingly, during this postinfection period, an increased number of macrophages was seen compared to week 10 (3- to 8-fold in WT and 5- to 10-fold in knockout [KO] mice), although no differences in the expression of activation markers were detected between the mouse strains (Fig. 6A). In addition, no marked differences in the number or activation profile of liver leukocytes were detected. WT mice, however, presented increased numbers of CD11b+CD80+ macrophages (Fig. 6D) and CD4+CD25+ lymphocytes (Fig. 6E), although no differences were detected in CD8+ T cells (Fig. 6F). Importantly, augmented numbers of CD4+CD25+Foxp3+ Treg cells were present in the cell exudates of the lungs and livers of WT mice (Fig. 6G). As a whole, these results demonstrated that the increased number and activation of macrophages and CD4+ and CD8+ T cells detected in WT mice at week 10 postinfection were lost during this later postinfection period. The same was true for the augmented number of naïve CD8+ T cells from CD28KO mice. In contrast, the regulatory T cells were maintained in expanded numbers in the livers and lungs of the WT strain.
FIG. 6.

At week 16, the numbers and levels of activation of inflammatory cells from lungs and livers of CD28−/− and WT mice were similar, but increased numbers of regulatory T cells were detected in the latter strain. Characterization of leukocyte subsets and the activation profile of cells by flow cytometry in the lung- and liver-infiltrating leukocytes (LIL) from CD28−/− and WT mice inoculated i.t. with 1 × 106 P. brasiliensis yeast cells. At week 16 after infection, lung and liver cell suspensions were obtained and stained as described in Materials and Methods. The acquisition and analysis gates were restricted to macrophages or lymphocytes. (A, B, and C) Lung macrophages and CD4+ and CD8+ T cells, respectively. (D, E, and F) Liver macrophages and CD4+ and CD8+ T cells, respectively. (G) To characterize the expansion of regulatory T cells in lungs and livers, surface staining of CD25+ and intracellular FoxP3 expression were back-gated on the CD4+ T-cell population. The data represent the mean ± SEM of the results from 6 to 8 mice per group and are representative of one experiment. *, P < 0.05; **, P < 0.01; ***, P < 0.001, compared with WT controls.
CD8+ T lymphocytes are more protective in the WT than in CD28-deficient mice.
In a study of the influence of CD28 costimulation in the pulmonary infection caused by Pneumocystis carinii, Rose et al. (66) observed an increased expansion of naïve CD8+ T cells in the lungs of CD28-deficient mice. Because the same phenomenon was observed in our study, we asked whether CD8+ T lymphocytes exerted a protective role in the CD28-deficient mouse strain. Thus, CD28−/− and WT mice were depleted of CD8+ T cells in vivo by a monoclonal antibody and were infected with P. brasiliensis. CFU counts were determined 2 weeks later. As shown in Fig. 7 A, a slight but significant increase in the number of fungal cells was seen in the lungs of both mouse strains. Depletion of CD8+ T cells, however, abolished the differences in fungal loads observed between the mouse strains. Thus, CD8+ T cells appear to exert a more efficient protective role in CD28-sufficient mice compared to CD28-deficient mice, and this agrees with the activated phenotype of the cells observed in WT mice. As expected, the anti-CD8 treatment selectively depleted CD8+ T cells but had no influence on the numbers of CD4+ T lymphocytes or CD11b+ macrophages (Fig. 7B).
FIG. 7.

In vivo depletion of CD8+ T cells abrogated the differences in fungal loads between WT and CD28-deficient mice. Both mouse strains were treated in vivo with H35 anti-CD8 monoclonal antibodies or control immunoglobulin and i.t. infected with 1 × 106 P. brasiliensis yeasts. (A) The number of viable yeasts recovered from lungs was assessed at week 2 postinfection. (B) Lung-infiltrating CD4+ and CD8+ T cells and CD11b+ macrophages were phenotyped at week 2 postinfection. *, P < 0.05; **, P < 0.01; ***, P < 0.001, compared with controls.
Early in infection, CD28 costimulation is protective and prevents fungal dissemination.
Histopathological examination of the lungs was performed at weeks 2, 10, and 26 after fungal infection. As early as week 2 of infection, increased inflammatory reaction mixtures composed of macrophages, lymphocytes, and epithelioid cells were detected as organized granulomas in the lungs of CD28-normal mice. In CD28-deficient mice, the histological architecture of lungs was more preserved, and inflammatory cells did not organize into typical granulomas (data not shown). However, at week 10 postinfection, the lower fungal loads of WT mice were controlled by a reduced number of well-organized granulomas scattered through the lung tissue. These granulomas were composed of giant and epithelioid cells. A wide halo of mononuclear cells containing some plasmocytes was seen surrounding the epithelioid cells and xanthomatous macrophages (Fig. 8 A and B). In contrast, CD28-deficient mice showed numerous well-organized epithelioid granulomas that generally presented central supurative necrosis surrounding a high number of budding yeasts. Hyperplasia of bronchoalveolar lymphoid tissue (BALT) was detected alongside the granulomas, which occupied an extensive area of lung parenchyma (Fig. 8C and D). No dissemination of yeast cells to the livers of WT mice was seen (Fig. 8E and F), but several granulomatous lesions were detected in CD28-deficient mice (Fig. 8G and H). Therefore, early in infection, WT mice appeared to develop an immune response able to control fungal growth in the lungs and dissemination to other organs.
FIG. 8.

Photomicrographs of pulmonary and hepatic lesions of WT and CD28−/− mice at week 10 postinfection with 1 × 106 P. brasiliensis yeasts. (A and B) WT mice showed well-organized granulomas containing an elevated number of yeasts surrounded by a halo of mononuclear cells containing a few plasmocytes; most of the lung tissue was preserved, with limited signs of inflammatory cell recruitment. (C and D) CD28-deficient mice presented an elevated number of well-defined epithelioid granulomas containing a large number of yeast budding cells; these lesions occupy a large area of lung tissue and were concomitant with an extensive expansion of the bronchoalveolar lymphoid tissue. No hepatic lesions were observed in WT mice (E and F), while CD28-deficient mice showed a large number of viable yeasts surrounded by granulomatous lesions scattered through the liver parenchyma (G and H). For panels A, C, E, and G: H&E, ×100; for panels B, D, F, and H: Groccot stain, ×100.
Late in infection, CD28-sufficient mice develop a severe, disseminated infection.
Histopathological analysis of the lungs at week 26 after infection did not show any differences in the pattern or intensity of the granulomatous lesions developed by CD28-deficient and WT control mice (Fig. 9 A to D). In contrast, compared with those of CD28−/− mice, the livers of WT mice showed elevated numbers of fungal cells contained in wide lesions, which replaced large areas of the liver parenchyma (Fig. 9E to H). Morphometric analysis confirmed the significantly increased areas of hepatic lesions developed by WT mice compared with those of CD28-deficient mice (Fig. 9I).
FIG. 9.

At week 26 after infection, WT mice present large numbers of lesions and P. brasiliensis yeasts in their livers. CD28−/− and WT mice were i.t. infected with 1 × 106 fungal cells, and histopathological analyses of lungs and livers were performed at week 26 after infection. (A to D) No differences in pulmonary lesions were noted between WT and CD28-deficient mice. (E to H) Increased numbers of yeast cells and more-severe hepatic lesions were detected in WT mice. For panels A, B, E, and F: HE, ×100; for C, D, G, and H, Groccot stain, ×100. (I) Morphometrical analysis confirmed the more extensive areas occupied by the liver lesions of WT mice. (J) Levels of liver cytokines at week 26 postinfection. *, P < 0.05; **, P < 0.01, compared with WT controls.
At week 26 postinfection, almost no differences in pulmonary cytokines were observed (data not shown), and this fact agrees with the similar numbers of fungal cells observed in the lungs. The intense growth of fungal cells in the livers of WT mice, however, was accompanied by increased levels of anti-inflammatory cytokines (IL-4, IL-10, TGF-β, and GM-CSF). Interestingly, IL-2, an important growth factor for Treg cells (31), was also present at elevated levels, but IL-12, IFN-γ, and TNF-α appeared in equivalent levels in both strains (Fig. 9J).
DISCUSSION
In this report, we examined the influence of CD28 costimulation on immunity to P. brasiliensis infection. Our results demonstrate that CD28 exerts both protective and deleterious effects in PCM and that P. brasiliensis-infected hosts can develop CD28-independent immunity.
The efficient immunological activation developed by WT mice was confirmed by the early control of fungal loads associated with the elevated production of antibody, NO, and cytokines. In addition, at week 10 postinfection, the inflammatory macrophages and T cells present in the lungs expressed high levels of activation/regulatory markers. In contrast, the absence of CD28 costimulation led to a severe pulmonary and hepatic infection and impaired the activation of the immune system, as revealed by the diminished levels of cytokines and NO in the lungs and the decreased production of all P. brasiliensis-specific IgG isotypes except IgG3. Interestingly, despite their more severe infection, CD28-deficient mice produced lower levels of P. brasiliensis-specific antibodies. This result is an unusual finding because in human and experimental PCM, the levels of antibodies have a direct correlation with disease severity (7, 16, 48). However, this result is in line with our previous demonstration that almost all P. brasiliensis antigens are T cell dependent (12). The lower levels of IgG1, IgG2a, and IgG2b isotypes regulated by IL-4, IFN-γ, and TGF-β, respectively (75), suggest that CD28 deficiency affected all of the T-helper subsets (Th1, Th2, and Th3) of CD4+ T-cell immunity.
Analysis of the survival times of infected mice suggests an immunological paradox. Why does the presence of CD28 costimulation, which induces an early and efficient pattern of immunity that is able to control fungal growth and induces regulatory mechanisms to avoid excessive tissue pathology, result in increased mortality of hosts? The corollary is also contradictory: why does the inefficient immunity associated with inefficient control of pathogen replication and underdeveloped regulatory mechanisms not lead to overwhelming disease characterized by high fungal burdens and enhanced tissue damage? Our results lead us to suggest that the fatal outcome of infection developed by WT mice was due to imperfect fungal clearance during the first phase of immunity followed by the activation of vigorous CD28-dependent suppressive mechanisms mediated by Treg cells, suppressive cytokines, and other T-cell dysfunctions. This would allow a late uncontrolled fungal growth that would have deleterious effects on infected tissues. In contrast, the weak, CD28-independent immunity developed by CD28−/− mice was not impaired by regulatory mechanisms but was sufficient to partially restrain fungal growth without causing damage to important tissues.
The early immune response of the WT mice was composed of several cells and mediators previously shown to exert a protective effect in P. brasiliensis-infected hosts. Thus, the increased production of pulmonary IL-2, IL-4, and GM-CSF at week 2 postinfection indicates a balanced and concomitant activation of type 1 and type 2 immunity. The presence of high levels of IL-2 appears to guarantee the late expansion of Treg cells, which consistently appeared in the lungs and livers of WT mice. The increased synthesis of TNF-α and IL-12 in the lungs and the increased levels of IFN-γ in the livers indicated the existence of effector mechanisms able to kill P. brasiliensis yeasts and control fungal infection. Both in human and experimental PCM, cellular immunity and activated macrophages have been shown to be the most important protective mechanisms. In addition, NO, IL-12, TNF-α, and IFN-γ have been implicated in the fungicidal mechanisms developed by phagocytes following the prevalent activation of Th1 cells (3, 22, 47, 58). The efficiency of these mechanisms can be inferred from the diminished fungal loads observed with the lungs and livers of WT mice up to week 10 after infection. Moreover, lung macrophages appeared in elevated numbers and were positive for the expression of costimulatory molecules (CD80, CD86, CD40, and MHC class II), indicating efficient effector and antigen-presenting cell activities. The latter activity could be confirmed by the increased influx of activated CD4+ and CD8+ T cells to the lungs. These inflammatory lymphocytes showed upregulated expression of activation/regulatory markers (CD44high, CD25, ICOS, CTLA4, and FasL), indicating that the significant activation of T cells was accompanied by the development of regulatory mechanisms to control excessive cell multiplication and inflammatory reactions (5, 86). Indeed, the high expression of CD25 and CTLA4 at week 10 postinfection was concomitant with an increased presence of Treg cells and cells undergoing apoptosis and necrosis (Fig. 5).
TGF-β, a cytokine involved in the induction of Th17 and Treg cells (42, 83), consistently appeared in elevated levels in the lungs and livers of WT mice. Because no Th17 polarization was observed in this strain, TGF-β appears to have preferentially driven Treg development. In CD28 KO mice, the production of TGF-β, probably by innate immune cells or Th3 lymphocytes, appears to have contributed to the control of inflammatory reactions but also to the constant and persistent fungal growth observed with this strain. As has been shown in murine candidiasis and aspergillosis (54, 55), our study demonstrates that in pulmonary PCM, CD28 signaling leads to Treg development, but in the absence of CD28 costimulation, production of IL-2 was impaired, and the commitment of T lymphocytes to the regulatory phenotype was practically abolished. Interestingly, with the opportunistic pathogen Candida albicans, CD28-dependent Treg cells were needed to restrain pathogen clearance, to control inflammatory reactions, and to ensure immunoprotection against a secondary challenge, but with Blastomyces dermatitidis, a primary pathogen, vaccine-induced memory was shown to be CD28 independent (54, 84). In our studies, WT mice developed a canonical pattern of immunity in which the activation process was tightly regulated by deactivation mechanisms that controlled excessive inflammatory reactions and consequent tissue damage. However, this was not observed in the chronic phase of infection developed by CD28-sufficient mice.
The data obtained at week 16 postinfection revealed the late deleterious effect mediated by CD28 costimulation in PCM. As can be seen in Fig. 1, at this time point, WT mice lost control of pulmonary fungal growth (CFU counts increased 65-fold), and fungal cells disseminated to the liver, reaching a value of 1 million fungal cells/g tissue. In addition, despite the equivalent number of fungal cells in the lungs of both mouse strains at week 26, the hepatic fungal loads of WT mice were 67-fold higher than those of CD28 KO mice. The inefficient late-T-cell immunity of WT mice was concomitant with Treg cell expansion, a fact not observed with CD28 KO mice, which maintained low-efficiency, but persistent, antifungal immunity. Furthermore, the inhibitory activity of Treg cells and anti-inflammatory cytokines was evidenced by the deactivated phenotype of the T cells and macrophages observed with the lungs and livers at week 16 postinfection (Fig. 6). Almost no CD4+CC44high and CD8+CD44high cells were detected in the lungs and livers of WT mice, and almost no significant differences in the number or activation status of macrophages were seen between the strains. The only activation/regulatory marker persistently expressed at elevated levels by WT mice was CD25, and this was consistent with the increased presence of Treg cells in the livers and lungs of WT mice.
As observed in chronic viral infections of the liver, the expression of programmed death-1 (PD-1, or CD279), a costimulatory receptor that inhibits T-cell receptor signaling, could have contributed to the inefficient late immunity and increased mortality of WT mice. Indeed, in cases of chronic viral infections, antigen-specific CD8+ T cells express high levels of PD-1 and assume an exhausted phenotype characterized by low secretion of IL-2 and IFN-γ (26, 32, 62). Importantly, recent investigations have shown that CD8+ T-cell exhaustion correlates with Treg expansion, and experimental ablation of these cells resulted in a significant increase of cytotoxic CD8+ T-cell responses, decreased viral loads, and no functional exhaustion of CD8+ T cells (57, 85). Because immune exhaustion has also been associated with increased expression of PD-1 for a fungal pathogen, Histoplasma capsulatum (41), it is tempting to suggest that the inefficient T-cell immunity developed by WT mice could be due to T-cell exhaustion controlled by increased expansion of Treg cells.
The immune response developed by CD28−/− mice was very different from that mounted by CD28-sufficient mice. The severe and disseminated infection in CD28−/− mice was only partially controlled by immunological mechanisms. However, these mechanisms were sufficient to organize granulomatous lesions (Fig. 8 and 9). CD28−/− mice secreted NO and produced pro- and anti-inflammatory cytokines in the lungs and livers, although at lower levels than WT mice. At week 10, the inflammatory exudates of the lungs were composed of diminished numbers of macrophages expressing low levels of costimulatory molecules and decreased numbers of activated CD4+ and CD8+ T cells compared to WT mice. The lymphocytes of CD28−/−-deficient mice expressed low levels of activation/regulatory markers, which were associated with a small number of necrotic and apoptotic cells. Furthermore, the number of CD4+CD25+Foxp3+ Treg cells was very low, which was likely due to the absence of CD28 costimulation (44, 79). Because Treg development involves a multistep process dependent on signals from the TCR, cytokines (IL-2 and IL- 15), and other receptors, the low levels of IL-2 produced by CD28-deficient cells appear to have contributed to the impaired expansion of Treg cells (44, 72, 79). Interestingly, as has been observed with Pneumocystis carinii infection (4, 66), the only cell population observed in increased numbers in the lungs of CD28−/− mice was of the CD8+ T-cell phenotype. Depletion experiments, however, indicated that CD8+ T cells were less protective in CD28-deficient mice than in WT mice, and this finding was consistent with the higher activation of CD8+ T cells developed by WT mice. Despite the impaired CD4+ T-cell immunity, T cells from CD28-deficient mice possibly used other members of the CD28 family of costimulatory molecules, such as ICOS, or other members of the tumor necrosis receptor superfamily, including CD40, OX40, 4-1BB, CD27, and CD30, to deliver costimulatory signals (27) and activate T-cell immunity.
Analysis of hepatic cytokines gave some clues to interpret the late massive fungal growth in the livers of WT mice. At week 10, in addition to increased levels of anti-inflammatory cytokines (IL-10 and TGF-β), augmented levels of proinflammatory cytokines were also present in the livers of WT mice (Fig. 4, IL-12, TNF-α, and IFN-γ). At week 26 (Fig. 9J), all of the anti-inflammatory cytokines (IL-4, IL-10, and TGF-β) remained at higher levels in WT mice, while the proinflammatory ones appeared in levels equivalent to those of KO mice. Thus, a prevalent anti-inflammatory pattern appears to progressively develop in WT mice, favoring P. brasiliensis growth. By week 26, WT mice showed, in addition to increased levels of IL-10 and TGF-β (effector cytokines of Treg cells), increased levels of IL-2, a fundamental growth factor for Treg cells (Fig. 9J), which were also found in elevated numbers at week 16 postinfection in the livers of this strain (Fig. 6G). Thus, the liver environment, particularly that of WT mice, appears to be heavily suppressive, leading us to suppose that the increased liver pathology observed (Fig. 9) was due much more to the uncontrolled fungal growth than to damage caused by inflammatory cells.
In summary, our work demonstrates for the first time the crucial importance of CD28 costimulation in adaptive immunity to P. brasiliensis infection. Early in infection, CD28 regulates the expansion of effector and regulatory T cells, restraining fungal growth without causing damage to infected tissues. Later in infection, however, suppressive mechanisms appear to prevail, allowing uncontrolled fungal growth and dissemination that induced increasing tissue damage and augmented the mortality of infected hosts. Our study also shows that P. brasiliensis is able to trigger CD28-independent immune responses that confer relative immunoprotection to the host.
Acknowledgments
This work was supported by Fundação de Amparo à Pesquisa (FAPESP) and Conselho Nacional de Pesquisas (CNPq).
We are grateful to Paulo Albee for his invaluable technical assistance and Anderson de Sá Nunes for the careful review of the manuscript.
Editor: G. S. Deepe, Jr.
Footnotes
Published ahead of print on 16 August 2010.
REFERENCES
- 1.Acuto, O., and F. Michel. 2003. CD28-mediated co-stimulation: a quantitative support for TCR signaling. Nat. Rev. Immunol. 3:939-951. [DOI] [PubMed] [Google Scholar]
- 2.Arruda, C., C. A. C. Vaz, and V. L. G. Calich. 2007. Aseptic cure of pulmonary paracoccidioidomycosis can be achieved after a previous subcutaneous immunization of susceptible but not resistance mice. Microbes Infect. 9:704-713. [DOI] [PubMed] [Google Scholar]
- 3.Arruda, C., S. S. Kashino, R. A. Fazioli, and V. L. G. Calich. 2007. A primary subcutaneous infection with Paracoccidioides brasiliensis leads to immunoprotection or exacerbated disease depending on the route of challenge. Microbes Infect. 9:308-316. [DOI] [PubMed] [Google Scholar]
- 4.Beck, J. M., M. B. Blackmon, C. M. Rose, S. L. Kimzey, A. M. Preston, and J. M. Green. 2003. T cell costimulatory molecule function determines susceptibility to infection with Pneumocystis carinii in mice. J. Immunol. 171:1969-1977. [DOI] [PubMed] [Google Scholar]
- 5.Belkaid, Y., and K. Tarbell. 2009. Regulatory T cells in the control of host-microorganism interactions. Annu. Rev. Immunol. 27:551-589. [DOI] [PubMed] [Google Scholar]
- 6.Benard, G., C. C. Romano, C. R. Cacere, M. Juvenale, M. J. Mendes-Giannini, and A. J. Duarte. 2001. Imbalance of IL-2, IFN-gamma and IL-10 secretion in the immunosuppression associated with human paracoccidioidomycosis. Cytokine 13:248-252. [DOI] [PubMed] [Google Scholar]
- 7.Benard, G., M. J. Mendes-Giannini, M. Juvenale, E. T. Miranda, and A. J. Duarte. 1997. Immunosuppression in paracoccidioidomycosis: T cell hyporesponsiveness to two Paracoccidioides brasiliensis glycoproteins that elicit strong humoral immune response. J. Infect. Dis. 175:1263-1267. [DOI] [PubMed] [Google Scholar]
- 8.Bertram, E. M., A. Tafuri, A. Shahinian, V. S. Chan, L. Hunziker, M. Recher, P. S. Ohashi, T. W. Mak, and T. H. Watts. 2002. Role of ICOS versus CD28 in antiviral immunity. Eur. J. Immunol. 32:3376-3385. [DOI] [PubMed] [Google Scholar]
- 9.Bour-Jordan, H., and J. A. Bluestone. 2009. Regulating the regulators: costimulatory signals control the homeostasis and function of regulatory T cells. Immunol. Rev. 229:41-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brummer, E., E. Castañeda, and A. Restrtepo. 1993. Paracoccidioidomycosis: an update. Clin. Microbiol. Rev. 6:89-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brummer, E., L. H. Hanson, and D. A. Stevens. 1988. Gamma-interferon activation of macrophages for killing of Paracoccidioides brasiliensis: evidence for non-oxidative mechanisms. Int. J. Immunopharmacol. 10:945-952. [DOI] [PubMed] [Google Scholar]
- 12.Burger, E., C. C. Vaz, A. Sano, V. L. G. Calich, L. M. Singer-Vermes, C. F. Xidieh, S. S. Kashino, K. Nishimura, and M. Miyaji. 1996. Paracoccidioides brasiliensis infection in nude mice: studies with isolates differing in virulence and definition of their T cell-dependent and T cell-independent components. Am. J. Trop. Med. Hyg. 55:391-398. [DOI] [PubMed] [Google Scholar]
- 13.Cacere, C. R., C. C. Romano, M. J. Mendes-Giannini, A. J. Duarte, and G. Benard. 2002. The role of apoptosis in the antigen-specific T cell hyporesponsiveness of paracoccidioidomycosis patients. Clin. Immunol. 105:215-222. [DOI] [PubMed] [Google Scholar]
- 14.Calich, V. L. G., and M. H. S. L. Blotta. 2005. Pulmonay paracoccidioidomycosis, p. 201-208. In P. L. Fidel and G. B. Huffnagle (ed.), Fungal immunology: from an organ perspective. Springer Press, New York, NY.
- 15.Calich, V. L. G., and S. S. Kashino. 1998. Cytokines produced by susceptible and resistance mice in the course of Paracoccidioides brasiliensis infection. Braz. J. Med. Biol. Res. 31:615-623. [DOI] [PubMed] [Google Scholar]
- 16.Calich, V. L. G., C. Vaz, and E. Burger. 1994. Immunogenetics in paracoccidioidomycosis, p. 151-166. In M. Franco, C. S. Lacaz, A. Restrepo, and G. Del Negro (ed.), Paracoccidioidomycosis. CRC Press, Boca Raton, FL.
- 17.Camargo, Z. P., and L. E. Cano. 1994. Humoral immunity, p. 187-197. In M. Franco, C. S. Lacaz, A. Restrepo, and G. Del Negro (ed.), Paracoccidioidomycosis. CRC Press, Boca Raton, FL.
- 18.Camargo, Z. P., C. P. Taborda, E. G. Rodrigues, and L. R. Travassos. 1991. The use of cell-free antigens of Paracoccidioides brasiliensis in serological tests. J. Med. Vet. Mycol. 29:31-38. [PubMed] [Google Scholar]
- 19.Campanelli, A. P., G. A. Martins, J. T. Souto, M. S. Pereira, M. C. Livonesi, R. Martinez, and J. S. Silva. 2003. Fas-Fas ligand (CD95-CD95L) and cytotoxic T lymphocyte antigen-4 engagement mediate T cell unresponsiveness in patients with paracoccidioidomycosis. J. Infect. Dis. 187:1496-1505. [DOI] [PubMed] [Google Scholar]
- 20.Cano, L. E., L. M. Singer-Vermes, C. A. C. Vaz, M. Russo, and V. L. G. Calich. 1995. Pulmonary paracoccidioidomycosis in resistant and susceptible mice: relationship among progression of infection, bronchoalveolar cell activation, cellular immune response and specific isotype patterns. Infect. Immun. 63:1777-1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cano, L. E., L. M. Singer-Vermes, T. A. Costa, J. O. Mengel, C. F. Xidieh, C. Arruda, D. C. André, C. A. Vaz, E. Burger, and V. L. G. Calich. 2000. Depletion of CD8(+) T cells in vivo impairs host defense of mice resistant and susceptible to pulmonary paracoccidioidomycosis. Infect. Immun. 68:352-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cano, L. E., S. S. Kashino, C. Arruda, D. André, C. F. Xidieh, L. M. Singer-Vermes, C. A. Vaz, E. Burger, and V. L. G. Calich. 1998. Protective role of gamma interferon in experimental pulmonary paracoccidioidomycosis. Infect. Immun. 66:800-806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Casadevall, A., M. Feldmesser, and L. A. Pirofski. 2002. Induced humoral immunity and vaccination against major human fungal pathogens. Curr. Opin. Microbiol. 5:386-391. [DOI] [PubMed] [Google Scholar]
- 24.Cavassani, K. A., A. P. Campanelli, A. P. Moreira, J. O. Vancim, L. H. Vitali, R. C. Mamede, R. Martinez, and J. S. Silva. 2006. Systemic and local characterization of regulatory T cells in a chronic fungal infection in humans. J. Immunol. 177:5811-5818. [DOI] [PubMed] [Google Scholar]
- 25.Chiarella, A. P., C. Arruda, A. Pina, T. A. Costa, R. C. Ferreira, and V. L. G. Calich. 2007. The relative importance of CD4+ and CD8+ T cells in immunity to pulmonary paracoccidioidomycosis. Microbes Infect. 9:1078-1088. [DOI] [PubMed] [Google Scholar]
- 26.Crispe, I. N. 2009. The liver as a lymphoid organ. Annu. Rev. Immunol. 27:147-163. [DOI] [PubMed] [Google Scholar]
- 27.Croft, M. 2003. Co-stimulatory members of the TNFR family: keys to effective T cell immunity? Nat. Rev. Immunol. 3:609-620. [DOI] [PubMed] [Google Scholar]
- 28.de Camargo, Z. P., and M. F. de Franco. 2000. Current knowledge on pathogenesis and immunodiagnosis of paracoccidioidomycosis. Rev. Iberoam. Micol. 17:41-48. [PubMed] [Google Scholar]
- 29.Fava Netto, C., V. S. Vegas, I. M. Sciannaméa, and E. D. B. Guarnieri. 1969. Antígeno polissacarídico do Paracoccidioides brasiliensis. Estudo do tempo de cultivo do P. brasiliensis necessário ao preparo do antígeno. Rev. Inst. Med. Trop. Sao Paulo 11:177-181. [PubMed] [Google Scholar]
- 30.Fazioli, R. A., L. M. Singer-Vermes, S. S. Kashino, E. Burger, M. F. Franco, M. Moscardi-Bacchi, and V. L. G. Calich. 1994. Delayed-type hypersensitivity response in an isogenic murine model of paracoccidioidomycosis. Mycopathology 126:137-146. [DOI] [PubMed] [Google Scholar]
- 31.Fontenot, J. D., J. P. Rasmussen, M. A. Gavin, and A. Y. Rudensky. 2005. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat. Immunol. 6:1142-1151. [DOI] [PubMed] [Google Scholar]
- 32.Greenwald, R. J., G. J. Freeman, and A. H. Sharpe. 2005. The B7 family revisited. Annu. Rev. Immunol. 23:515-548. [DOI] [PubMed] [Google Scholar]
- 33.Guo, F., C. Iclozan, W. K. Suh, C. Anasetti, and X. Z. Yu. 2008. CD28 controls differentiation of regulatory T cells from naïve CD4 T cells. J. Immunol. 181:2285-2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harris, N. L., and F. Ronchese. 1999. The role of B7 costimulation in T cell immunity. Immunol. Cell Biol. 77:304-311. [DOI] [PubMed] [Google Scholar]
- 35.Hogan, L. H., W. Markofski, A. Bock, B. Barger, J. D. Morrissey, and M. Sandor. 2001. Mycobacterium bovis BCG-induced granuloma formation depends on gamma interferon and CD40 ligand but does not require CD28. Infect. Immun. 69:2596-2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huffnagle, G. B., and G. S. Deepe. 2003. Innate and adaptive determinants of host susceptibility to medically important fungi. Curr. Opin. Microbiol. 6:344-350. [DOI] [PubMed] [Google Scholar]
- 37.Jenkins, M. K., P. S. Taylor, S. D. Norton, and K. B. Urdahl. 1991. CD28 delivers a costimulatory signal involved in antigen-specific IL-2 production by human T cells. J. Immunol. 147:2461-2466. [PubMed] [Google Scholar]
- 38.Kashino, S. S., L. M. Singer-Vermes, V. L. G. Calich, and E. Burger. 1990. Alterations in the pathogenicity of one Paracoccidioides brasiliensis isolate do not correlate with its in vitro growth. Mycopathology 111:173-180. [DOI] [PubMed] [Google Scholar]
- 39.Kashino, S. S., R. A. Fazioli, C. Cafalli-Favati, L. H. Meloni-Bruneri, C. A. Vaz, E. Burger, L. M. Singer, and V. L. G. Calich. 2000. Resistance to Paracoccidioides brasiliensis infection is linked to a preferential Th1 immune response, whereas susceptibility is associated with absence of IFN-γ production. J. Interferon Cytokine Res. 20:89-97. [DOI] [PubMed] [Google Scholar]
- 40.Kündig, T. M., A. Shahinian, K. Kaeai, H. W. Mittrücker, E. Sebzda, M. F. Bachmann, T. W. Mak, and P. S. Ohashi. 1996. Duration of TCR stimulation determines costimulatory requirement of T cells. Immunity 5:41-52. [DOI] [PubMed] [Google Scholar]
- 41.Lázár-Molnár, E., A. Gácser, G. J. Freeman, S. C. Almo, S. G. Nathenson, and J. D. Nosanchuk. 2008. The PD-1/PD-L costimulatory pathway critically affects host resistance to the pathogenic fungus Histoplasma capsulatum. Proc. Natl. Acad. Sci. U. S. A. 105:2658-2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee, Y. K., R. Mukasa, R. D. Hatton, and C. T. Weaver. 2009. Developmental plasticity of Th17 and Treg cells. Curr. Opin. Immunol. 21:274-280. [DOI] [PubMed] [Google Scholar]
- 43.Lenschow, D. J., T. L. Walunas, and J. A. Bluestone. 1996. CD28/B7 system of T cell costimulation. Annu. Rev. Immunol. 14:233-258. [DOI] [PubMed] [Google Scholar]
- 44.Lio, C. W., L. F. Dodson, C. M. Deppong, C. S. Hsieh, and J. M. Green. 2010. CD28 facilitates the generation of Foxp3(−) cytokine responsive regulatory T cell precursors. J. Immunol. 184:6007-6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Loures, F. V., A. Pina, M. Felonato, E. F. Araújo, K. R. Leite, and V. L. Calich. 2010. Toll-like receptor 4 signaling leads to severe fungal infection associated with enhanced proinflammatory immunity and impaired expansion of regulatory T cells. Infect. Immun. 78:1078-1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Loures, F. V., A. Pina, M. Felonato, and V. L. Calich. 2009. TLR2 is a negative regulator of Th17 cells and tissue pathology in a pulmonary model of fungal infection. J. Immunol. 183:1279-1290. [DOI] [PubMed] [Google Scholar]
- 47.Mamoni, R. L., and M. H. Blotta. 2006. Flow-cytometric analysis of cytokine production in human paracoccidioidomycosis. Cytokine 35:207-216. [DOI] [PubMed] [Google Scholar]
- 48.Mamoni, R. L., S. A. Nouér, S. J. Oliveira, C. C. Musatti, C. L. Rossi, Z. P. Camargo, and M. H. Blotta. 2002. Enhanced production of specific IgG4, IgE, IgA and TGF-beta in sera from patients with the juvenile form of paracoccidioidomycosis. Med. Mycol. 40:153-159. [DOI] [PubMed] [Google Scholar]
- 49.McKinney, M. M., and A. Parkinson. 1987. A simple non-chromatographic procedure to purify immunoglobulins from serum and ascites fluid. J. Immunol. Methods 96:271-278. [DOI] [PubMed] [Google Scholar]
- 50.Mesturini, R., S. Nicola, A. Chiocchetti, I. S. Bernardone, L. Castelli, T. Bensi, M. Ferretti, C. Comi, C. Dong, J. M. Rojo, J. Yagi, and U. Dianzani. 2006. ICOS cooperates with CD28, IL-2 and IFN-gamma and modulates activation of human naïve CD4+ T cells. Eur. J. Immunol. 36:2601-2612. [DOI] [PubMed] [Google Scholar]
- 51.Mittrücker, H. W., A. Köhler, T. W. Mak, and S. H. Kaufmann. 1999. Critical role of CD28 in protective immunity against Salmonella typhimurium. J. Immunol. 163:6769-6776. [PubMed] [Google Scholar]
- 52.Mittrücker, H. W., M. Kursar, A. Köhler, R. Hurwitz, and S. H. Kaufmann. 2001. Role of CD28 for the generation and expansion of antigen-specific CD8(+) T lymphocytes during infection with Listeria monocytogenes. J. Immunol. 167:5620-5627. [DOI] [PubMed] [Google Scholar]
- 53.Miyahira, Y., M. Katae, S. Kobayashi, T. Takeuchi, Y. Fukuchi, R. Abe, K. Okumura, H. Yagita, and T. Aoki. 2003. Critical contribution of CD28-CD80/CD86 costimulatory pathway to protection from Trypanosoma cruzi infection. Infect. Immun. 71:3131-3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Montagnoli, C., A. Bacci, S. Bozza, R. Gaziano, P. Mosci, A. H. Sharpe, and L. Romani. 2002. B7/CD28-dependent CD4+CD25+ regulatory T cells are essential components of the memory-protective immunity to Candida albicans. J. Immunol. 169:6298-6308. [DOI] [PubMed] [Google Scholar]
- 55.Montagnoli, C., F. Fallarino, R. Gaziano, S. Bozza, S. Bellocchio, T. Zelante, W. P. Kurup, L. Pitzurra, P. Puccetti, and L. Romani. 2006. Immunity and tolerance to Aspergillus involve functionally distinct regulatory T cells and tryptophan catabolism. J. Immunol. 176:1712-1723. [DOI] [PubMed] [Google Scholar]
- 56.Moreira, A. P., K. A. Cavassani, F. S. Massafera Tristão, A. P. Campanelli, R. Martinez, M. A. Rossi, and J. S. Silva. 2008. CCR5-dependent regulatory T cell migration mediates fungal survival and severe immunosuppression. J. Immunol. 180:3049-3056. [DOI] [PubMed] [Google Scholar]
- 57.Myers, L., R. J. Messer, A. B. Carmody, and K. J. Hasenkrug. 2009. Tissue-specific abundance of regulatory T cells correlates with CD8+ T cell dysfunction and chronic retrovirus loads. J. Immunol. 183:1636-1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nascimento, F. R., V. L. G. Calich, D. Rodrigues, and M. Russo. 2002. Dual role for nitric oxide in paracoccidioidomycosis: essential for resistance, but overproduction associated with susceptibility. J. Immunol. 168:4593-4600. [DOI] [PubMed] [Google Scholar]
- 59.Oliveira, S. J., R. L. Mamoni, C. C. Musatti, P. M. Papaiordanou, and M. H. Blotta. 2002. Cytokines and lymphocyte proliferation in juvenile and adult forms of paracoccidioidomycosis: comparison with infected and non-infected controls. Microbes Infect. 4:139-144. [DOI] [PubMed] [Google Scholar]
- 60.Pina, A., P. H. Saldiva, L. E. Restrepo, and V. L. G. Calich. 2006. Neutrophil role in pulmonary paracoccidioidomycosis depends on the resistance pattern of hosts. J. Leukoc. Biol. 79:1202-1213. [DOI] [PubMed] [Google Scholar]
- 61.Pina, A., S. Bernardino, and V. L. G. Calich. 2008. Alveolar macrophages from susceptible mice are more competent than those of resistant mice to control initial Paracoccidioides brasiliensis infection. J. Leukoc. Biol. 83:1088-1099. [DOI] [PubMed] [Google Scholar]
- 62.Radziewicz, H., C. C. Ibegbu, M. L. Fernandez, K. A. Workowski, K. Obideen, M. Wehbi, H. L. Hanson, J. P. Steinberg, D. Masopust, E. J. Wherry, J. D. Altman, B. T. Rouse, G. J. Freeman, R. Ahmed, and A. Grakoui. 2007. Liver-infiltrating lymphocytes in chronic human hepatitis C virus infection display an exhausted phenotype with high levels of PD-1 and low levels of CD127 expression. J. Virol. 81:2545-2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reiser, H., and M. J. Stadecker. 1996. Costimulatory B7 molecules in the pathogenesis of infectious and autoimmune diseases. N. Engl. J. Med. 335:1369-1377. [DOI] [PubMed] [Google Scholar]
- 64.Romani, L. 2004. Immunity to fungal infections. Nat. Rev. Immunol. 4:1-23. [DOI] [PubMed] [Google Scholar]
- 65.Romani, L. 2008. Parasites and autoimmunity: the case of fungi. Autoimmun. Rev. 8:129-133. [DOI] [PubMed] [Google Scholar]
- 66.Rose, C. M., S. L. Kimzey, and J. M. Green. 2006. The host response of CD28-deficient mice to Pneumocystis infection. Microb. Pathog. 40:23-28. [DOI] [PubMed] [Google Scholar]
- 67.Rudd, C. E., and H. Schneider. 2003. Unifying concepts in CD28, ICOS and CTLA-4 co-receptor signaling. Nat. Rev. Immunol. 3:544-556. [DOI] [PubMed] [Google Scholar]
- 68.Sakaguchi, S. 2005. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat. Immunol. 6:345-352. [DOI] [PubMed] [Google Scholar]
- 69.Sakaguchi, S., N. Sakaguchi, M. Asano, M. Itoh, and M. Toda. 1995. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 155:1151-1164. [PubMed] [Google Scholar]
- 70.Sakaguchi, S., T. Yamaguchi, T. Nomura, and M. Ono. 2008. Regulatory T cells and immune tolerance. Cell 133:775-787. [DOI] [PubMed] [Google Scholar]
- 71.Sansom, D. M., and L. S. Walker. 2006. The role of CD28 and cytotoxic T-lymphocyte antigen-4 (CTLA-4) in regulatory T cell biology. Immunol. Rev. 212:131-148. [DOI] [PubMed] [Google Scholar]
- 72.Seddon, B., and D. Mason. 1999. Peripheral auto-antigen induces regulatory T cells that prevent autoimmunity. J. Exp. Med. 189:877-882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shahinian, A., K. Pfeffer, K. P. Lee, T. M. Kündig, K. Kishihara, A. Wakeham, K. Kawai, P. S. Ohashi, C. B. Thompson, and T. W. Mak. 1993. Differential T cell costimulatory requirements in CD28-deficient mice. Science 261:609-612. [DOI] [PubMed] [Google Scholar]
- 74.Singer-Vermes, L. M., M. C. Ciavaglia, S. S. Kashino, E. Burger, and V. L. G. Calich. 1992. The source of the growth-promoting factor(s) affects the plating efficiency of Paracoccidioides brasiliensis. J. Med. Vet. Mycol. 30:261-264. [DOI] [PubMed] [Google Scholar]
- 75.Snapper, C. M., K. B. Marcu, and P. Zelazowsky. 1997. Immunoglobulin class switch: beyond accessibility. Immunity 6:217-223. [DOI] [PubMed] [Google Scholar]
- 76.Souto, J. T., F. Figueiredo, A. Furlanetto, K. Pfeffer, M. A. Rossi, and J. S. Silva. 2000. Interferon-gamma and tumor necrosis factor-alpha determine resistance to Paracoccidioides brasiliensis infection in mice. Am. J. Pathol. 156:1811-1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tai, X., M. Cowan, L. Feigenbaum, and A. Singer. 2005. CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nat. Immunol. 6:152-162. [DOI] [PubMed] [Google Scholar]
- 78.Tang, Q., and J. A. Bluestone. 2008. The Foxp3 regulatory T cell: a jack of all trades, master of regulation. Nat. Immunol. 9:239-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tang, Q., K. J. Henriksen, E. K. Boden, A. J. Tooley, J. Ye, S. K. Subudhi, X. X. Zheng, T. B. Strom, and J. A. Bluestone. 2003. Cutting edge: CD28 controls peripheral homeostasis of CD4+CD25+ regulatory T cells. J. Immunol. 171:3348-3352. [DOI] [PubMed] [Google Scholar]
- 80.Teder, P., R. W. Vandivier, D. Jiang, J. Liang, L. Cohn, E. Puré, P. M. Henson, and P. W. Noble. 2002. Resolution of lung inflammation by CD44. Science 296:155-158. [DOI] [PubMed] [Google Scholar]
- 81.Teh, H. S., and S. J. Teh. 1997. High concentrations of antigenic ligand activate and do not tolerize naïve CD4 T cells in the absence of CD28/B7 costimulation. Cell Immunol. 179:74-83. [DOI] [PubMed] [Google Scholar]
- 82.Vermes, I., C. Haanen, H. Steffens-Nakken, and C. Reutelingsperger. 1995. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. Immunol. Methods 184:39-51. [DOI] [PubMed] [Google Scholar]
- 83.Weaver, C. T., and R. D. Hatton. 2009. Interplay between the TH17 and Treg cell lineages: a (co-)evolutionary perspective. Nat. Rev. Immunol. 9:883-889. [DOI] [PubMed] [Google Scholar]
- 84.Wüthrich, M., T. Warner, and B. S. Klein. 2005. CD28 is required for optimal induction, but not maintenance, of vaccine-induced immunity to Blastomyces dermatitidis. Infect. Immun. 73:7436-7441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zelinskyy, G., K. K. Dietze, Y. P. Hüsecken, S. Schimmer, S. Nair, T. Werner, K. Gibbert, O. Kershaw, A. D. Gruber, T. Sparwasser, and U. Dittmer. 2009. The regulatory T-cell response during acute retroviral infection is locally defined and controls the magnitude and duration of the virus-specific cytotoxic T-cell response. Blood 114:3199-3207. [DOI] [PubMed] [Google Scholar]
- 86.Ziegler, S. F., F. Ramsdell, and M. R. Alderson. 1994. The activation antigen CD69. Stem Cells 92:456-465. [DOI] [PubMed] [Google Scholar]