

Abstract
The bacterial pathogen Listeria monocytogenes causes food-borne illnesses culminating in gastroenteritis, meningitis, or abortion. Listeria induces its internalization into some mammalian cells through binding of the bacterial surface protein InlB to the host receptor tyrosine kinase Met. Interaction of InlB with the Met receptor elicits host downstream signaling pathways that promote F-actin cytoskeletal changes responsible for pathogen engulfment. Here we show that the mammalian signaling protein ARAP2 plays a critical role in cytoskeletal remodeling and internalization of Listeria. Depletion of ARAP2 through RNA interference (RNAi) caused a marked inhibition of InlB-mediated F-actin rearrangements and bacterial entry. ARAP2 contains multiple functional domains, including a GTPase-activating protein (GAP) domain that antagonizes the GTPase Arf6 and a domain capable of binding the GTPase RhoA. Genetic data indicated roles for both the Arf GAP and RhoA binding domains in Listeria entry. Experiments involving Arf6 RNAi or a constitutively activated allele of Arf6 demonstrated that one of the ways in which ARAP2 promotes bacterial uptake is by restraining the activity of Arf6. Conversely, Rho activity was dispensable for Listeria internalization, suggesting that the RhoA binding domain in ARAP2 acts by engaging a host ligand other than Rho proteins. Collectively, our findings indicate that ARAP2 promotes InlB-mediated entry of Listeria, in part, by antagonizing the host GTPase Arf6.
Listeria monocytogenes is a Gram-positive, food-borne bacterial pathogen capable of causing gastroenteritis, meningitis, or abortions (39, 32). Listeria induces its own internalization (entry) into nonphagocytic mammalian cells, a process that likely plays an important role in traversal of the intestinal, placental, and blood-brain barriers (7, 14, 16, 23). One of the pathways of Listeria entry is mediated by interaction of the bacterial surface protein InlB with its host receptor, the Met receptor tyrosine kinase (16, 37).
InlB-Met interaction triggers activation (tyrosine phosphorylation) of the Met receptor and subsequent rearrangements in the F-actin cytoskeleton of the mammalian cell (16, 29). These cytoskeletal changes remodel the host cell surface, resulting in engulfment of adherent Listeria. One of the host mammalian proteins that acts downstream of Met to promote F-actin rearrangements and bacterial entry is type IA phosphoinositide (PI) 3-kinase (8, 17, 18). This PI 3-kinase is a heterodimeric enzyme comprised of an 85-kDa regulatory subunit and a 110-kDa catalytic subunit (4, 11). p85-p110 generates phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2)] and phosphatidylinositol 3,4,5-trisphosphate [PI(3,4,5)P3], which are lipid second messengers that regulate a variety of biological processes, including growth, survival, and motility of mammalian cells. A plethora of downstream “target” proteins that bind PI(4,5)P2 and/or PI(3,4,5)P3 and mediate the biological effects of p85-p110 have been identified (4, 24). However, mammalian proteins that act downstream of type IA PI 3-kinase to control Listeria uptake have yet to found.
In this work, we demonstrate that the human GTPase-activating protein (GAP) ARAP2 is required for InlB-mediated cytoskeletal changes and entry of Listeria. ARAP2 is known to bind PI(3,4,5)P3, resulting in upregulation of a GAP domain that inactivates the mammalian GTPase Arf6 (42). We provide genetic evidence indicating that the ArfGAP domain of ARAP2 stimulates Listeria entry by antagonizing Arf6. ARAP2 has several functional domains in addition to its ArfGAP domain. One of these domains in ARAP2 interacts with the mammalian GTPase RhoA (42). Our genetic data demonstrate that this “RhoA binding” (RB) domain also plays a critical role in bacterial entry. Surprisingly, pharmacological experiments indicate that the RB domain controls Listeria uptake through an unknown mechanism that does not involve Rho proteins. Our work indicates a key role for host ARAP2 in InlB-mediated entry of Listeria. One of the likely ways that type IA PI 3-kinase controls entry of Listeria is through regulation of ARAP2.
MATERIALS AND METHODS
Bacterial strains, mammalian cell lines, and media.
The wild-type Listeria monocytogenes strain EGD and the isogenic ΔinlB mutant strain containing an in-frame deletion of the inlB gene were previously described (10). These strains were grown in brain heart infusion (BHI) broth (Difco) and prepared for infection as described previously (10, 18).
The human epithelial cell line HeLa (ATTC CCL-2) was grown in Dulbecco's modified Eagle's medium (DMEM) with 4.5 g of glucose per liter and 2 mM glutamine (11995-065; Invitrogen), supplemented with 10% fetal bovine serum (FBS). Cell growth, cell stimulation, and bacterial infections were performed at 37°C with 5% CO2.
Antibodies, inhibitors, siRNAs, and other reagents.
The polyclonal antibodies used were anti-ARAP2 1185 (42), anti-Met C-12 (sc-10; Santa Cruz Biotechnology), rabbit anti-L. monocytogenes R11 (13), and polyclonal anti-InlB antiserum (8). The monoclonal antibodies used were antitubulin (T5168; Sigma), anti-Flag M2 (F3165), anti-Arf6 clone 3A1 (Santa Cruz Biotechnology, sc-7971), and anti-green fluorescent protein (anti-GFP) clone B-2 (sc-9996; Santa Cruz Biotechnology). Secondary antibody horseradish peroxidase (HRPO) or Cy5 conjugates were from Jackson Immunolabs. Secondary antibody or phalloidin conjugates coupled to Texas Red or BODYP FL were from Molecular Probes. Recombinant hepatocyte growth factor (HGF) (294-HGN) was purchased from R&D Systems. The PI 3-kinase inhibitor LY294002 (LL9908) was from Sigma, and the cell-permeative C3 toxin (CT04) was from Cytoskeleton. Recombinant InlB protein was expressed in Escherichia coli and purified essentially as described previously (18). The negative, “nontargeting” control small interfering RNA (siRNA) molecule 1 (catalog no. D-001210-01) was purchased from Dharmacon. This siRNA contains two or more mismatches with all sequences in the human genome, indicating that it should not target host mRNAs. Compared with cells treated with transfection reagent alone, control siRNA 1 did not affect entry of the wild-type Listeria strain EGD into HeLa cells (see Fig. 1B, panel i). siRNA molecules targeting mRNA encoding Arf6 (5′-GCACCGCAUUAUCAAUGACCGUU-3′) or ARAP2 (ARAP2-1, (5′-GUAAGAAGACAUUGGGUUAUU-3′; ARAP2-2, 5′-GGCUGGUUUGCUAUGGACAUU-3′) were also from Dharmacon. ARAP2-1 siRNA targets a sequence in the 3′ untranslated region (UTR) of ARAP2 mRNA (42), whereas ARAP2-2 siRNA is complementary to a sequence in the coding region of the mRNA.
FIG. 1.

InlB-mediated entry of Listeria requires the host GAP ARAP2. (A) Effect of siRNA molecules on ARAP2 expression. HeLa cells were left untreated (−) or transfected with a control siRNA or either of two siRNA molecules targeting ARAP2 (ARAP2-1 or ARAP2-2). At about 48 h posttransfection, cells were solubilized for measurement of ARAP2 expression by Western blotting. (B) Effect of siRNAs targeting ARAP2 on entry or association of Listeria. Transfected HeLa cells were infected with wild-type (Wt) Listeria or an isogenic mutant strain with inlB deleted (ΔinlB). Panel i, bacterial entry was assessed using a gentamicin protection assay (37). Panel ii, association of bacteria with host cells was measured as described previously (8). Values are averages ± standard errors of the means (SEM) from three or four experiments. *, P < 0.01 compared to the no-siRNA control (−). **, P < 0.001. (C) Effect of ARAP2 depletion on internalization of Listeria, as determined by a fluorescence microscopy-based assay (38). Values are averages ± standard deviations (SD) from three experiments. *, P < 0.01 compared to control siRNA conditions. **, P < 0.001.
Plasmids.
Mammalian expression plasmids encoding Flag-tagged wild-type, R728K, and K1190P alleles of ARAP2 were previously described (42). These plasmids were based on the expression vector pCI (42) and were kindly provided by Paul Randazzo and Hye-Young Yoon (NIH, Bethesda, MD). As a control plasmid for experiments for Fig. 2B, pCI harboring Flag-tagged firefly luciferase was constructed using standard PCR-based techniques. The final sequence of the Flag-luciferase insert was verified by DNA sequencing.
FIG. 2.

Restoration of ARAP2 expression allows normal entry of Listeria in the presence of ARAP2 siRNA. (A) Expression of an ARAP2 cDNA that is resistant to siRNA-mediated depletion. HeLa cells were transfected with either control (C) siRNA or ARAP2-1 (A2-1) siRNA, which targets the 3′ untranslated region (UTR) present in endogenous ARAP2 mRNA. Cells were transfected again with plasmids expressing either Flag-tagged luciferase (L) or ARAP2 (AR2) encoded by a cDNA that lacks the 3′ UTR. At about 24 h after plasmid DNA transfection, ARAP2 expression was evaluated by Western blotting. Note that ectopic expression of ARAP2 lacking the 3′ UTR (AR2) restored normal ARAP2 levels to cells treated with ARAP2-1 (A2-1) siRNA. (B) Levels of Flag-tagged luciferase or ARAP2 expression. Confocal microscopy was used to measure relative expression of tagged luciferase or ARAP2 proteins in transfected HeLa cells. P = 0.19 (not significant). (C) Restoration of bacterial entry in cells expressing ARAP2 resistant to siRNA-mediated depletion. A fluorescence microscopy-based assay (38) was used to measure entry of Listeria in cells transfected with siRNAs and expressing Flag-tagged luciferase or ARAP2. Values are averages ± SD from three experiments. *, P < 0.001 compared to the luciferase (L) plus control siRNA (C) condition.
Transfection of mammalian cells with siRNA or plasmid DNA.
Seeding of HeLa cells, transfection with siRNA and the lipid reagent LF2000 (Invitrogen), and preparation of samples for Western blotting were as described previously (8). Cells were used for bacterial infections or stimulation with InlB or HGF at approximately 48 h after transfection with siRNA. Control experiments employing an 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay (28) or enumeration of CFU indicated that the control, ARAP2-1, ARAP2-2, and Arf6 siRNAs did not affect viability of mammalian cells or bacteria.
In some experiments (see Fig. 2 and 5), HeLa cells were first transfected with siRNA and then approximately 24 h later transfected a second time with plasmid DNA expressing various Flag-tagged proteins. In these experiments, transfections of siRNA and plasmid DNA with LF2000 were performed as described previously (8).
FIG. 3.

ARAP2 is dispensable for activation of the Met receptor. HeLa cells were transfected with 30 nM control or ARAP2-2 (A2-2) siRNA for ∼48 h. Cells were then treated with either InlB protein (4.5 nM) or HGF (1 nM) for 1 min (A) or infected with wild-type (wt) or ΔinlB (ΔB) strains of Listeria for 5 min (B). Top panels, tyrosine phosphorylation of Met was evaluated by Western blotting of anti-Met immunoprecipitates (IP) (8, 37). Middle panels, depletion of ARAP2 was assessed by Western blotting of total cell lysates. Bottom panels, equivalent loading of total cell lysates was confirmed by probing stripped membranes with antitubulin antibodies. The results are representative of two experiments.
FIG. 4.

The ArfGAP and Rho binding domains of ARAP2 promote entry of Listeria. (A) Domains present in ARAP2 and locations of R728K and K1190P mutations. SAM, sterile α motif; PH, pleckstrin homology domain; ArfGAP, Arf GTPase-activating protein; A, ankyrin repeat; RB, Rho binding domain; RA, Ras-associating domain. Top diagram, wild-type (wt) ARAP2. Bottom two diagrams, ARAP2 alleles R728K and K1190P, containing single amino acid substitutions that inactivate the ArfGAP and RhoA binding domains, respectively. (B) Effect of mutations in the ArfGAP or Rho binding domain on bacterial internalization. HeLa cells were transfected with plasmids expressing Flag-tagged wild-type (wt) ARAP2 or ARAP2 alleles containing the R728K or K1190 mutation. Panel i, at approximately 24 h posttransfection, entry of Listeria was assessed using a fluorescence microscopy-based assay (38). Data are averages ± SD from four experiments. *, P < 0.001 compared to the ARAP2wt control. Panel ii, expression of the Flag-tagged ARAP2 alleles was measured using confocal microscopy (see Materials and Methods). P = 0.32 (not significant).
FIG. 5.
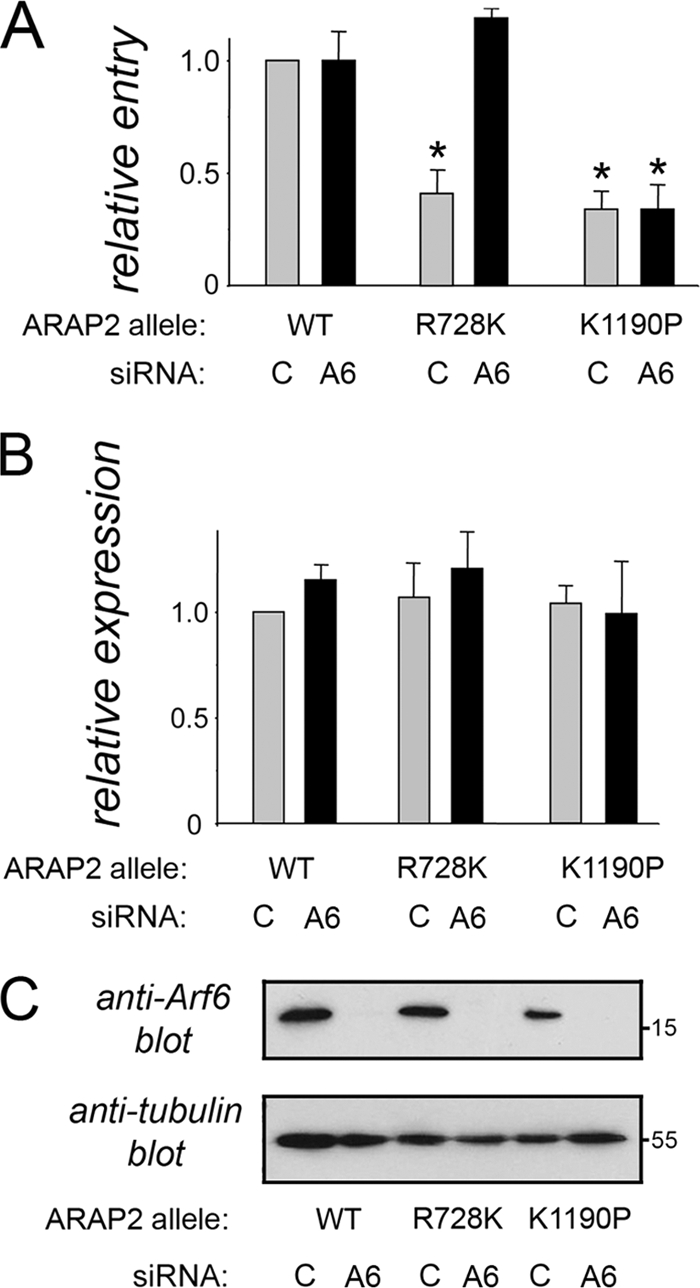
ARAP2 promotes entry of Listeria in part by inactivating host Arf6 GTPase. (A) Depletion of Arf6 restores bacterial internalization to cells expressing ARAP2.R278K. HeLa cells were transfected with control or Arf6 (A6) siRNA and then with plasmids expressing Flag-tagged wild-type (WT), R728K, or K1190P alleles of ARAP2. Entry of Listeria was measured using a fluorescence microscopy-based assay (38) (see Materials and Methods). Data are averages ± SD from three experiments. *, P < 0.001 compared to the ARAP2wt plus control siRNA condition. (B) Expression of ARAP2 alleles. Expression of Flag-tagged ARAP2 proteins was measured using confocal microscopy (see Materials and Methods). P = 0.59 (not significant). (C) Depletion of Arf6. siRNA-mediated depletion of Arf6 was confirmed by Western blotting.
Bacterial entry or association assays.
Gentamicin protection assays to measure entry of Listeria strains (see Fig. 1B, 6B, and 7C) were performed by infecting cells for 1 h in the absence of gentamicin and then incubating them in DMEM with 20 μg/ml gentamicin for 2 h as described previously (8, 37). In experiments involving cell-permeative C3 toxin (see Fig. 7C), HeLa cells were pretreated with 2 μg/ml C3 in DMEM supplemented with 0.1% bovine serum albumin (BSA) for 5 h prior to addition of bacteria. C3 toxin was left in contact with cells during the 1-h infection period. Toxin was washed away just prior to the 2-h period involving gentamicin treatment. Cell association assays involving a 30-min infection period without subsequent gentamicin treatment were performed as described previously (8).
FIG. 6.

Constitutively activated Arf6 blocks internalization of Listeria. HeLa cells were infected with retrovirus expressing either EGFP alone, EGFP-tagged wild-type (wt) Arf6, or tagged constitutively activated (Q67L) Arf6. At about 24 h after addition of retrovirus, cells were solubilized for assessment of EGFP protein expression by Western blotting. (B) Cells were infected with wild-type or ΔinlB strains of Listeria, and entry was measured by a gentamicin protection assay (37). Data are averages ± SD from three experiments. *, P < 0.001 compared to the EGFP control.
FIG. 7.

Host Rho activity is dispensable for InlB-mediated entry of Listeria. (A) Disruption of F-actin stress fibers by C3 toxin. HeLa cells left untreated (−) or treated with 2 μg/ml cell-permeative C3 for 5 h (+) were fixed and labeled with phalloidin coupled to Texas Red in order to detect the F-actin cytoskeleton. Untreated cells exhibit pronounced stress fibers, whereas stress fibers are largely absent in cells treated with C3. Scale bars, 10 μm. (B). Lack of inhibition of bacterial internalization by C3 exoenzyme treatment. HeLa cells pretreated (+) or not (−) with cell-permeative C3 toxin were infected with wild-type (Wt) or ΔinlB strains of Listeria, and entry was measured using gentamicin protection assay (37). Despite the strong inhibitory effect of C3 on the F-actin cytoskeleton (A), the toxin did not impair entry of Listeria. Data are averages ± SD from three experiments. *, P < 0.01 compared to untreated cells infected with wild-type Listeria.
Experiments measuring bacterial entry by a fluorescence microscopy-based assay (see Fig. 1C, 2C, 4B [panel i], and 5A) were carried out essentially as described previously (8, 38). Briefly, extracellular bacteria in fixed monolayers were labeled by incubation with antiserum R11 at a dilution of 1:500 for 1 h, followed by three washes and a second 1-h incubation with Cy5-conjugated antibodies against rabbit IgG. As Texas Red-X-succinimidyl ester (TRXSE)-labeled bacteria were used for infection, both extracellular and intracellular bacteria were Texas Red positive. After labeling, intracellular bacteria were negative for Cy5, whereas extracellular bacteria were Cy5 positive. Infected mammalian cells were then permeabilized in phosphate-buffered saline (PBS) containing 0.4% Triton X-100 for 5 min. In the case of HeLa cells expressing tagged ARAP2 alleles (see Fig. 4B and 5A), permeabilized cells were labeled with anti-Flag antibodies, followed by incubation with fluorescein isothiocyanate (FITC)-conjugated secondary antibodies. This procedure typically resulted in ∼10% of the population subjected to transfection appearing brightly fluorescent (Flag positive). Samples were mounted using Mowiol 4-88 (475904; Calbiochem). A Zeiss LSM 510 confocal microscope equipped with argon (488 nm), helium-neon 1 (543 nm), and helium-neon 2 (633 nm) lasers was used for analysis of bacterial entry. In the case of experiments for Fig. 2C, 4B (panel i), and 5A, only cells that were positive for Flag-tagged protein expression were analyzed. Images of five or six serial optical sections of 1 μm in thickness were captured. Optical sections started just above the host cell and ended below the cell. For each section, images of Cy5, Texas Red, and FITC fluorescence were acquired. Individual serial sections corresponding to Cy5 or Texas Red fluorescence were superimposed so as to create a composite image containing all Cy5- or Texas Red-positive bacteria throughout the host cell. The Cy5 and Texas Red images (artificially colored in red and green, respectively) were then overlaid, resulting in extracellular bacteria appearing yellow and intracellular bacteria red. Finally, in experiments involving Flag-tagged ARAP2 alleles, the FITC fluorescent image (artificially colored in blue) was superimposed on the Cy5/Texas Red overlay to allow identification of bacteria associated with transfected cells. Typically, 50 to 100 intracellular bacteria were examined for the control condition in each experiment. Control conditions for the various figures involved infection with wild-type Listeria of the following cells: for Fig. 1C, cells treated with control siRNA; for Fig. 2C, cells transfected with control siRNA and expressing Flag-luciferase; for Fig. 4B, panel i, cells expressing Flag-tagged wild-type ARAP2 (ARAP2wt); and for Fig. 5A, cells transfected with control siRNA and expressing Flag-tagged wild-type ARAP2. The number of host cells analyzed under control conditions was noted, and similar numbers of cells subjected to the other experimental conditions were evaluated for Listeria entry.
Measurement of expression of Flag-tagged proteins by confocal microscopy.
Transfection of HeLa cells with plasmids expressing Flag-tagged luciferase, ARAP2wt, ARAP2.R728K, or ARAP2.K1190P resulted in a subpopulation of cells expressing the tagged transgene. Moreover, the proportions of cells expressing the various Flag-tagged proteins were not identical. For this reason, confocal microscopy was used to quantify the level of expression of Flag-tagged proteins in subpopulations of HeLa cells that were positive for Flag protein expression (see Fig. 2B, 4B [panel ii], and 5B). In this way, expression levels in the subpopulation of cells that were analyzed for bacterial entry (see the description of fluorescence-based measurement of bacterial entry above) were assessed. When acquiring images by confocal microscopy, care was taken to ensure that identical laser and gain settings were used when analyzing samples with different Flag-tagged proteins. Quantification of pixel intensities in cells positive for Flag staining was performed using Image J (version 1.43r) software. Briefly, the “ROI” function was used to select individual cells, and the “measure” function was employed to obtain average pixel intensities. Typically, average pixel intensities of about 50 Flag-positive cells were quantified for each condition in each experiment. The mean pixel intensities were then determined. Data in Fig. 2B, 4B (panel ii), and 5B are average pixel intensities ± standard deviations (SD) from three experiments.
Retrovirus-mediated expression of Arf6 alleles.
Murine leukemia virus (MLV) pseudotypes bearing the envelope protein of vesicular stomatitis virus (VSV) were used to express enhanced GFP (EGFP) alone or EGFP-tagged wild-type or Q67L alleles of human Arf6. These retroviral vectors are incapable of replication, as they lack the majority of the virus genome, but express the transgene after integration into the cell chromosome. Transcription is driven by the viral long terminal repeat (LTR) promoter/enhancer elements that flank the transgene and is dependent solely on normal cellular transcription mechanisms. Pseudotyped viruses were made essentially as described previously (20). Briefly, 293FT cells (Invitrogen) were transfected with plasmids encoding MLV Gag-Pol and VSV G together with a plasmid encoding the EGFP-tagged construct flanked by MLV LTRs and a 5′ packaging sequence. The transfection was by calcium phosphate precipitation. After 2 days, the culture supernatant was passed through a 0.45-μm filter to remove cell debris. The virus-containing filtrate was then used immediately or frozen at −80C. The virus titer was determined by infecting HEK293 cells and counting EGFP-expressing cells 2 days later. Virus was concentrated by centrifugation through a 20% sucrose cushion and resuspension of the pellet in DMEM plus 10% FBS.
HeLa cells were infected with retroviruses, and the effect of retrovirus-mediated expression of Arf6 alleles was determined as follows. First, 4 × 104 HeLa cells per well were seeded in 24-well plates and grown for ∼24 h. EGFP, Arf6wt-EGFP, or Arf6Q67L-EGFP transgenes were introduced into cells by incubation with recombinant virus at a multiplicity of infection (MOI) of 5:1. Cells were incubated with virus or control medium for 48 h before analysis of protein expression or Listeria internalization. Fluorescence microscopy analysis indicated that the retroviruses allow expression of EGFP, Arf6wt-EGFP, or Arf6Q67L-EGFP in 95 to 99% of HeLa cells. Before infection with bacterial strains, retrovirus- or mock-infected cells were washed twice with DMEM. Gentamicin protection assays were performed as described above. For Western blotting, retrovirus-infected cells were solubilized in hot sample buffer, and samples were migrated on 10% SDS-polyacrylamide gels and transferred to polyvinylidene difluoride (PVDF) membranes. Membranes were probed with commercially available anti-EGFP antibodies.
Western blotting and immunoprecipitation.
Western blotting and detection using enhanced chemiluminescence (ECL) or ECL Plus reagents (GE Health Care) and film were performed as described previously (18, 37).
In experiments assessing tyrosine phosphorylation of Met (see Fig. 3), 8 × 104 HeLa cells per well were seeded in six-well plates. One six-well plate was used for each stimulation condition. About 24 h later, cells were transfected with 30 nM control or ARAP2-2 siRNA. Approximately 48 h after transfection, cells were washed twice in PBS and serum starved in DMEM without FBS for ∼3 h. Incubation of cells with InlB, HGF, or wild-type or ΔinlB strains of Listeria; preparation of cell lysates; immunoprecipitation of Met with antibody C-12; and detection of tyrosine phosphorylation by Western blotting were all as described previously (8, 37).
Real-time PCR analysis.
HeLa cells (1.5 × 104) were seeded in wells of 24-well plates. About 24 h later, cells were transfected with 10 nM control, ARAP2-1, or ARAP2-2 siRNA as described previously (8). Approximately 48 h after transfection, cells were washed twice in PBS and lysed, and a TaqMan Gene Expression Cells to Ct kit (Applied Biosystems) was used to prepare cDNA. Real-time PCR was performed using an ABI7900 instrument and TaqMan gene expression assays for ARAP2 (Hs00367391_m1; Applied Biosystems) and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (Hs99999905_m1; Applied Biosystems) as an endogenous control. Data were analyzed by the comparative threshold cycle (CT method (25), normalizing CT values for ARAP2 expression to those for GAPDH. Relative quantity (RQ) values were calculated by the formula RQ = 2−ΔΔCT.
Assessing effects of cell C3 toxin on the actin cytoskeleton.
HeLa cells (2 × 105) were seeded onto sterile 22- by 22-mm glass coverslips in six-well plates. Cells were grown for approximately 2 days and then treated for 5 h with 2 μg/ml cell-permeative C3 toxin in DMEM supplemented with 0.1% BSA. After treatment, cells were washed once in PBS, fixed in PBS with 3% paraformaldehyde, and permeabilized by a 10-min incubation in PBS with 0.4% Triton X-100. Samples were labeled for F-actin using phalloidin coupled to Texas Red (Molecular Probes). After mounting on coverslips using Mowiol, samples were analyzed using a Zeiss LSM confocal microscope with the helium-neon 1 (543 nm) laser.
Measurement of internalization of InlB-coated beads and F-actin recruitment.
Carboxylate-modified latex beads 3.0 μm in diameter (09850; Polysciences) were used for coating with InlB protein, as described previously (8). HeLa cells were incubated with InlB-coated beads for 10 min at 37°C with 5% CO2, fixed, labeled, and analyzed by confocal microscopy as described previously (8). The data in Fig. 8A are averages ± SD from three experiments. In each experiment, 100 to 300 intracellular beads were scored for control siRNA-treated cells. The number of host cells analyzed was noted, and a similar number of ARAP2-2 siRNA-treated cells was analyzed for entry of beads. The data in Fig. 8A, panel ii, are expressed as the percentage of total cell-associated beads that were internalized. The data in Fig. 8A, panel i, are relative values obtained by normalizing percent internalization values in Fig. 8A, panel ii, to values for control siRNA-treated cells.
FIG. 8.

ARAP2 is needed for cytoskeletal rearrangements during InlB-mediated entry. (A) Inhibition of internalization of InlB-coated beads by siRNA-mediated depletion of ARAP2. HeLa cells treated with control or ARAP2-2 siRNA were incubated with InlB-coated beads for 10 min. Entry of beads was measured using a fluorescence microscopy-based assay (8). Panel i, entry expressed as internalization relative to that in control siRNA-treated cells. Panel ii, entry expressed as absolute values (percentage of beads internalized). Values are averages ± SD from three experiments. P = 0.0054. (B) Inhibition of F-actin cytoskeletal changes by depletion of ARAP2. HeLa cells treated with control or ARAP2-2 siRNA were incubated with InlB-coated beads for 2.5 min, followed by fixation and labeling for beads (red) or F-actin (green). Panel i, images of several cells showing labeling of both F-actin and beads or F-actin only. Arrows indicate beads encircled by F-actin. Arrowheads show cell-associated beads that do not display obvious F-actin recruitment. Scale bars indicate 5 μm. Panel ii, enlarged images of selected beads (numbered 1 to 4) and associated F-actin indicated in panel i. In the case of beads that recruit F-actin, recruitment in control siRNA-treated cells (bead 1) is typically more intense than that in cells depleted for ARAP2 (bead 3). (C) Quantification of the effect of ARAP2 depletion on cytoskeletal changes. Panel i, confocal microscopy was used to quantify the percentage of beads associated with F-actin. For examples of beads recruiting or not recruiting F-actin, see beads 1 to 4 in panel ii of panel B. HeLa cells treated with ARAP2-2 siRNA had fewer actin-associated beads than control siRNA-treated cells. P = 0.0007. Panel ii, confocal microscopy was used to quantify the degree of F-actin enrichment around beads in cells treated with control or ARAP2-2 siRNA. This analysis included only beads that displayed obvious association with F-actin. For examples, see beads 1 and 3 in panel ii of panel B. Beads bound to cells treated with ARAP2-2 siRNA displayed reduced enrichment of F-actin compared to beads adhering to control siRNA-treated cells. P = 0.0002. The data in both panel i and ii are averages ± SD from three experiments.
Recruitment of F-actin to adherent (extracellular) InlB-coated beads (see Fig. 8B and C) was assessed essentially as described previously (8). Images from serial sections spaced 1 μm apart were used to ensure that all cell-associated beads were detected. Any beads that exhibited obvious recruitment of F-actin in “ring-like” structures at the site of bead attachment (see bead 1 in Fig. 8B for an example) were scored as positive. Beads that were not associated with rings containing F-actin (see bead 2 in Fig. 8B) were considered to be negative. In the case of cells treated with ARAP2-2 siRNA, InlB-coated beads were typically not associated with cups containing F-actin and were scored as “negative.” More rarely, weak accumulation of F-actin was observed, in which case beads were scored as “positive.” Approximately 200 extracellular particles were scored for F-actin recruitment for each condition in each experiment. Data showing the percentage of cell-associated beads that were positive or negative for F-actin recruitment (see Fig. 8C, panel i) are shown as the average ± SD from three experiments. In the case of beads that scored positive for recruitment, we also quantified enrichment of F-actin around the bead relative to F-actin throughout the cell (see Fig. 8C, panel ii). The thresholding function of Image J (version 1.43r) software was used to select F-actin rings surrounding a bead. The average pixel intensity of F-actin around the bead was then determined. This average pixel intensity surrounding the bead was normalized to the average F-actin pixel intensity throughout the cell. The corresponding ratio was termed the “F-actin enrichment factor,” where a value greater than 1.0 indicates enrichment of F-actin around the bead compared to in the entire cell. The data in Fig. 8C, panel ii, represent averages ± SD from three experiments. In each of these experiments, 30 to 50 beads were used under each condition for calculation of F-actin enrichment values.
Statistical analysis.
Statistical analysis was performed using InStat (version 2.2; GraphPad Software). When comparing data sets from two conditions, a Student t test was used. When comparing data from three or more conditions, analysis of variance (ANOVA) was performed. The Tukey-Kramer test was used as a posttest. A P value of 0.05 or lower was considered significant.
RESULTS
The human GAP ARAP2 is needed for InlB-dependent entry of Listeria.
Host type IA PI 3-kinase (here abbreviated as p85-p110) plays a critical role in entry of Listeria downstream of the Met receptor (8, 16, 17, 18, 37). In order to better understand the molecular mechanism by which p85-p110 promotes bacterial entry, we sought to identify human proteins that act downstream of p85-p110 in this process. p85-p110 produces PI(3,4)P2 and PI(3,4,5)P3, two phosphoinositides that bind to a multitude of signaling proteins and control a variety of biological events in mammalian cells (4, 11, 24). We performed a literature search to identify mammalian proteins reported to be regulated by p85-p110. A list of approximately 60 human proteins was compiled (S. Jiwani and K. Ireton, unpublished data). Most of these PI 3-kinase-regulated proteins interact directly with PI(3,4)P2 or PI(3,4,5)P3 or with the PI 3-kinase subunit p85 or p110. In a few cases, host proteins that are indirectly regulated by p85-p110 were included. The list of p85-p110-regulated proteins was used to construct an siRNA library, and this library was screened for effects on host target gene expression and entry of Listeria. The siRNA-based screen will be the subject of a future publication. Here, we describe the role of one of the human proteins identified from the siRNA screen in InlB-mediated entry of Listeria. This protein is the GTPase-activating protein (GAP) ARAP2 (27, 42). ARAP2 contains pleckstrin homology (PH) domains (24, 34) that bind to PI(3,4,5)P3, resulting in upregulation of the GAP activity of ARAP2 (42). In this way, p85-p110 regulates the enzymatic activity of ARAP2.
We found that siRNA-mediated depletion of ARAP2 caused a marked inhibition of entry of Listeria into HeLa cells (Fig. 1). Inhibition of entry was observed using two different assays, involving gentamicin protection (Fig. 1B, panel i) or fluorescence microscopy (Fig. 1C). Two lines of evidence indicated that the inhibitory effect of ARAP2 siRNA on Listeria internalization was genuine and not due to an “off-target” effect of the siRNA. First, two different siRNA molecules targeting distinct regions in ARAP2 mRNA caused depletion of ARAP2 and impairment of Listeria uptake (Fig. 1A and B). Inhibition of ARAP2 gene expression was confirmed by both Western blotting (Fig. 1A) and real-time PCR analysis. The latter approach indicated that ARAP2-1 and ARAP2-2 siRNAs reduced ARAP2 mRNA to 16 and 10%, respectively, of control levels. The second line of evidence that excluded the possibility of an off-target effect involved a “rescue” experiment. The defect in bacterial entry normally caused by an siRNA targeting the 3′ untranslated region (UTR) of ARAP2 mRNA was complemented by expression of an ARAP2 cDNA lacking the 3′ UTR (Fig. 2). Thus, by expressing a resistant ARAP2 cDNA, it was possible to restore normal internalization of Listeria in the presence of the ARAP2-1 siRNA.
Importantly, the ability of Listeria to associate with HeLa cells was unaffected by ARAP2 depletion (Fig. 1B, panel ii). This finding suggests that ARAP2 is not needed for binding of bacteria to host cells but rather affects postbinding events. In agreement with this idea, ARAP2 depletion did not affect the ability of InlB protein or bacteria to induce activation (tyrosine phosphorylation) of the Met receptor (Fig. 3). We also tested the ability of the cellular ligand of Met, hepatocyte growth factor (HGF) (12), to stimulate phosphorylation of the receptor. Similar to the results observed with InlB and bacteria, HGF-induced phosphorylation was unaffected by ARAP2 depletion (Fig. 3A). The results in Fig. 3 indicate that ARAP2 controls entry of Listeria by acting downstream of Met.
The ArfGAP and RhoGAP domains in ARAP2 play important roles in Listeria entry.
ARAP2 has several functional domains, including an ArfGAP domain that inactivates the GTPase Arf6 and a domain that associates with the activated form of the GTPase RhoA (Fig. 4 A) (27, 33, 42). Interestingly, the ArfGAP domain is activated by PI(3,4,5)P3 (42), suggesting that type IA PI 3-kinase enhances ARAP2-mediated inhibition of Arf6. The domain in ARAP2 that binds RhoA-GTP has sequence similarity with GAP domains for Rho GTPases and hence has been termed a “RhoGAP” domain (27, 33, 42). However, this domain in ARAP2 is different from true RhoGAP domains in that it lacks a key catalytic arginine residue and is consequently unable to stimulate GTP hydrolysis (27, 42). Since the RhoGAP domain functions as a Rho-interacting module rather than as a GAP, we here refer to this region as a “RhoA binding” (RB) domain.
In order to test the roles of the ARAP2 ArfGAP and RB domains in internalization of Listeria, we used mutant alleles containing single amino acid substitutions that inactivate these domains (Fig. 4A). The ARAP2.R728K allele contains a mutation that abolishes the ability of ARAP2 to stimulate GTP hydrolysis on Arf6 (42). The K1190P allele of ARAP2 is defective for interaction with RhoA-GTP. We transiently expressed Flag-tagged wild-type ARAP2, ARAP2.R728K, or ARAP2.K1190P in HeLa cells and measured bacterial entry using a previously described fluorescence microscopy-based approach (see Materials and Methods) (38). Importantly, compared to tagged wild-type ARAP2, the ARAP2.R728K or ARAP2.K1190P allele inhibited internalization of Listeria by ∼60% or ∼75%, respectively (Fig. 4B, panel i). Quantitative confocal microscopy (see Materials and Methods) indicated that Flag-tagged wild-type, R728K, and K1190P ARAP2 alleles were expressed at similar levels (Fig. 4B, panel ii). Taken together, the results in Fig. 4 indicate that both the ArfGAP and RB domains in ARAP2 have important functions in entry of Listeria.
The ArfGAP domain in ARAP2 promotes Listeria entry by inactivating the GTPase Arf6.
The bulk of evidence supports the idea that the in vivo substrate of the ARAP2 ArfGAP domain is the GTPase Arf6 (42). However, in vitro, both Arf6 and the related GTPase Arf5 can be inactivated by the ARAP2 ArfGAP domain (42). In order to determine if the ArfGAP activity in ARAP2 controls Listeria entry by inhibiting Arf6, we investigated the effect of RNA interference (RNAi)-mediated depletion of Arf6 in cells expressing the ARAP2.R728K allele. If the ArfGAP domain affects bacterial entry through Arf6, then depletion of Arf6 would be expected to restore normal entry to cells expressing ARAP2.R728K. Indeed, we found that RNAi-mediated knockdown of Arf6 suppressed the defect in Listeria internalization normally caused by the ARAP2.R728K allele (Fig. 5). Importantly, depletion of Arf6 did not affect bacterial entry in cells expressing tagged wild-type or K1190P ARAP2. Collectively, the results in Fig. 5 indicate that the ArfGAP domain in ARAP2 stimulates uptake of Listeria by inactivating host Arf6.
The results in Fig. 5 imply that activated (GTP-bound) Arf6 antagonizes Listeria entry. In order to further investigate this idea, we tested the effect of expression of a constitutively activated form of Arf6 (Arf6Q67L) on bacterial internalization. The Arf6Q67L protein is locked in the GTP-bound state because of a defect in GTP hydrolysis (9, 22). Hence, GAPs, such as ARAP2, cannot help inactivate the Arf6Q67L protein. In support of the data in Fig. 5, expression of the Arf6Q67L allele caused a marked inhibition in uptake of Listeria into HeLa cells (Fig. 6).
The RB domain in ARAP2 controls entry of Listeria independently of host Rho proteins.
The RB (formerly RhoGAP) domain in ARAP2 is known to interact with GTP-bound RhoA (42). Given the importance of the RB domain in entry of Listeria (Fig. 4B), we investigated the role of host Rho proteins (19) in this process. HeLa cells were treated with C3, a toxin known to inactivate RhoA and other Rho proteins (5, 40). Rho protein activity was monitored by assessing the integrity of stress fibers, which are F-actin structures regulated by RhoA and related GTPases. Treatment of HeLa cells with C3 toxin (2 mg/liter for 5 h) resulted in a marked disappearance of stress fibers (Fig. 7 A), indicating strong inhibition of Rho proteins. Despite the severe inhibitory effect of C3, entry of Listeria was not impaired by toxin treatment (Fig. 7B). In fact, bacterial uptake was enhanced by about 2-fold, suggesting an antagonistic role for Rho proteins in entry. Taken together, the results in Fig. 7 indicate that InlB-mediated entry of Listeria does not require activated host Rho proteins. These findings suggest that the RB domain in ARAP2 likely promotes internalization of Listeria through a mechanism independent of Rho proteins.
ARAP2 is needed for F-actin cytoskeletal changes during InlB-mediated entry.
InlB-mediated entry of Listeria requires rearrangements in the F-actin cytoskeleton (8, 16, 17, 29). These cytoskeletal changes are thought to elicit host cell surface changes that drive Listeria engulfment. Importantly, type IA PI 3-kinase (p85-p110) plays a critical role in F-actin rearrangements during InlB-mediated uptake (8).
Given that ARAP2 ArfGAP activity is stimulated by lipid products of p85-p110 (42), we wondered if ARAP2 is needed for cytoskeletal changes during InlB-mediated internalization. In order to investigate this possibility, we used 3-μm latex beads coated with InlB protein. Our previous work indicated that these InlB-coated beads are internalized by HeLa cells through a mechanism that is indistinguishable from that utilized by Listeria (8). Moreover, F-actin cytoskeletal changes are more readily detected with InlB-coated beads than with bacteria, perhaps because of a greater amount of InlB on the surface of the beads (8). Importantly, we found that RNAi-mediated depletion of ARAP2 caused a marked inhibition of entry of InlB-coated beads (Fig. 8 A) and of accompanying F-actin rearrangements (Fig. 8B and C). In HeLa cells treated with control siRNA and incubated with InlB-coated beads for 2.5 min, ∼45% of beads were encircled by structures enriched in F-actin (Fig. 8B and C, panel i). Quantitative confocal microscopy analysis indicated that the degree of F-actin enrichment around beads, compared to the average F-actin distribution through the entire cell (see Materials and Methods), was roughly 4-fold (Fig. 8C, panel ii). We note that, even under control conditions, not every HeLa cell displayed obvious F-actin accumulation (Fig. 8B, panel i). On average, ∼50% of cells treated with control siRNA recruited F-actin near adherent beads. While the reason for this heterogeneity in F-actin accumulation is not known, it is consistent with the fact that ∼40 or ∼50% of HeLa cells internalized beads or Listeria, respectively. Importantly, in cells treated with ARAP2 siRNA, only ∼11% of cell-associated beads displayed obvious recruitment of F-actin (Fig. 8B and C, panel i). Of these beads that were encircled by F-actin, the intensity of actin was diminished relative to that in control siRNA-treated cells (Fig. 8C, panel ii). Collectively, the data in Fig. 8 indicate that host ARAP2 plays an important role in F-actin rearrangements during InlB-mediated entry.
DISCUSSION
In this work, we show that the human GTPase-activating protein (GAP) ARAP2 plays a critical role in InlB-mediated entry of Listeria. ARAP2 is a large, multidomain protein that controls bacterial uptake through at least two distinct pathways (Fig. 9). The ArfGAP domain in ARAP2 promotes internalization of Listeria by inactivating the host GTPase Arf6. The ARAP2 RhoA binding (RB) domain likely contributes to entry by binding an unidentified host ligand. Importantly, ARAP2 is needed for actin cytoskeletal rearrangements that drive InlB-mediated internalization. Altogether, our findings have identified ARAP2 as a crucial host factor that mediates F-actin changes and pathogen engulfment downstream of the Met receptor tyrosine kinase.
FIG. 9.

Model for the role of ARAP2 in InlB-mediated entry of Listeria. ARAP2 promotes bacterial internalization through at least two distinct pathways. One pathway involves the inactivation of host GTPase Arf6 through the ArfGAP domain in ARAP2. The other pathway involves the RhoA binding domain (RB) of ARAP2, which likely mediates uptake of Listeria by interacting with an unidentified host ligand (X). The ArfGAP activity in ARAP2 is stimulated by binding of the type IA PI 3-kinase product PI(3,4,5)P3 (PIP3) to PH domains in ARAP2 (42). Based on these data, ARAP2 is one of the likely downstream targets of PI 3-kinase promoting Listeria entry.
ARAP2 is probably coupled to the Met receptor through the host type IA PI 3-kinase p85-p110 (Fig. 9). Binding of Listeria or InlB protein to Met results in activation of p85-p110 and accumulation of its lipid product phosphatidylinositol 3,4,5-trisphosphate [PI(3,4,5)P3] (8, 17, 18). Importantly, PI(3,4,5)P3 stimulates the ArfGAP activity in ARAP2 (42), indicating that ARAP2 is regulated by p85-p110. The sites of binding of PI(3,4,5)P3 are predicted to be the first, fifth, and perhaps third pleckstrin homology (PH) domains in ARAP2 (27, 42). In combination with previous findings on ARAP2 (27, 42), our work indicates that this GAP is one of the likely downstream targets of p85-p110 involved in Listeria entry.
How does ARAP2 contribute to internalization of Listeria? Our work indicates that both the ArfGAP and RhoA binding (RB) domains of ARAP2 play important roles in bacterial entry. Although the RB domain in ARAP2 is known to control actin stress fiber formation by interacting with the GTPase RhoA (42), our results indicate that Rho proteins were dispensable for Listeria uptake. Treatment of host cells with C3 exoenzyme, a toxin that inactivates RhoA and other Rho proteins (5, 40), failed to impair bacterial entry. This finding with C3 is in agreement with previous results indicating that expression of a dominant negative allele of RhoA did not interfere with InlB-mediated bacterial entry (2). Since Listeria internalization does not require host Rho activity, it is likely that the RB domain in ARAP2 acts by binding a cellular factor apart from Rho proteins. In future work, it will be interesting to identify novel ligands of the RB domain. Finding such ligands will help us to understand how ARAP2 controls entry of Listeria and may also provide new information on the normal cellular functions of ARAP2.
Our genetic data indicated that the ArfGAP domain in ARAP2 stimulates uptake of Listeria by antagonizing the human GTPase Arf6. In cells expressing normal levels of Arf6, ectopic expression of ARAP2 mutated in its ArfGAP domain (ARAP2.R728K) inhibited bacterial entry. Importantly, this impairment in entry was suppressed by RNAi-mediated depletion of Arf6. In addition, a constitutively activated allele of Arf6 (Arf6Q67L) blocked internalization of Listeria. Collectively, these genetic data indicate that in the absence of ARAP2, Arf6-GTP inhibits internalization of Listeria. In normal situations where ARAP2 is present, this GAP relieves Arf6-GTP-mediated inhibition. Based on our results, we postulate that ARAP2 restrains accumulation of Arf6-GTP, which would otherwise interfere with entry of Listeria (Fig. 9). A prediction from these genetic studies is that Arf6-GTP amounts would increase in HeLa cells depleted of ARAP2. Using a previously described coprecipitation approach (31, 35), we made several unsuccessful attempts to measure endogenous Arf6-GTP in control and ARAP2-depleted cells. The inability to detect Arf6-GTP could reflect a low abundance of activated Arf6 in HeLa cells or could be due to limited sensitivity of the anti-Arf6 antibodies used in Western blotting. Regardless of technical issues encountered in the biochemical experiments, our genetic data are consistent with the mechanism of action of ARAP2 and Arf6 depicted in Fig. 9.
How unrestrained accumulation of Arf6-GTP might interfere with entry of Listeria is not known. One possibility is that deregulation of Arf6 through depletion of ARAP2 elicits membrane trafficking events that sequester one or more host factors needed for internalization of Listeria. Previous studies indicate that the Arf6Q67L constitutively activated mutant causes the intracellular accumulation of cholesterol and phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] in enlarged vacuoles (3, 30). Interestingly, cholesterol-rich lipid rafts at the plasma membrane are needed for InlB-mediated uptake of Listeria (36). PI(4,5)P2 plays a critical role in actin polymerization in mammalian cells and also is required for endocytosis and phagocytosis in macrophages (34). Although the role of PI(4,5)P2 in uptake of Listeria has yet to be addressed, an important function for this phosphoinositide in bacterial entry seems likely. In future studies, it will be interesting to investigate if ARAP2 controls Listeria entry by maintaining proper levels of cholesterol and PI(4,5)P2 at the plasma membrane.
Interestingly, in addition to affecting entry of Listeria, Arf6 regulates internalization of several other bacterial, viral, and protozoan pathogens, including Yersinia (41), Chlamydia (1), baculovirus (21), coxsackievirus A9 (15), Leishmania (26), and Toxoplasma (6). However, in the cases of these other microbial pathogens, Arf6 positively contributes to entry into host cells. Notably, RNAi-mediated silencing of Arf6 and/or expression of Arf6 interfering mutants impairs entry of these pathogens. In contrast to the situation with these microbes, our findings with Listeria indicate a negative role for Arf6 that must be overcome by ARAP2 in order to allow efficient bacterial entry. The collective data on Arf6 suggest that the GTPase can use distinct mechanisms to control internalization of different microbial pathogens. The specific mechanism employed by Arf6 may depend on the cellular surface receptor recognized by the pathogen.
In summary, our studies have uncovered a critical role for the human signaling protein ARAP2 in InlB-dependent entry of Listeria. Future work with Listeria has the potential to identify novel ligands of ARAP2 domains, such as the Rho binding (RB) domain, and may also contribute to a better understanding of the cellular consequences of unrestrained activation of Arf6 GTPase. Finally, our findings that ARAP2 controls bacterial entry mediated by the Met receptor raise the possibility that ARAP2 might also contribute to some of the normal biological functions of Met. Such functions controlled by Met and its cellular ligand HGF include mammalian cell growth, motility, and morphogenesis (12).
Acknowledgments
We thank Paul Randazzo and H. Y. Yoon (NIH) for technical advice and generous gifts of ARAP2 expression plasmids and antibodies. We acknowledge Shahanawaz Jiwani for performing real-time PCR analysis. We thank Curtis Lanier for help with Western blotting.
This work was supported, in part, through an “in-house” grant awarded to K.I. from the University of Central Florida and by National Institutes of Health grant 5R01AI063513-02 to R.A.D.
Editor: S. M. Payne
Footnotes
Published ahead of print on 7 September 2010.
REFERENCES
- 1.Balana, M. E., F. Niedergang, A. Subtil, A. Alcover, P. Chavrier, and A. Dautry-Varsat. 2005. Arf6 GTPase controls bacterial invasion by actin remodeling. J. Cell Sci. 118:2201-2210. [DOI] [PubMed] [Google Scholar]
- 2.Bierne, H., E. Gouin, P. Roux, P. Caroni, H. L. Yin, and P. Cossart. 2001. A role for cofilin and LIM kinase in Listeria-induced phagocytosis. J. Cell Biol. 155:101-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown, F. D., A. L. Rozelle, H. L. Yin, R. Balla, and J. G. Donaldson. 2001. Phosphatidylinositol 4,5-bisphosphate and Arf6-regulated membrane traffic. J. Cell Biol. 154:1007-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cantley, L. C. 2002. The phosphoinositide 3-kinase pathway. Science 296:1655-1657. [DOI] [PubMed] [Google Scholar]
- 5.Chardin, P., P. Boquet, P. Madaule, M. R. Popoff, E. J. Rubin, and D. M. Gill. 1989. The mammalian G protein RhoC is ADP-ribosylated by Clostridium botulinum exoenzyme C3 and affects actin microfilaments in Vero cells. EMBO J. 8:1087-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.da Silva, V. C., E. A. da Silva, M. C. Cruz, P. Charvier, and R. A. Mortara. 2009. Arf6, PI 3-kinase and host cell actin cytoskeleton in Toxoplasma gondii cell invasion. Biochem. Biophys. Res. Commun. 378:656-661. [DOI] [PubMed] [Google Scholar]
- 7.Disson, O., S. Grayo, E. Huillet, G. Nikitas, F. Langa-Vives, O. Dussurget, M. Ragon, A. Le Monnier, C. Babinet, P. Cossart, and M. Lecuit. 2008. Congugated action of two species-specifc invasion proteins for fetoplacental listeriosis. Nature 455:1114-1118. [DOI] [PubMed] [Google Scholar]
- 8.Dokainish, H., B. Gavicherla, Y. Shen, and K. Ireton. 2007. The carboxyl-terminal SH3 domain of the mammalian adaptor CrkII promotes internalization of Listeria monocytogenes through activation of host phosphoinositide 3-kinase. Cell. Microbiol. 10:2497-2516. [DOI] [PubMed] [Google Scholar]
- 9.Donaldson, J. G., N. Porat-Shliom, and L. A. Cohen. 2009. Clathrin-independent endocytosis: a unique platform for cell signaling and PM remodeling. Cell. Signal. 21:1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dramsi, S., I. Biswas, E. Maguin, L. Braun, P. Mastroeni, and P. Cossart. 1995. Entry of Listeria monocytogenes into hepatocytes requires expression of inIB, a surface protein of the internalin multigene family. Mol. Microbiol. 16:251-261. [DOI] [PubMed] [Google Scholar]
- 11.Engelman, J. A., J. Luo, and L. C. Cantley. 2006. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 7:606-619. [DOI] [PubMed] [Google Scholar]
- 12.Gentile, A., L. Trusolino, and P. M. Comoglio. 2008. The Met tyrosine kinase receptor in development and cancer. Cancer Metastasis Rev. 27:85-94. [DOI] [PubMed] [Google Scholar]
- 13.Gouin, E., P. Dehoux, J. Mengaud, C. Kocks, and P. Cossart. 1995. iactA of Listeria ivanovii, although distantly related to Listeria monocytogenes actA, restores actin tail formation in an L. monocytogenes actA mutant. Infect. Immun. 63:2729-2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamon, M., H. Bierne, and P. Cossart. 2006. Listeria monocytogenes: a multifaceted model. Nat. Rev. Microbiol. 4:423-434. [DOI] [PubMed] [Google Scholar]
- 15.Heikkila, O., P. Susi, T. Tevaluoto, H. Harma, V. Marjomaki, T. Hyypia, and S. Kiljunen. 2010. Internalization of coxsackievirus A9 is mediated by beta 2-microglobulin, dynamin, and Arf6 but not by caveolin-1 or clathrin. J. Virol. 84:3666-3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ireton, K. 2007. Entry of the bacterial pathogen Listeria monocytogenes into mammalian cells. Cell. Microbiol. 9:1365-1375. [DOI] [PubMed] [Google Scholar]
- 17.Ireton, K., B. Payrastre, H. Chap, W. Ogawa, H. Sakaue, M. Kasuga, and P. Cossart. 1996. A role for phosphoinositide 3-kinase in bacterial invasion. Science 274:780-782. [DOI] [PubMed] [Google Scholar]
- 18.Ireton, K., B. Payrastre, and P. Cossart. 1999. The Listeria monocytogenes protein InlB is an agonist of mammalian phosphoinositude-3-kinase. J. Biol. Chem. 274:17025-17032. [DOI] [PubMed] [Google Scholar]
- 19.Jaffe, A. B., and A. Hall. 2005. Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 21:247-269. [DOI] [PubMed] [Google Scholar]
- 20.Kolokoltsov, A. A., S. C. Weaver, and R. A. Davey. 2005. Efficient functional pseudotyping of oncoretroviral and lentiviral vectors by Venezuelan equine encephalitis virus envelope proteins. J. Virol. 79:756-763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laakkonen, J. P., A. R. Makela, E. Kakkonen, P. Turkki, S. Kukkonen, J. Peranen, S. Yla-Herttuala, K. J. Airenne, C. Oker-Blom, M. Vihinen-Ranta, and V. Marjomaki. 2009. Clathrin-independent entry of baculovirus triggers uptake of E. coli in non-phagocytic human cells. PLoS One 4:e5093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Larsen, J. E., R. H. Massol, T. J. Nieland, and T. Kirchhausen. 2004. HIV Nef-mediated major histocompatibility complex class I down-modulation is independent of Arf6 activity. Mol. Biol. Cell 15:323-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lecuit, M., S. Vandormael-Pournin, J. Lefort, M. Huerre, P. Gounon, C. Dupuy, C. Babinet, and P. Cossart. 2001. A transgenic model for Listeriosis: role of inernalin in crossing the intestinal barrier. Science 292:1722-1724. [DOI] [PubMed] [Google Scholar]
- 24.Lemmon, M. A. 2003. Phosphoinositide recognition domains. Traffic 4:201-213. [DOI] [PubMed] [Google Scholar]
- 25.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real time quantitative PCR and the 2(−ΔΔC(t)) method. Methods 25:402-408. [DOI] [PubMed] [Google Scholar]
- 26.Lodge, R., and A. Descoteaux. 2006. Phagocytosis of Leishmania donovani amastigotes is Rac1 dependent and occurs in the absence of oxidase activation. Eur. J. Immunol. 36:2735-2744. [DOI] [PubMed] [Google Scholar]
- 27.Miura, K., K. M. Jacques, S. Stauffer, A. Kubosaki, K. Zhu, D. S. Hirsch, J. Reseau, Y. Zheng, and P. A. Randazzo. 2002. ARAP1: a point of convergence for Arf and Rho signaling. Mol. Cell 9:109-119. [DOI] [PubMed] [Google Scholar]
- 28.Mosmann, T. 1983. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65:55-63. [DOI] [PubMed] [Google Scholar]
- 29.Mostowy, S., and P. Cossart. 2009. Cytoskeleton rearrangements during Listeria infection: clathrin and septins as new players in the game. Cell Motil. Cytoskeleton 66:816-823. [DOI] [PubMed] [Google Scholar]
- 30.Naslavsky, N., R. Weigert, and J. G. Donaldson. 2003. Convergence of non-clathrin- and clathrin-derived endosomes involves Arf6 inactivation and changes in phosphoinositides. Mol. Biol. Cell 14:417-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niedergang, F., E. Colucci-Guyon, T. Dubois, G. Raposo, and P. Chavrier. 2003. ADP ribosylation factor 6 is activated and controls membrane delivery during phagocytosis in macrophages. J. Cell Biol. 161:1143-1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Posfay-Barbe, K. M., and E. R. Wald. 2009. Listeriosis. Semin. Fetal Neonatal Med. 14:228-233. [DOI] [PubMed] [Google Scholar]
- 33.Randazzo, P. A., H. Inoue, and S. Bharti. 2007. Arf GAPs as regulators of the actin cytoskeleton. Biol. Cell 99:583-600. [DOI] [PubMed] [Google Scholar]
- 34.Saarikangas, J., H. Zhao, and P. Lappalainen. 2010. Regulation of the actin cytoskeleton-plasma membrane interplay by phosphoinositides. Physiol. Rev. 90:259-289. [DOI] [PubMed] [Google Scholar]
- 35.Santy, L. C., and J. E. Casanova. 2001. Activation of Arf6 by ARNO stimulates epithelial cell migration through downstream activation of both Rac and phospholipase D. J. Cell Biol. 154:599-610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seveau, S., H. Bierne, S. Giroux, M. C. Prevost, and P. Cossart. 2004. Role of lipid rafts in E-cadherin- and HGF-R/Met-mediated entry of Listeria monocytogenes into host cells. J. Cell Biol. 166:743-753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen, Y., M. Naujokas, M. Park, and K. Ireton. 2000. InlB-dependent internalization of Listeria is mediated by the Met receptor tyrosine kinase. Cell 103:501-510. [DOI] [PubMed] [Google Scholar]
- 38.Sun, H., Y. Shen, H. Dokainish, M. Holgado-Madruga, A. Wong, and K. Ireton. 2005. Host adaptor proteins Gab1 and CrkII promote InlB-dependent entry of Listeria monocytogenes. Cell. Microbiol. 7:443-457. [DOI] [PubMed] [Google Scholar]
- 39.Vazquez-Boland, J. A., M. Kuhn, P. Berche, T. Chakraborty, G. Dominguez-Bernal, W. Goebel, B. Gonzalez-Zorn, J. Wehland, and J. Kreft. 2001. Listeria pathogenesis and molecular virulence determinants. Clin. Microbiol. Rev. 14:584-640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Visvikis, O., M. P. Maddugoda, and E. Lemichez. 2010. Direct modifications of Rho proteins: deconstructing GTPase regulation. Biol. Cell 102:377-389. [DOI] [PubMed] [Google Scholar]
- 41.Wong, K. W., and R. R. Isberg. 2003. Arf6 and phosphoinositol-4-phosphate-5-kinase activities permit bypass of the Rac1 requirement for b1 integrin-mediated bacterial uptake. J. Exp. Med. 198:603-614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoon, H. Y., K. Miura, E. J. Cuthbert, K. K. Davis, B. Ahvazi, J. E. Casanova, and P. A. Randazzo. 2006. ARAP2 effects on the actin cytoskeleton are dependent on Arf6-specific GTPase-activating-protein activity and binding to RhoA-GTP. J. Cell Sci. 119:4650-4666. [DOI] [PubMed] [Google Scholar]