

Abstract
Deletion of the chemokine receptor CXCR2 prevents the recruitment of neutrophils into tissues and subsequent development of experimental Lyme arthritis. Following footpad inoculation of Borrelia burgdorferi, the agent of Lyme disease, expression of the CXCR2 ligand KC (CXCL1) is highly upregulated in the joints of arthritis-susceptible mice and is likely to play an important role in the recruitment of neutrophils to the site of infection. To test this hypothesis, we infected C3H KC−/− mice with B. burgdorferi and followed the development of arthritis and carditis. Ankle swelling was significantly attenuated during the peak of arthritis in the KC−/− mice. Arthritis severity scores were significantly lower in the KC−/− mice on days 11 and 21 postinfection, with fewer neutrophils present in the inflammatory lesions. Cardiac lesions were also significantly decreased in KC−/− mice at day 21 postinfection. There were, however, no differences between C3H wild-type and KC−/− mice in spirochete clearance from tissues. Two other CXCR2 ligands, LIX (CXCL5) and MIP-2 (CXCL2), were not increased to compensate for the loss of KC, and the production of several innate cytokines was unaltered. These results demonstrate that KC plays a critical nonredundant role in the development of experimental Lyme arthritis and carditis via CXCR2-mediated recruitment of neutrophils into the site of infection.
The recruitment of neutrophils to the site of infection is a critical first step in the immune response to pathogens. Neutrophils are highly phagocytic and are capable of engulfing and destroying many microbial invaders; however, they are also linked to tissue damage, which can occur upon the extracellular release of neutrophil granule contents. In experimental Lyme arthritis, neutrophils are the primary cell type present during the acute inflammatory phase, while macrophages predominate during the resolution phase of the disease (6). The presence of live Borrelia burgdorferi spirochetes is required for the development of disease, as indicated by the failure of injecting dead spirochetes or borrelial antigen to cause arthritis (7, 26). The presence of spirochetes in the joint alone, however, is not sufficient to induce disease, as some inbred mouse strains are resistant to the development of Lyme arthritis even though they harbor as many spirochetes in the joint as arthritis-susceptible strains (12, 34). Thus, experimental Lyme arthritis is an immunopathology, most likely resulting from an overly exuberant or inappropriately regulated inflammatory response.
Neutrophils are required for the development of arthritis in many if not most experimental animal models. Depletion of neutrophils has been shown to ameliorate or attenuate disease severity in adjuvant-induced arthritis (AIA) (46), collagen-induced arthritis (CIA) (47), streptococcal cell wall-induced arthritis (48), K/BxN serum transfer arthritis (54), and collagen antibody-induced arthritis (CAIA) (56). Similarly, treatment modalities that alter the recruitment of neutrophils or their ability to extravasate into the joint tissue can also have profound effects on both the incidence and severity of arthritis. For example, blocking E-selectin or junctional adhesion molecule C (JAM-C) can inhibit AIA (23, 42), and blocking VCAM-1 or CD147 or inhibiting granulocyte colony-stimulating factor (G-CSF)-mediated upregulation of adhesion molecules can reduce the development of CIA (13, 16, 18). Thus, the development of arthritis in general appears to require the presence of neutrophils within the joint tissue, and inhibition of neutrophil extravasation, and perhaps activation, can prevent the onset and progression of disease.
Chemokines are chemotactic cytokines that guide the homeostatic movement of cells, as well as leukocyte recruitment during immune responses. Chemokines mediate their biologic effects via G protein-coupled receptors, including the receptors CXCR1 and CXCR2, which appear to play major roles in neutrophil recruitment during arthritis (27). The human neutrophil chemoattractant ligands for these receptors, GROα (CXCL1) (29), ENA-78 (CXCL5) (28), and interleukin-8 (IL-8; CXCL8) (30), are abundantly present in the synovial fluid of rheumatoid arthritis (RA) patients. Studies in animal models have demonstrated that antagonism or genetic depletion of CXCR2 can block the development of arthritis in CAIA (36), AIA (5, 15, 20), and K/BxN serum-induced arthritis (24); however, studies targeting CXCR2 ligands have been less successful at blocking disease progression (8). This may stem from the perceived redundancy of chemokines and their receptors or from the difficulty of targeting the chemokines themselves in vivo, e.g., via antibody-mediated blockade or depletion.
In a previous study, we measured the production over time of 12 cytokines and chemokines directly from the ankle joints of B. burgdorferi-infected mouse strains in order to gain insight into the mechanisms of disease resistance and susceptibility to experimental Lyme arthritis (10). Of the cytokines and chemokines tested, only the production of KC and monocyte chemoattractant protein 1 (MCP-1; CCL2) correlated with the development of arthritis. KC is a major neutrophil chemoattractant and binds to CXCR2 in mice, while MCP-1 is a major monocyte chemoattractant and binds to CCR2 in mice. Infection of C3H CCR2−/− mice had little effect on the development of Lyme arthritis, but C3H CXCR2−/− mice were fully protected from the development of disease. However, whether KC alone was responsible for the CXCR2-mediated recruitment of neutrophils into the infected joint or whether other chemokines or neutrophil chemoattractants also played a role was unclear. In the current study, we have used mice genetically deficient in KC (KC−/−) to determine the role of KC-mediated CXCR2 signaling in the pathogenesis of Lyme borreliosis. Our results demonstrate that KC plays a critical role in the development of both arthritis and carditis in the murine model of Lyme disease.
MATERIALS AND METHODS
Animals.
Male mice heterozygous for expression of KC on a mixed B6/129 genetic background were obtained from Sergio Lira (Mount Sinai School of Medicine) and backcrossed for 5 generations onto the C3H/HeJ background using speed congenics. Male and female heterozygotes were then crossed to create C3H KC−/− mice. Male and female KC−/− mice and littermate controls were 6 to 10 weeks old at the time of infection. In some experiments, age-matched, wild-type C3H/HeJ mice were purchased from the Jackson Laboratory and used in experiments. The mice were housed in a specific-pathogen-free facility and given food and water ad libitum. All experimental protocols were approved by the Animal Care and Use Committee of the University of Missouri.
Bacteria and infections.
A virulent, low-passage isolate of the B. burgdorferi N40 strain was used for all experiments. Frozen aliquots were thawed and grown in 7.5 ml of Barbour-Stoenner-Kelly II (BSK II) medium supplemented with 6% rabbit serum (Sigma-Aldrich) for 5 days at 32°C. Viable spirochetes were enumerated using a Petroff-Hausser counting chamber and dark-field microscopy. Mice were inoculated in both hind footpads with 1 × 105 or 5 × 104 B. burgdorferi spirochetes contained in 50 μl of medium.
Assessment of pathology.
The progression of arthritis development was monitored by measuring ankle swelling through the thickest craniocaudal portion of the ankle joints using a metric caliper. Joint thickness was determined prior to infection and then twice weekly during the course of infection. Ankle diameter increases were determined by subtracting the initial baseline measurement from the subsequent measurements. Carditis and arthritis severity scores were determined as previously described (11). Briefly, for carditis the hearts were sagittally bisected through both ventricles and atria, fixed in 10% buffered zinc-formalin, mounted, and stained with hematoxylin and eosin (H&E). The samples were then evaluated independently by two experienced veterinary pathologists for the extent of atrial inflammation in a blind manner. Grade 0 represents no change from uninfected controls, grade 1 represents minimal scattered inflammation covering <1% of the sample, grade 2 represents mild multifocal inflammation over 1 to 25% of the sample, grade 3 represents moderate focally extensive inflammation covering 25 to 50% of the sample, and grade 4 represents marked confluent areas of inflammation over 50% of the sample. For arthritis, the severity scores were graded on a scale from 0 to 4, with 0 representing no inflammation and 4 representing severe inflammation involving more than 50% of the sample. Inflammation type scores for both arthritis and carditis were also graded on a scale from 0 to 4 and represent the ratios of neutrophils to macrophages within the inflammatory lesion. A grade of 0 represents scant neutrophils and scant macrophages, grade 1 represents <5% neutrophils and >90% macrophages, grade 2 represents 5 to 25% neutrophils and >75% macrophages, grade 3 represents 25 to 50% neutrophils and 50 to 75% macrophages, and grade 4 represents >50% neutrophils and <50% macrophages. Immunohistochemical staining of paraffin-embedded sections was completed as described previously (10) using monoclonal antibodies RB6-8C5 (BD Pharmingen, San Jose, CA) and F4/80 (Serotec, Raleigh, NC).
Assessment of B. burgdorferi numbers in tissues by qPCR.
Quantitative multiplex real-time PCR (qPCR) was used to determine the numbers of spirochetes present in the tissues as previously described (10). Briefly, one ankle joint, one-half of the heart cut as described above, and a section of ear tissue were each removed from the animal shortly after its sacrifice, immediately frozen in liquid nitrogen, and stored at −80°C until extraction. Each sample was then homogenized in TRIzol (Invitrogen), and DNA was extracted according to the manufacturer's instructions. The extracted DNA was quantified and diluted to 50 ng/ml using Tris-EDTA buffer. One microliter of the DNA was then used in PCRs. Murine DNA was quantified using the single-copy nidogen gene as described previously (39), and B. burgdorferi DNA in the samples was quantified using the flagellin gene as described previously (41). Quantitative multiplex real-time PCR was performed in duplicate, and levels of flagellin were normalized to levels of nidogen in the same tube.
Chemokine and cytokine quantification from tissues.
Direct measurement of chemokines and cytokines from tissue homogenates was performed as described previously (10). Tissues were excised and immediately snap-frozen in liquid nitrogen. The frozen samples were individually wrapped in aluminum foil, and the still-frozen tissue was pulverized with a hammer and immediately placed into 1 ml ice-cold homogenization buffer (Hanks balanced salt solution [HBSS] containing 0.2% protease inhibitor [Sigma-Aldrich] and 0.4% Triton X-100). The samples were sonicated (3 times for 20 s on ice), cleared via centrifugation (2,000 × g for 20 min, 4°C), and filtered through a 0.45-μm filter. Finally, the filtrate was diluted to 1.5 ml using cold homogenization buffer. The total protein concentration in the sample was determined using a bicinchoninic acid (BCA) assay (Pierce), and cytokine concentrations were expressed in picograms per milligram protein. Murine IL-1β, IL-6, and tumor necrosis factor alpha (TNF-α) were quantified using OptEIA kits (BD Pharmingen). KC, LIX, and MIP-2 were quantified using DuoSet enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems).
Statistical analysis.
Results are expressed as means ± standard errors of the means (SEM). Group means were analyzed for statistical significance using Student's t test or the Mann-Whitney rank sum test using GraphPad Prism software. Significance levels were set at an α of 0.05.
RESULTS
Pathogenesis of Lyme borreliosis is attenuated in both joints and hearts of KC−/− mice.
The recruitment of neutrophils into joint tissue has been shown to be important for the development of disease in several models of arthritis, including CIA in rats (47), serum transfer models in mice (54, 56), and experimental Lyme arthritis (10). KC is a powerful chemoattractant that recruits neutrophils to sites of infection and is expressed at high levels in the joints of arthritis-susceptible mice infected with B. burgdorferi (10). To determine the role of KC in the pathogenesis of experimental Lyme arthritis, we infected wild-type C3H and C3H KC−/− mice in both hind footpads with 5 × 104 virulent N40 strain B. burgdorferi organisms. In some experiments the mice were infected with 1 × 105 B. burgdorferi organisms; this dose level had no measurable effect on disease parameters. Disease development was monitored by twice-weekly measurements of ankle diameters. In the experimental Lyme arthritis model, ankle swelling is generally indicative of the underlying inflammatory response, although some exceptions have been reported (2). Infection of wild-type C3H mice resulted in a typical increase in ankle diameters beginning around 10 days postinfection, which peaked during the second week of infection and then began to resolve (Fig. 1). In contrast, ankle swelling in KC−/− mice was attenuated, with significantly less swelling on days 14 and 17 postinfection (the peak of arthritis severity in wild-type mice; P < 0.04). Thus, a lack of KC signaling, presumably through CXCR2, decreased the level of edema typically seen in the joints of susceptible mice during infection with B. burgdorferi.
FIG. 1.

Ankle swelling following infection of C3H wild-type (control) and KC−/− mice with B. burgdorferi. Mice (8/group) were 6 to 10 weeks old at the time of infection and were inoculated in the hind footpads. Data are means ± SEM and are representative of 4 separate experiments. *, significant differences from control values at the same time point (P < 0.04).
To further characterize the development of disease, we used histology and immunohistochemistry to assign disease severity scores as described previously (11). Examination of H&E-stained histologic sections revealed that arthritis severity scores were significantly reduced in the KC−/− mice at both days 11 and 21 postinfection (Table 1) (P < 0.02). This was accompanied by a significant decrease in the recruitment of neutrophils into the ankle joint, as revealed by the inflammation type scores (Table 1) (P < 0.04) and a lack of specific staining with the neutrophil-specific monoclonal antibody RB6-8C5 in KC−/− ankle joints (Fig. 2). In addition, immunohistochemical staining with the monoclonal antibody F4/80 demonstrated markedly reduced macrophage recruitment into the joints of KC−/− mice (Fig. 2). Together these data demonstrate that a lack of KC expression in the joints of C3H mice results in decreased recruitment of neutrophils and macrophages and an amelioration of arthritis following infection with B. burgdorferi.
TABLE 1.
Disease severity scoresa for C3H wild-type and KC−/− mice
| C3H strain | Day | Heart |
Ankle |
||
|---|---|---|---|---|---|
| Severity score | Type score | Severity score | Type score | ||
| Wild type | 11 | 0.7 ± 0.2 | 0.4 ± 0.2 | 0.9 ± 0.1 | 1.3 ± 0.3 |
| KC−/− | 11 | 0.4 ± 0.1 | 0.4 ± 0.1 | 0.4 ± 0.2b | 0.4 ± 0.2b |
| Wild type | 21 | 2.2 ± 0.3 | 1.8 ± 0.3 | 2.2 ± 0.2 | 1.7 ± 0.2 |
| KC−/− | 21 | 1.1 ± 0.3b | 0.9 ± 0.3b | 1.2 ± 0.2b | 1.0 ± 0.2b |
Values are means ± SEM (eight mice per group) of inflammation severity and inflammation type scores (scales of 0 to 4) in hearts or ankles of control and KC−/− mice on days 11 and 21 postinfection.
Significantly different (P < 0.05) from the C3H values for the same time point.
FIG. 2.

Histologic comparison of inflammatory responses in hearts and ankles of C3H and KC−/− mice infected 21 days prior with B. burgdorferi. Representative sections were stained with H&E or subjected to immunohistochemistry to identify infiltrating neutrophils (RB6) or macrophages (F4/80). Magnification, ×100. Bar = 50 μm.
In addition to arthritis, carditis is a significant sequela which develops in Lyme disease patients (50) and is also recapitulated in the mouse model of Lyme disease. The mechanisms controlling the development of carditis are thought to be different than those controlling development of arthritis (1, 9, 21), as macrophages appear to be the dominant cell type in cardiac lesions, while neutrophils predominate during acute Lyme arthritis (45). To determine the effect of KC deficiency on the development of Lyme carditis, we examined histologic sections of hearts from B. burgdorferi-infected C3H control and KC−/− mice for inflammatory lesions and scored them for severity and inflammation type. By day 11 postinfection there was little evidence of carditis in the hearts of either the wild-type or KC−/− mice (Table 1). This may reflect the fact that few spirochetes have colonized the hearts by this time point (see Fig. 3). On day 21 postinfection, the KC−/− mice had significantly lower levels of carditis severity than the C3H mice (Table 1) (P < 0.03), with significantly fewer neutrophils recruited into the inflammatory lesions as revealed by the type scores (P < 0.01). Immunohistochemical staining revealed an almost complete absence of neutrophils in cardiac tissues of KC−/− mice, as well as greatly reduced numbers of macrophages (Fig. 2). These results suggest that, while macrophages dominate in cardiac lesions, it is neutrophil recruitment that controls the overall intensity of the inflammatory response. Together, these results demonstrate that the KC/CXCR2 signaling interaction is critical for the recruitment of neutrophils into the infected joints and hearts of B. burgdorferi-infected animals and is responsible for the subsequent development of Lyme arthritis and carditis.
FIG. 3.
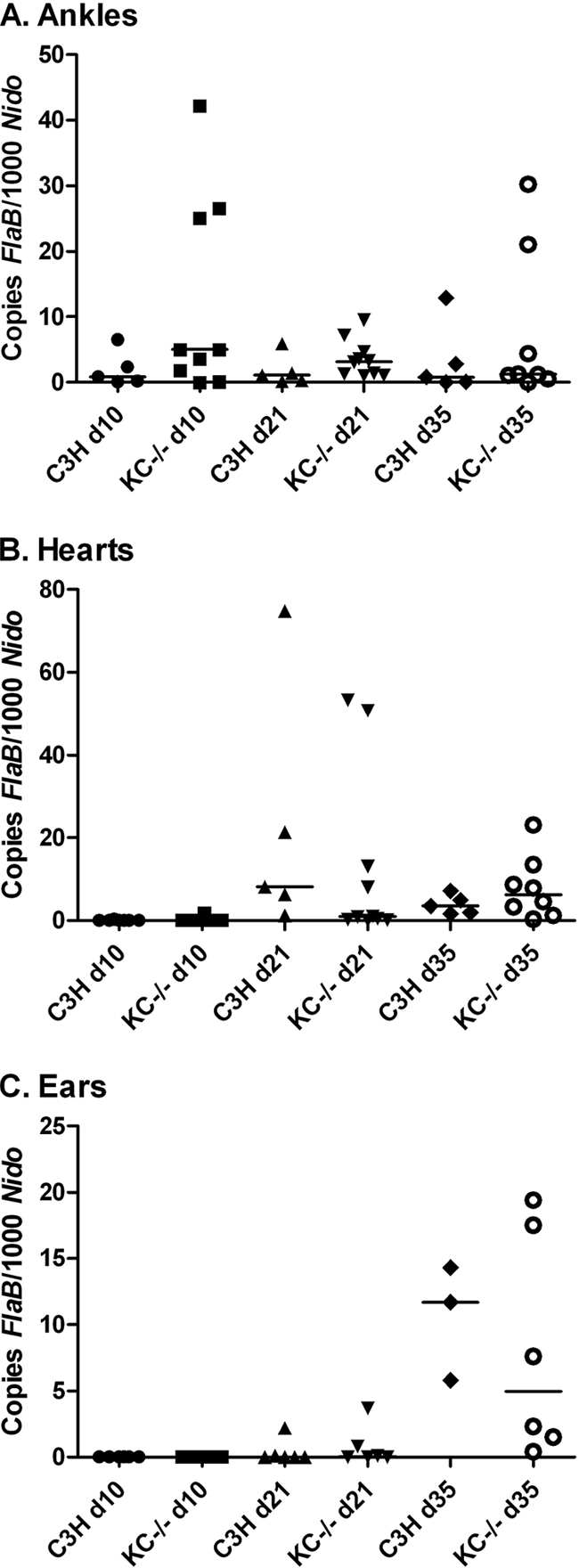
B. burgdorferi burdens in tissues of C3H wild-type and KC−/− mice. The mice were infected via footpad inoculation, and ankles (A), hearts (B), and ears (C) were harvested on days 10, 21, and 35 postinfection. Spirochete DNA was quantified using real-time PCR for flab and normalized to murine nidogen. Data are representative of two separate experiments. Bars represent median values.
Spirochete levels in tissues are not altered in KC−/− mice.
Neutrophils are thought to make significant contributions to the clearance of spirochetes from infected tissues (33, 40, 55). We were therefore interested in determining if KC deficiency and lack of neutrophil recruitment might impact B. burgdorferi loads in tissues. C3H wild-type and KC−/− mice were sacrificed on days 10, 21, and 35 postinfection, and DNA was harvested from ankle, heart, and ear samples. Numbers of copies of the spirochete flab gene were determined by quantitative real-time PCR and normalized to 1,000 copies of the single-copy murine gene nidogen, as described previously (10). Levels of spirochete DNA in ankle joints from KC−/− mice were slightly higher than those in wild-type control mice at all time points analyzed (Fig. 3 A), although none of the differences reached statistical significance. This could reflect a minor defect in spirochete clearance in the KC−/− mice. Spirochete DNA levels were not different between control and KC−/− mice in the hearts (Fig. 3B) or ears (Fig. 3C) at any time point and peaked on days 21 and 35, respectively. This most likely reflects the increased time required for dissemination to these tissues from the footpad inoculation site. These results demonstrate there is no significant defect in spirochete dissemination or clearance by innate immune mechanisms in mice deficient in KC.
Production of chemokines and cytokines is not altered in KC−/− mice.
Neutrophils are thought to contribute to innate immunity not only through phagocytosis and the killing of microbes but also by guiding the developing immune response through the secretion of cytokines and other immune modulators (14). Thus, the cytokine and chemokine milieu may be significantly altered in KC−/− mice and contribute to the change in disease progression observed in these mice. We first examined the production of CXCR2-binding chemokines since these are known to be potent chemoattractants for neutrophils. As we previously reported (10), KC is expressed in both ankle (Fig. 4 A) and heart (Fig. 5 A) tissue following infection with B. burgdorferi. We also investigated whether there were compensatory increases in the production LIX and MIP-2 in the KC−/− mice. We found no differences in the expression of LIX or MIP-2 in either the ankles (Fig. 4B and C) or hearts (Fig. 5B and C) between C3H wild-type and KC−/− mice. Thus, the expression of these two CXC chemokines was not increased during B. burgdorferi infection to compensate for the absence of KC. Interestingly, however, while the expression levels of KC in ankle and heart tissues of wild-type C3H mice were similar, the production of LIX and MIP-2 showed differential tissue expression, with 10-fold-higher levels of LIX in the hearts and 10-fold-higher levels of MIP-2 in the ankles of infected mice than in the reciprocal tissue. This suggests distinct mechanisms of neutrophil recruitment to these tissues during B. burgdorferi infection.
FIG. 4.

Chemokine and cytokine production in the ankles of C3H wild-type and KC−/− mice at 11 and 21 days after infection with B. burgdorferi. Ankle joints were harvested at the indicated time points, and protein was extracted as described previously (10). Levels of KC (A), LIX (B), MIP-2 (C), IL-6 (D), IL-1β (E), and TNF-α (F) were quantified by ELISA. With the exception of KC, there were no significant differences in any of the cytokine or chemokine levels between the C3H wild-type and KC−/− mice at either time point.
FIG. 5.

Chemokine and cytokine production in the hearts of C3H wild-type and KC−/− mice at 11 and 21 days after infection with B. burgdorferi. Hearts were harvested at the indicated time points, and protein was extracted as described previously (10). Levels of KC (A), LIX (B), MIP-2 (C), IL-6 (D), IL-1β (E), and TNF-α (F) were quantified by ELISA. With the exception of KC there were no significant differences in any of the cytokine or chemokine levels between the C3H wild-type and KC−/− mice at either time point.
Neutrophils are known to produce several cytokines that can modulate the developing immune response and subsequently influence the production of cytokines by tissue macrophages and other cell types (14). Since the KC−/− mice demonstrated defective neutrophil recruitment into the sites of infection, we wanted to determine if this affected the local cytokine microenvironment. We measured three cytokines that are classically important in the developing inflammatory response. The levels of IL-6, IL-1β, and TNF-α from the ankles (Fig. 4D to F) and hearts (Fig. 5D to F) of control and KC−/− mice were measured on days 11 and 21 postinfection. We found no differences in the production of these cytokines between the C3H and KC−/− mice in either tissue or at either time point studied. This suggests that these cytokines are produced independently of the recruitment of neutrophils to the site of infection and are not influenced by the subsequent production of neutrophil inflammatory mediators. Again, it is interesting that, similar to the chemokine results, the production of IL-6 was 30-fold higher in the heart tissue than in the joints in both KC−/− and control mice. This further supports the premise that cytokines and chemokines are regulated in a tissue-specific manner during B. burgdorferi infection.
DISCUSSION
The critical role of neutrophils in mediating the development of RA has been recognized for quite some time (43, 53), although the mechanism(s) underlying neutrophil-mediated damage to the joint remains unclear. Neutrophil entry into joint tissue has been shown to be a required step in various animal models of arthritis (10, 46-48, 54, 56). Thus, neutrophil chemoattractants are thought to provide attractive targets for not only prevention of inflammatory diseases but also the development of therapies to alleviate the symptoms of chronic inflammation (8, 27). Indeed, treatments targeting either the adhesion molecules that neutrophils utilize to extravasate from the blood vessel into the joint tissue or neutrophil chemoattractants and/or their receptors have proven to be efficacious in blocking or reducing arthritis severity in numerous animal models (51). However, despite success in animal models, clinical trials of antichemokine therapies in RA patients have had limited success (8, 35, 52). Although this may be attributable to the perceived promiscuity and redundancy of chemokine receptors and their ligands, it was recently reported that a single amino acid change in a chemoattractant ligand could alter the functional outcome of binding to a single receptor (25). Further studies are necessary to define the functional in vivo specificity of chemokine ligand/receptor interactions.
Previous work from our lab demonstrated high levels of KC and MCP-1 production in the ankle joints of Lyme arthritis-susceptible, but not -resistant, mice (10). Infection of C3H CCR2−/− mice with B. burgdorferi had little effect on Lyme arthritis development but did result in differential development of Lyme carditis (38). In contrast, infection of C3H CXCR2−/− mice resulted in protection against Lyme arthritis development and prevented entry of neutrophils into the infected joint (10). In the mouse, MIP-2 and LIX, in addition to KC, bind to CXCR2 and are neutrophil chemoattractants. Thus, these chemokines could also play a role in the neutrophil response to B. burgdorferi infection, as suggested by the effect of CXCR2 deficiency. Initial experiments to determine the role of KC in Lyme arthritis development were unsuccessful, as injection of an anti-KC polyclonal antibody during B. burgdorferi infection failed to inhibit neutrophil recruitment and actually increased the expression of KC in the infected joint (C. Brown, unpublished observation). In the present study, infection of KC−/− mice resulted in a defect in neutrophil recruitment into the infected joint and prevented the development of experimental Lyme arthritis in C3H mice. Thus, it appears that during B. burgdorferi infection KC alone is solely responsible for the recruitment of neutrophils into the infected joint via its binding to CXCR2. These results are similar to studies in which intra-articular injections of anti-KC antibodies could inhibit neutrophil recruitment into the joint during AIA (20, 31) or during monosodium urate crystal-induced arthritis in rabbits (19). In rat AIA, antibodies against ENA-78-like protein were effective in attenuating arthritis severity (22). However, it is impossible to determine if the partial effects of the antibody treatments on arthritis severity were due to incomplete target blockades or to redundant chemokine function. Our study is the first to use KC−/− mice to conclusively demonstrate a role for the KC/CXCR2 axis in the development of experimental Lyme arthritis.
The mechanisms responsible for the development of Lyme carditis are not entirely clear but are thought to be different from those controlling arthritis (1, 9, 21). Murine Lyme carditis is characterized by bacterial invasion and inflammation at the heart base and in tissue surrounding the major vessels and valves (3). The inflammatory infiltrate is dominated by macrophages, with smaller numbers of neutrophils (45), but like arthritis, disease arises independently of adaptive immunity. We have previously demonstrated that mice deficient in CCR2 develop a carditis predominated by neutrophils, suggesting a role for neutrophils in disease development (38). In the current study we found that carditis severity was greatly attenuated in mice deficient in KC. Despite the relative lack of inflammatory cells in cardiac tissue, spirochete numbers were effectively controlled by tissue macrophages or other mechanisms present at the site of infection. Thus, while macrophages clearly dominate the inflammatory infiltrate during Lyme carditis and are likely important for controlling spirochete numbers, neutrophils appear to control the overall level of the inflammatory response. This is most likely through the induction of monocyte chemoattractant signals and adhesion molecules and the subsequent recruitment of monocytes from the blood (49). Further studies are required to determine the precise mechanism by which neutrophils orchestrate this response.
The importance of phagocyte-mediated clearance of B. burgdorferi from tissues remains unclear. In vitro, macrophages can be demonstrated to take up and kill spirochetes quite efficiently, while neutrophil uptake and killing of spirochetes appear to require opsonization (37). However, in vivo neutrophil killing of spirochetes engineered to secrete KC is highly efficient, and a 100,000-fold increase in spirochete numbers is required to infect the murine host (55). Thus, during an inflammatory response (i.e., in the presence of KC), neutrophil killing of B. burgdorferi would appear to be independent of opsonization. In the current study, we found no differences in spirochete tissue burdens in the presence (wild-type mice) or absence (KC−/− mice) of inflammation. Similarly, arthritis-resistant and -susceptible mouse strains are known to harbor similar numbers of spirochetes within their tissues even though they have distinctly different inflammatory phenotypes (12, 34). Thus, there is no correlation between the intensity of the inflammatory response and clearance of spirochetes from tissues. This issue remains a conundrum.
The chemokine system appears to be highly redundant, with several ligands capable of binding to a single receptor and, in some instances, a single ligand capable of binding to several receptors. In the mouse, KC, MIP-2, and LIX are all thought to bind exclusively to CXCR2 and are known to mediate neutrophil recruitment. In the current study, we demonstrate that all three are produced in murine ankles and hearts during B. burgdorferi infection. However, they do not appear to be functionally redundant since KC−/− mice were resistant to the development of both Lyme arthritis and carditis, with a lack of neutrophil recruitment into both tissues. In addition, levels of LIX and MIP-2 in ankles and hearts were unaltered in the KC−/− tissues, demonstrating a lack of compensatory changes during KC deficiency. Our results are in agreement with other studies suggesting that, despite apparent functional redundancy in vitro, in vivo these chemokines are under differentially regulated control, as illustrated by distinct tissue expression profiles, and likely have unique biological functions (4, 17, 32, 44). Although functional redundancy is likely to exist under certain conditions in vivo, neutrophil recruitment and subsequent disease development during murine Lyme borreliosis appear to be mediated primarily via the KC/CXCR2 axis.
Finally, neutrophils are the first cells recruited to sites of injury or infection and are therefore positioned to guide the developing immune response through the production of various cytokines and other bioactive molecules. Murine Lyme arthritis and carditis are mediated by innate immune responses that are likely modulated by the entry of neutrophils into the site of infection. We found no differences in the tissue expression of TNF-α, IL-1β, or IL-6 between wild-type and KC−/− mice. This result suggests that these cytokines are produced by resident sentinel cells following their encounter with B. burgdorferi spirochetes, and thus their expression is not altered by the presence or absence of neutrophils, nor are they directly linked to the development of disease. Interestingly, the levels of several chemokines and cytokines were 7- to 30-fold higher in infected hearts than in ankle joints. These differences may be due to differing levels of inflammation or to differential regulation of inflammation in the two tissues, contributing to qualitative differences in the resulting type of inflammatory response produced. However, caution must be used in interpreting these results as the hearts in our study were not perfused and may contain blood and circulating cytokines not contained within the tissue itself. Further studies are necessary to clarify the biological significance of these differences.
Acknowledgments
This work was supported by Public Health Service grants AI059292 and AR052748 from the National Institutes of Health.
We thank Sergio Lira for providing the KC−/− mice.
Editor: R. P. Morrison
Footnotes
Published ahead of print on 7 September 2010.
REFERENCES
- 1.Anguita, J., S. W. Barthold, S. Samanta, J. Ryan, and E. Fikrig. 1999. Selective anti-inflammatory action of interleukin-11 in murine Lyme disease: arthritis decreases while carditis persists. J. Infect. Dis. 179:734-737. [DOI] [PubMed] [Google Scholar]
- 2.Anguita, J., D. H. Persing, M. Rincon, S. W. Barthold, and E. Fikrig. 1996. Effect of anti-interleukin 12 treatment on murine lyme borreliosis. J. Clin. Invest. 97:1028-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Armstrong, A. L., S. W. Barthold, D. H. Persing, and D. S. Beck. 1992. Carditis in Lyme disease susceptible and resistant strains of laboratory mice infected with Borrelia burgdorferi. Am. J. Trop. Med. Hyg. 47:249-258. [DOI] [PubMed] [Google Scholar]
- 4.Armstrong, D. A., J. A. Major, A. Chudyk, and T. A. Hamilton. 2004. Neutrophil chemoattractant genes KC and MIP-2 are expressed in different cell populations at sites of surgical injury. J. Leukoc. Biol. 75:641-648. [DOI] [PubMed] [Google Scholar]
- 5.Barsante, M. M., T. M. Cunha, M. Allegretti, F. Cattani, F. Policani, C. Bizzarri, W. L. Tafuri, S. Poole, F. Q. Cunha, R. Bertini, and M. M. Teixeira. 2008. Blockade of the chemokine receptor CXCR2 ameliorates adjuvant-induced arthritis in rats. Br. J. Pharmacol. 153:992-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barthold, S. W., D. S. Beck, G. M. Hansen, G. A. Terwilliger, and K. D. Moody. 1990. Lyme borreliosis in selected strains and ages of laboratory mice. J. Infect. Dis. 162:133-138. [DOI] [PubMed] [Google Scholar]
- 7.Barthold, S. W., K. D. Moody, G. A. Terwilliger, P. H. Duray, R. O. Jacoby, and A. C. Steere. 1988. Experimental Lyme arthritis in rats infected with Borrelia burgdorferi. J. Infect. Dis. 157:842-846. [DOI] [PubMed] [Google Scholar]
- 8.Bizzarri, C., A. R. Beccari, R. Bertini, M. R. Cavicchia, S. Giorgini, and M. Allegretti. 2006. ELR+ CXC chemokines and their receptors (CXC chemokine receptor 1 and CXC chemokine receptor 2) as new therapeutic targets. Pharmacol. Ther. 112:139-149. [DOI] [PubMed] [Google Scholar]
- 9.Brown, C. R., V. A. Blaho, K. L. Fritsche, and C. M. Loiacono. 2006. Stat1 deficiency exacerbates carditis but not arthritis during experimental Lyme borreliosis. J. Interferon Cytokine Res. 26:390-399. [DOI] [PubMed] [Google Scholar]
- 10.Brown, C. R., V. A. Blaho, and C. M. Loiacono. 2003. Susceptibility to experimental Lyme arthritis correlates with KC and monocyte chemoattractant protein-1 production in joints and requires neutrophil recruitment via CXCR2. J. Immunol. 171:893-901. [DOI] [PubMed] [Google Scholar]
- 11.Brown, C. R., A. Y. C. Lai, S. T. Callen, V. A. Blaho, J. M. Hughes, and W. J. Mitchell. 2008. Adenoviral delivery of interleukin-10 fails to attenuate experimental Lyme disease. Infect. Immun. 76:5500-5507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown, C. R., and S. L. Reiner. 1998. Clearance of Borrelia burgdorferi may not be required for resistance to experimental Lyme arthritis. Infect. Immun. 66:2065-2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carter, R. A., I. K. Campbell, K. L. O'Donnel, and I. P. Wicks. 2002. Vascular cell adhesion molecule-1 (VCAM-1) blockade in collagen-induced arthritis reduces joint involvement and alters B cell trafficking. Clin. Exp. Immunol. 128:44-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cassatella, M. A. 1999. Neutrophil-derived proteins: selling cytokines by the pound. Adv. Immunol. 73:369-509. [DOI] [PubMed] [Google Scholar]
- 15.Coelho, F. M., V. Pinho, F. A. Amaral, D. Sachs, V. V. Costa, D. H. Rodrigues, A. T. Vieira, T. A. Silva, D. G. Souza, R. Bertini, A. L. Teixeira, and M. M. Teixeira. 2008. The chemokine receptors CXCR1/CXCR2 modulate antigen-induced arthritis by regulating adhesion of neutrophils to the synovial microvasculature. Arthritis Rheum. 58:2329-2337. [DOI] [PubMed] [Google Scholar]
- 16.Damsker, J. M., I. Okwumabua, T. Pushkarsky, K. Arora, M. I. Bukrinsky, and S. L. Constant. 2009. Targeting the chemotactic function of CD147 reduces collagen-induced arthritis. Immunology 126:55-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Endlich, B., D. Armstrong, J. Brodsky, M. Novotny, and T. A. Hamilton. 2002. Distinct temporal patterns of macrophage-inflammatory protein-2 and KC chemokine gene expression in surgical injury. J. Immunol. 168:3586-3594. [DOI] [PubMed] [Google Scholar]
- 18.Eyles, J. L., M. J. Hickey, M. U. Norman, B. A. Croker, A. W. Roberts, S. F. Drake, W. G. James, D. Metcalf, I. K. Campbell, and I. P. Wicks. 2008. A key role for G-CSF-induced neutrophil production and trafficking during inflammatory arthritis. Blood 112:5193-5201. [DOI] [PubMed] [Google Scholar]
- 19.Fujiwara, K., S. Ohkawara, K. Takagi, M. Yoshinaga, and A. Matsukawa. 2002. Involvement of CXC chemokine growth-related oncogene-alpha in monosodium urate crystal-induced arthritis in rabbits. Lab. Invest. 82:1297-1304. [DOI] [PubMed] [Google Scholar]
- 20.Grespan, R., S. Y. Fukada, H. P. Lemos, S. M. Vieira, M. H. Napimoga, M. M. Teixeira, A. R. Fraser, F. Y. Liew, I. B. McInnes, and F. Q. Cunha. 2008. CXCR2-specific chemokines mediate leukotriene B4-dependent recruitment of neutrophils to inflamed joints in mice with antigen-induced arthritis. Arthritis Rheum. 58:2030-2040. [DOI] [PubMed] [Google Scholar]
- 21.Guerau-de-Arellano, M., J. Alroy, D. Bullard, and B. T. Huber. 2005. Aggravated Lyme carditis in CD11a−/− and CD11c−/− mice. Infect. Immun. 73:7637-7643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Halloran, M. M., J. M. Woods, R. M. Strieter, Z. Szekanecz, M. V. Volin, S. Hosaka, G. K. Haines III, S. L. Kunkel, M. D. Burdick, A. Walz, and A. E. Koch. 1999. The role of an epithelial neutrophil-activating peptide-78-like protein in rat adjuvant-induced arthritis. J. Immunol. 162:7492-7500. [PubMed] [Google Scholar]
- 23.Issekutz, A. C., J. Y. Mu, G. Liu, J. Melrose, and E. L. Berg. 2001. E-selectin, but not P-selectin, is required for development of adjuvant-induced arthritis in the rat. Arthritis Rheum. 44:1428-1437. [DOI] [PubMed] [Google Scholar]
- 24.Jacobs, J. P., A. Ortiz-Lopez, J. J. Campbell, C. J. Gerard, D. Mathis, and C. Benoist. 2010. Deficiency of CXCR2, but not other chemokine receptors, attenuates autoantibody-mediated arthritis in a murine model. Arthritis Rheum. 62:1921-1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jia, N., U. Semba, H. Nishiura, A. Kuniyasu, T. K. Nsiama, N. Nishino, and T. Yamamoto. 2010. Interconversion between pure chemotactic ligands and chemoattractant/secretagogue ligands of neutrophil C5a receptor by a single amino acid substitution. J. Leukoc. Biol. 87:965-975. [DOI] [PubMed] [Google Scholar]
- 26.Kenefick, K. B., J. A. Lederer, R. F. Schell, and C. J. Czuprynski. 1992. Borrelia burgdorferi stimulates release of interleukin-1 activity from bovine peripheral blood monocytes. Infect. Immun. 60:3630-3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koch, A. E. 2005. Chemokines and their receptors in rheumatoid arthritis: future targets? Arthritis Rheum. 52:710-721. [DOI] [PubMed] [Google Scholar]
- 28.Koch, A. E., S. L. Kunkel, L. A. Harlow, D. D. Mazarakis, G. K. Haines, M. D. Burdick, R. M. Pope, A. Walz, and R. M. Strieter. 1994. Epithelial neutrophil activating peptide-78: a novel chemotactic cytokine for neutrophils in arthritis. J. Clin. Invest. 94:1012-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koch, A. E., S. L. Kunkel, M. R. Shah, S. Hosaka, M. M. Halloran, G. K. Haines, M. D. Burdick, R. M. Pope, and R. M. Strieter. 1995. Growth-related gene product α. A chemotactic cytokine for neutrophils in rheumatoid arthritis. J. Immunol. 155:3660-3666. [PubMed] [Google Scholar]
- 30.Kraan, M., D. Patel, J. Haringman, M. Smith, H. Weedon, M. Ahern, F. Breedveld, and P. Tak. 2001. The development of clinical signs of rheumatoid synovial inflammation is associated with increased synthesis of the chemokine CXCL8 (interleukin-8). Arthritis Res. 3:65-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lemos, H. P., R. Grespan, S. M. Vieira, T. M. Cunha, W. A. Verri, K. S. S. Fernandes, F. O. Souto, I. B. McInnes, S. H. Ferreira, F. Y. Liew, and F. Q. Cunha. 2009. Prostaglandin mediates IL-23/IL-17-induced neutrophil migration in inflammation by inhibiting IL-12 and IFNγ production. Proc. Natl. Acad. Sci. U. S. A. 106:5954-5959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lomas, J. L., C. S. Chung, P. S. Grutkoski, B. W. LeBlanc, L. Lavigne, J. Reichner, S. H. Gregory, L. A. Doughty, W. G. Cioffi, and A. Ayala. 2003. Differential effects of macrophage inflammatory chemokine-2 and keratinocyte-derived chemokine on hemorrhage-induced neutrophil priming for lung inflammation: assessment by adoptive cells transfer in mice. Shock 19:358-365. [DOI] [PubMed] [Google Scholar]
- 33.Lusitani, D., S. E. Malawista, and R. R. Montgomery. 2003. Calprotectin, an abundant cytosolic protein from human polymorphonuclear leukocytes, inhibits the growth of Borrelia burgdorferi. Infect. Immun. 71:4711-4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma, Y., K. P. Seiler, E. J. Eichwald, J. H. Weis, C. Teuscher, and J. J. Weis. 1998. Distinct characteristics of resistance to Borrelia burgdorferi-induced arthritis in C57BL/6N mice. Infect. Immun. 66:161-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mackay, C. R. 2008. Moving targets: cell migration inhibitors as new anti-inflammatory therapies. Nat. Immunol. 9:988-998. [DOI] [PubMed] [Google Scholar]
- 36.Min, S. H., Y. Wang, W. Gonsiorek, G. Anilkumar, J. Kozlowski, D. Lundell, J. S. Fine, and E. P. Grant. 2010. Pharmacological targeting reveals distinct roles for CXCR2/CXCR1 and CCR2 in a mouse model of arthritis. Biochem. Biophys. Res. Commun. 391:1080-1086. [DOI] [PubMed] [Google Scholar]
- 37.Montgomery, R. R., D. Lusitani, A. B. Chevance Ad, and S. E. Malawista. 2002. Human phagocytic cells in the early innate immune response to Borrelia burgdorferi. J. Infect. Dis. 185:1773-1779. [DOI] [PubMed] [Google Scholar]
- 38.Montgomery, R. R., C. J. Booth, X. Wang, V. A. Blaho, S. E. Malawista, and C. R. Brown. 2007. Recruitment of macrophages and polymorphonuclear leukocytes in Lyme carditis. Infect. Immun. 75:613-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morrison, T. B., Y. Ma, J. H. Weis, and J. J. Weis. 1999. Rapid and sensitive quantification of Borrelia burgdorferi-infected mouse tissues by continuous fluorescent monitoring of PCR. J. Clin. Microbiol. 37:987-992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morrison, T. B., J. H. Weis, and J. J. Weis. 1997. Borrelia burgdorferi outer surface protein A (OspA) activates and primes human neutrophils. J. Immunol. 158:4838-4845. [PubMed] [Google Scholar]
- 41.Pahl, A., U. Kuhlbrandt, K. Brune, M. Rollinghoff, and A. Gessner. 1999. Quantitative detection of Borrelia burgdorferi by real-time PCR. J. Clin. Microbiol. 37:1958-1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Palmer, G., N. Busso, M. Aurrand-Lions, D. Talabot-Ayer, V. Chobaz-Peclat, C. Zimmerli, P. Hammel, B. Imhof, and C. Gabay. 2007. Expression and function of junctional adhesion molecule-C in human and experimental arthritis. Arthritis Res. Ther. 9:1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pillinger, M. H., and S. B. Abramson. 1995. The neutrophil in rheumatoid arthritis. Rheum. Dis. Clin. North Am. 21:691-714. [PubMed] [Google Scholar]
- 44.Rovai, L. E., H. R. Herschman, and J. B. Smith. 1998. The murine neutrophil-chemoattractant chemokines LIX, KC, and MIP-2 have distinct induction kinetics, tissue distributions, and tissue-specific sensitivities to glucocorticoid regulation in endotoxemia. J. Leukoc. Biol. 64:494-502. [DOI] [PubMed] [Google Scholar]
- 45.Ruderman, E. M., J. S. Kerr, S. R. Telford III, A. Spielman, L. H. Glimcher, and E. M. Gravallese. 1995. Early murine Lyme carditis has a macrophage predominance and is independent of major histocompatibility complex class II-CD4+ T cell interactions. J. Infect. Dis. 171:362-370. [DOI] [PubMed] [Google Scholar]
- 46.Santos, L. L., E. F. Morand, P. Hutchinson, N. W. Boyce, and S. R. Holdsworth. 1997. Anti-neutrophil monoclonal antibody therapy inhibits the development of adjuvant arthritis. Clin. Exp. Immunol. 107:248-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schrier, D., R. B. Gilbertsen, M. Lesch, and J. Fantone. 1984. The role of neutrophils in type II collagen-induced arthritis in rats. Am. J. Pathol. 117:26-29. [PMC free article] [PubMed] [Google Scholar]
- 48.Schrier, D. J., R. C. Schimmer, C. M. Flory, D. K. Tung, and P. A. Ward. 1998. Role of chemokines and cytokines in a reactivation model of arthritis in rats induced by injection with streptococcal cell walls. J. Leukoc. Biol. 63:359-363. [DOI] [PubMed] [Google Scholar]
- 49.Soehnlein, O., L. Lindbom, and C. Weber. 2009. Mechanisms underlying neutrophil-mediated monocyte recruitment. Blood 114:4613-4623. [DOI] [PubMed] [Google Scholar]
- 50.Steere, A. C., W. P. Batsford, M. Weinberg, J. Alexander, H. J. Berger, S. Wolfson, and S. E. Malawista. 1980. Lyme carditis: cardiac abnormalities of Lyme disease. Ann. Intern. Med. 93:8-16. [DOI] [PubMed] [Google Scholar]
- 51.Szekanecz, Z., and A. Koch. 2008. Vascular involvement in rheumatic diseases: ‘vascular rheumatology.’ Arthritis Res. Ther. 10:224-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vergunst, C. E., D. M. Gerlag, L. Lopatinskaya, L. Klareskog, M. D. Smith, F. van den Bosch, H. J. Dinant, Y. Lee, T. Wyant, E. W. Jacobson, D. Baeten, and P. P. Tak. 2008. Modulation of CCR2 in rheumatoid arthritis: a double-blind, randomized, placebo-controlled clinical trial. Arthritis Rheum. 58:1931-1939. [DOI] [PubMed] [Google Scholar]
- 53.Weissmann, G., and H. Korchak. 1984. Rheumatoid arthritis. The role of neutrophil activation. Inflammation 8(Suppl.):S3-S14. [DOI] [PubMed] [Google Scholar]
- 54.Wipke, B. T., and P. M. Allen. 2001. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J. Immunol. 167:1601-1608. [DOI] [PubMed] [Google Scholar]
- 55.Xu, Q., S. V. Seemanapalli, K. E. Reif, C. R. Brown, and F. T. Liang. 2007. Increasing the recruitment of neutrophils to the site of infection dramatically attenuates Borrelia burgdorferi infectivity. J. Immunol. 178:5109-5115. [DOI] [PubMed] [Google Scholar]
- 56.Zhou, J. S., W. Xing, D. S. Friend, K. F. Austen, and H. R. Katz. 2007. Mast cell deficiency in KitW-sh mice does not impair antibody-mediated arthritis. J. Exp. Med. 204:2797-2802. [DOI] [PMC free article] [PubMed] [Google Scholar]