

Abstract
Group A Streptococcus (GAS) is a common causative agent of pharyngitis, but the role of GAS in otitis media is underappreciated. In this study, we sought to test the hypothesis that GAS colonizes the middle ear and establishes itself in localized, three-dimensional communities representative of biofilms. To test this hypothesis, the middle ears of chinchillas were infected with either a strain of GAS capable of forming biofilms in vitro (MGAS5005) or a strain deficient in biofilm formation due to the lack of the transcriptional regulator Srv (MGAS5005 Δsrv). Infection resulted in the formation of large, macroscopic structures within the middle ears of MGAS5005- and MGAS5005 Δsrv-infected animals. Plate counts, scanning electron microscopy, LIVE/DEAD staining, and Gram staining revealed a difference in the distributions of MGAS5005 versus MGAS5005 Δsrv in the infected samples. High numbers of CFU of MGAS5005 Δsrv were isolated from the middle ear effusion, and MGAS5005 Δsrv was found randomly distributed throughout the excised macroscopic structure. In contrast, MGAS5005 was found in densely packed microcolonies indicative of biofilms within the excised material from the middle ear. CFU levels of MGAS5005 from the effusion were significantly lower than that of MGAS5005 Δsrv early during the course of infection. Allelic replacement of the chromosomally encoded streptococcal cysteine protease (speB) in the MGAS5005 Δsrv background restored biofilm formation in vivo. Interestingly, our results suggest that GAS naturally forms a biofilm during otitis media but that biofilm formation is not required to establish infection following transbullar inoculation of chinchillas.
Group A Streptococcus (GAS) is a Gram-positive pathogen that is capable of causing a variety of infections in the human host. These infections range from mild illnesses such as pharyngitis and impetigo to diseases of greater severity, including necrotizing fasciitis and streptococcal toxic shock syndrome (11, 19, 32, 41, 43). Thus, the organism has evolved to colonize a number of physiologically distinct host sites. The role of GAS in otitis media (OM) is often underappreciated due to the effectiveness of beta-lactam antibiotics in elimination of GAS; however, there have been studies showing GAS to be one of the top 4 causative agents of OM infection and, more importantly, of development of OM complications such as acute mastoiditis (19, 35, 43). Mastoiditis is a severe bacterial infection of the mastoid bone and air cell system, often requiring surgical intervention and aggressive antibiotic treatment (40). Various studies have reported GAS to be one of the leading causes of severe mastoiditis (28, 40, 42), and an increase in the number of GAS mastoiditis cases has occurred since administration of the heptavalent pneumococcal conjugate vaccine in the years 2001 to 2005 (40). A 5-year study of 11,311 acute OM episodes found that 3.1% were positive for GAS and that 4 of the 346 GAS acute OM cases developed into acute mastoiditis (41). The mechanism by which GAS transitions from colonizing the middle ear to colonizing the mastoid process is unclear.
One mechanism which may contribute to GAS colonization of the middle ear is the ability to form biofilms. As hypothesized by Donlan and Costerton, a biofilm is a bacterial sessile community encased in a matrix of extracellular polymeric substances and attached to a substratum or interface (15). Biofilms are believed to be inherently tolerant to host defenses and antibiotic therapies and are often linked to chronic illness due to impaired clearance (17, 18). There are some estimates suggesting that nearly 60% of all bacterial infections, including those resulting in dental caries, periodontitis, otitis media, chronic tonsillitis, endocarditis, necrotizing fasciitis, and other disease manifestations, involve biofilms (12, 15).
There is a growing understanding of the importance of biofilm formation to GAS (1, 4, 6, 8, 14, 26, 29, 30). Recently, we sought insight into how GAS regulates biofilm formation by testing the hypothesis that inactivation of the streptococcal regulator of virulence (srv) would alter the GAS biofilm phenotype (14). We found that loss of Srv resulted in a significant reduction in the ability of MGAS5005, an invasive serotype M1T1 clone of GAS, to form biofilms under static and continuous-flow conditions in vitro (14). One of the phenotypes of MGAS5005 Δsrv, the srv mutant strain, is the constitutive production of an active cysteine protease, SpeB (13, 14, 36). Chemical inhibition of SpeB restored the biofilm phenotype of MGAS5005 Δsrv to wild-type levels in vitro (14). Furthermore, MGAS5005 Δsrv ΔspeB formed biofilms at wild-type levels (A. L. Roberts, submitted for publication). Taken together, these data led us to hypothesize that Srv-regulated production of SpeB may be one mechanism by which GAS can actively disperse the biofilm.
We still have much to learn about the makeup of the GAS biofilm, how it is regulated, and how it contributes to virulence at physiologically distinct host sites. In this study, we sought to examine the role of GAS biofilm formation in vivo in a chinchilla model of otitis media. Based on our previous findings, we hypothesized that MGAS5005 Δsrv would not form biofilms in vivo. Furthermore, we hypothesized that the lack of biofilm formation would lead to enhanced clearance of the infection. The majority of animals inoculated with either MGAS5005 or MGAS5005 Δsrv developed an infection that resulted in inflammation and was accompanied by effusion and the formation of a dense macroscopic structure within the middle ear chamber. Counter to our hypothesis, MGAS5005 Δsrv-infected animals had a significantly higher bacterial load present in the effusion early in the course of the infection. Microscopic analysis identified MGAS5005 present in microcolonies reminiscent of biofilms within the macroscopic structure, while MGAS5005 Δsrv was found to be dispersed throughout the infected samples. Interestingly, we found that allelic replacement of the chromosomally encoded cysteine protease (speB) in the MGAS5005 Δsrv background (MGAS5005 Δsrv ΔspeB) restored biofilm formation in vivo. Taken together, our results suggest that GAS naturally forms a biofilm during otitis media but that biofilm formation is not required to establish infection following transbullar inoculation of chinchillas. Furthermore, these data provide additional support for a role for Srv in the establishment of GAS biofilms in vivo and suggest that SpeB is involved in biofilm dispersal.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
MGAS5005, a clinical group A Streptococcus M1T1 isolate obtained from an invasive form of infection, was utilized in this study (33, 37). MGAS5005 Δsrv, the isogenic srv mutant strain, was generated by allelic replacement (39). MGAS5005 Δsrv ΔspeB was generated by allelic replacement of speB in the MGAS5005 Δsrv background (Roberts, submitted). Cultures were grown in Todd-Hewitt broth (Becton-Dickinson) supplemented with 2% yeast extract (THY) (Fisher Scientific) at 37°C in a 5% CO2 atmosphere overnight and diluted in pyrogen-free phosphate-buffered saline (PBS) prior to infection. Serial dilutions were plated onto THY agar plates.
Chinchilla infections.
Studies were approved by the Animal Care and Use Committee of Wake Forest University Health Sciences. Healthy adult chinchillas (Chinchilla lanigera) were purchased from Rauscher's Chinchilla Ranch and allowed to acclimate to the animal holding facility for 5 or 6 days prior to infection. None of the animals exhibited any signs of otitis media or any other overt disease. The infectious dose was verified through bacterial plate counts. Chinchillas were anesthetized with isoflurane and inoculated via transbullar injection with 0.2 ml of bacterial suspension containing a total range of 5 × 104 to 2.5 × 105 CFU.
Otoscopy images were captured by digital imaging prior to infection and at 2, 4, and 7 days postinfection (dpi). Images were assessed for indicators of inflammation, including vessel dilation, fluid accumulation, and opacity of the tympanic membrane (22). Groups of animals were euthanized at 2, 4, or 7 dpi, and the superior bullae were opened to expose the middle ear cavity as previously described (22). The presence of surface-attached material was assessed by visual inspection and photographed. When present, this material was removed and analyzed as described below. Subsets of the macroscopic structures were weighed, suspended in 1 ml of sterile PBS, and homogenized using a FastPrep-24 lysing matrix D system (MP Biomedicals) for 40 s at speed 6 in a FastPrep FP-120 cell disruptor (Thermo Electron Corp.). The homogenized suspension was serially diluted and plated. Effusion material was removed from the middle ear cavity, and the middle ear cavity was then washed with 0.5 ml of sterile PBS. Wash and effusion fluids were combined and equalized to form a total volume of 1 ml and serially diluted. Bacterial loads (measured in numbers of CFU per milliliter) were determined by plate counts.
Scanning electron microscopy (SEM).
The excised surface-attached communities were fixed for 1 h with 2.5% glutaraldehyde-PBS and then rinsed twice (for 10 min per wash) in PBS prior to dehydration in a graded ethanol series. The samples were then subjected to critical point drying, mounted onto stubs, and sputter coated with palladium prior to being viewed with a Philips SEM-515 scanning electron microscope.
LIVE/DEAD staining.
The material isolated from the middle ear was analyzed for viability by LIVE/DEAD BacLight bacterial viability kit (Molecular Probes) staining at 7 dpi. The surface-attached communities were incubated in 0.5 ml of PBS containing a mixture of equal amounts of SYTO 9 and propidium iodide for 25 min and rinsed in PBS 3 times. Samples were visualized using a Zeiss LSM 510 confocal laser-scanning microscope (CLSM) and Zeiss LSM Image Browser software.
H&E staining and Gram staining.
Fresh macroscopic structures were snap-frozen in OCT resin (Sakura Finetek) and stored at −80°C. Samples were acclimated to −20°C, cut into 10-μm-thick sections with a cryotome, and placed onto positively charged microscope slides (Fisher Scientific). The sample sections were stored at −20°C until processing was performed with a standard hematoxylin and eosin (H&E) protocol, utilizing Harris's hematoxylin formula and eosin-phloxine. Gram-stain processing was conducted utilizing Taylor's Brown-Brenn-modified Gram stain. Samples were analyzed with a Nikon Eclipse TE300 light microscope (Nikon) and were blind scored. Images were taken using a QImaging Retiga-EXi camera (AES) and stored using ImageJ software.
RESULTS
Defining the GAS biofilm.
To assist in the interpretation of the results that follow, we briefly discuss how a GAS biofilm may be defined. We consider a GAS biofilm to be a three-dimensional clustering of the bacteria into microcolonies that are nonrandomly distributed at the infected host site. While there are undoubtedly a matrix of bacterial components and possibly host-derived components necessary for the formation of these microcolonies, the microcolonies themselves may be present in a larger macroscopic structure that primarily consists of host-derived products such as neutrophils, macrophages, fibrin, fibronectin, etc. For the purposes of this study, we consider the presence of a microcolony to be evidence of GAS biofilm formation. As our understanding of the GAS biofilm evolves, our GAS biofilm definition will expand.
GAS infection of the chinchilla middle ear resulted in inflammation, effusion, and the formation of visible macroscopic structures.
To begin to study GAS biofilm formation in an in vivo model of OM, cohorts of chinchillas were infected with MGAS5005 or MGAS5005 Δsrv. A subset of 20 animals was assessed by digital otoscopy prior to infection and at 2, 4, and 7 dpi. Prior to infection, each of the animals was observed to be free of middle ear disease. At each of the time points postinfection, the majority of ears showed visible signs of tympanic membrane and inner ear inflammation (Fig. 1). Removal of the superior bulla revealed that this inflammation coincided with the presence of macroscopic structures within the middle ear cavity (Fig. 2, Table 1). In addition, the presence of serous middle ear fluid, or effusion, was observed. In all cases, the macroscopic structures were dense enough to permit physical removal with forceps. Structures were removed, the effusion was collected, and the middle ears were washed with PBS. The effusion and wash, hereafter referred to as the effusion, were combined, brought up to a 1-ml total volume, and plated. A statistically higher number of CFU per milliliter (P < 0.05) was recovered from the effusion material from MGAS5005 Δsrv-infected ears at 2 and 4 dpi (Fig. 3 A). The average numbers of CFU recovered from the effusion by 7 dpi were roughly equivalent (∼1 × 108) for MGAS5005- and MGAS5005 Δsrv-infected animals (Fig. 3A). However, more animals had cleared the MGAS5005 from the effusion by 7 dpi (Fig. 3A). Homogenization and plating of excised macroscopic structures revealed a trend in bacterial counts similar to that observed for the numbers of CFU recovered from the effusion, although the bacterial load decreased over time (Fig. 3B). While the results were not statistically significant, survival analysis indicated a larger number of MGAS5005 Δsrv-infected animals had succumbed to infection (Fig. 3C).
FIG. 1.

Digital otoscopy of representative MGAS5005- and MGAS5005 Δsrv-infected animals at time 0 (uninfected) and at 2, 4, and 7 dpi. MGAS5005-infected ears showed an inflammatory response accompanied by inflammation by 2 dpi. Redness and swelling remained and effusion developed over the course of the infection. By 2 dpi, MGAS5005 Δsrv-infected ears showed signs of redness and swelling accompanied by effusion and an air-fluid level at the tympanic membrane. Otorrhoea was visible at 7 dpi.
FIG. 2.

GAS infection leads to the development of macroscopically visible surface-attached structures in the chinchilla middle ear. Images show representative middle ear chambers of euthanized animals. (A) PBS (mock)-infected middle ear; (B) MGAS5005; (C) MGAS5005 Δsrv. Arrows indicate the visible structures for panels B and C.
TABLE 1.
Frequency of macroscopic structure formation over time following infection with GAS
| dpia | No. (%) of infected ears showing formation of visible structure/total no. of infected ears |
|
|---|---|---|
| MGAS5005 | MGAS5005 Δsrv | |
| 2 | 12/20 (60) | 16/17 (94) |
| 4 | 20/24 (83) | 21/22 (95) |
| 7 | 22/33 (66) | 20/25 (80) |
dpi, day postinfection.
FIG. 3.

Increased clearance of MGAS5005, but not MGAS5005 Δsrv, was observed over the course of experimental chinchilla otitis media within effusion samples. (A) Higher concentrations (CFU per milliliter) of MGAS5005 Δsrv were recovered from middle ear effusions and washes. Conversely, more middle ear effusion samples were cleared of MGAS5005 infection over time. Statistics were determined by using an unpaired Student t test. (B) Bacterial concentrations (CFU per gram) recovered from middle ear macroscopic structures. Higher concentrations of MGAS5005 Δsrv were recovered from homogenized macroscopic structures at 2 and 4 dpi. At 7 dpi, macroscopic structures recovered from all 8 MGAS5005-infected ears were positive for GAS. Only 2 structures recovered from MGAS5005 Δsrv-infected ears were positive for GAS at 7 dpi. Of note, 2 MGAS5005 Δsrv-infected animals had to be euthanized before the day 7 time point was reached. *, P ≤ 0.05 at 4 dpi. (C) Kaplan-Meier survival curves showing the relative rates of chinchilla mortality caused by MGAS5005 and MGAS5005 Δsrv. Two groups of 21 (MGAS5005) and 21 (MGAS5005 Δsrv) animals received ∼2 × 105 CFU/ml by transbullar inoculation. While the results were not statistically significant (Wilcoxon test; P value ≤ 0.0755), more animals succumbed to MGAS5005 Δsrv infection. All remaining animals were euthanized at day 7.
GAS and host components are detected within macroscopic structures.
As an initial step toward understanding the composition of the material observed, scanning electron microscopy was used to analyze the macroscopic structures excised from the middle ear. As a comparison, we also analyzed a 24-h in vitro MGAS5005 biofilm grown under conditions of continuous flow (14). The in vitro biofilm consisted of densely packed chains of GAS, with each cell measuring approximately 0.5 to 1.0 μm in diameter (Fig. 4 A). Close examination revealed that the chains were coated with a matrix-like material of various thicknesses (Fig. 4A). In the MGAS5005 in vivo samples, we also observed three-dimensional communities or groupings of chains of cocci that morphologically resembled GAS microcolonies (Fig. 4B and C). These MGAS5005 communities were reminiscent of the biofilm observed in vitro. In contrast, we were able to identify only individual chains or pairs of cocci in the MGAS5005 Δsrv samples (Fig. 4D and E). We take this as evidence of MGAS5005 biofilms embedded within the macroscopic structures.
FIG. 4.

SEM reveals GAS biofilms embedded within the macroscopic structures recovered from MGAS5005-infected middle ears. (A) GAS biofilm grown in an in vitro continuous-flow cell. Chains of cocci coated in a matrix material are visible. (B and C) White arrows demonstrate chains of cocci that are consistent in size and length with the presence of GAS. Note the seemingly complex matrix, three-dimensional community structure, and other cellular features present. Images were taken at 2 dpi (B) and 7 dpi (C). (D and E) Only isolated chains of cocci were identified in MGAS5005 Δsrv-infected samples at 2 (D) and 7 (E) dpi. Scale bars, 10 μm.
Viability staining revealed differences in the distributions of MGAS5005 and MGAS5005 Δsrv.
To further examine the distribution of GAS within the macroscopic structures, unfixed material was recovered 7 dpi and stained using a LIVE/DEAD BacLight bacterial viability kit (Molecular Probes). In this method, live cells with intact membranes were stained green with SYTO 9, while dead cells, nuclei, and DNA were stained red with propidium iodide. Colocalization of both signals was represented by yellow staining. In both MGAS5005- and MGAS5005 Δsrv-infected samples, dead cells and associated materials were distributed throughout the field of view and stained at various intensities (Fig. 5 A and D). However, living cells in the MGAS5005 sample were nonrandomly distributed and localized to a distinct community resembling an intact biofilm. Vertically stacked Z-slice images (Fig. 5C, margins) revealed that this community had a three-dimensional structure with living cells present throughout. In the MGAS5005 Δsrv-infected sample, a dense structure of cells was also observed (Fig. 5E), but it was not as organized. Overlap of the two fluorescent signals indicated the structure was composed of more dead than living cells (Fig. 5F). However, a large number of living cells was randomly distributed throughout the field of view in the MGAS5005 Δsrv-infected sample (Fig. 5F). The images provided here are representative of what was observed.
FIG. 5.

LIVE/DEAD viability staining of material recovered from the chinchilla middle ear following GAS infection. Material was recovered 7 dpi with MGAS5005 (A to C) or MGAS5005 Δsrv (D to F) and stained for a two-color fluorescence assay of bacterial viability (LIVE/DEAD BacLight; Molecular Probes). Nonviable cells with damaged membranes are depicted in red (A and D), and cells with intact membranes stained green (B and E). Overlap of both signals appears as yellow staining (C and F). A horizontal Z-slice image (center) and vertically stacked images (margins) show living MGAS5005 bacteria in a concentrated community suggestive of a biofilm (C); alternatively, MGAS5005 Δsrv was found dispersed throughout the macroscopic structure depicted in Fig. 2 (F). Zeiss LSM Image Browser software three-dimensional distance analysis predicted the thickness of the MGAS5005 community to be ∼680 μm (C) and that the MGAS5005 Δsrv community to be ∼696 (F) μm, as indicated by the lines in the respective panels.
Gram staining revealed differences between MGAS5005- and MGAS5005 Δsrv-infected samples with respect to microcolony formation.
To further investigate the apparent differences between the structures of the MGAS5005 and MGAS5005 Δsrv populations in vivo, H&E staining and Gram staining were used. H&E staining revealed abundant polymorphonuclear leukocytes (blue) and fibrin (pink) within sections of the macroscopic material removed from the chinchilla middle ear (Fig. 6 A, C, E, and G). Blinded analysis revealed that the degrees of cellularity and fibrin composition exhibited by the MGAS5005 and MGAS5005 Δsrv samples examined were approximately equivalent. However, Gram staining revealed densely packed and darkly stained microcolonies of bacteria within the MGAS5005-infected sample at 7 dpi (Fig. 6B and D). Such microcolonies have previously been interpreted as evidence of the presence of a biofilm (9, 10, 15, 45). In comparison, Gram-stained MGAS5005 Δsrv was diffused throughout the samples examined (Fig. 6F and H). While microcolony formation was not absent, fewer and less-dense microcolonies were observed, as is consistent with the differences observed by SEM and LIVE/DEAD staining.
FIG. 6.

H&E staining (A, C, E, and G) and Gram staining (B, D, F, and H) of macroscopic structures removed at 7 dpi from the chinchilla middle ear cavity. MGAS5005 is represented in the top two rows, and MGAS5005 Δsrv is represented in the bottom two rows. H&E staining allows detection of host fibrin and polymorphonuclear leukocyte infiltration, whereas Gram staining allows detection of the Gram-staining phenotype, shape, and groupings of the bacteria. Magnification, ×20. Note the abundant microcolonies present in the MGAS5005 Gram-stained samples (B and D), whereas the bacteria appear more dispersed in the MGAS5005 Δsrv Gram-stained samples (F and H).
Allelic replacement of speB in the MGAS5005 Δsrv background restored biofilm formation.
Previously, we provided evidence that constitutive production of SpeB by MGAS5005 Δsrv led to a decrease in biofilm formation in vitro (14). More recently, we have shown that allelic replacement of speB in the MGAS5005 Δsrv background restores biofilm formation in vitro (Roberts, submitted). Given the observations described above, we sought to directly address whether loss of speB would restore the ability of MGAS5005 Δsrv to form biofilms. Macroscopic structure development and effusion were observed in the middle ears of animals infected with MGAS5005 Δsrv ΔspeB (Fig. 7 A). As seen with the MGAS5005-infected samples, SEM revealed the presence of three-dimensional MGAS5005 Δsrv ΔspeB microcolonies embedded within the macroscopic structures (Fig. 7B).
FIG. 7.

SEM and Gram staining reveals GAS biofilms embedded within the macroscopic structures recovered from MGAS5005 Δsrv ΔspeB-infected middle ears. (A) Middle ear chamber of a euthanized chinchilla infected with MGAS5005 Δsrv ΔspeB filled with a visible macroscopic structure (black arrow). (B) SEM of a representative 2 dpi macroscopic structure. Notice the clustering of chains of cocci into microcolonies (white arrow). (C) H&E staining (left column) and Gram staining (right column) of sections from two representative 7 dpi macroscopic structures. Notice the presence of host fibrin and numerous Gram-positive microcolonies (white and black arrows). Magnification, ×20.
Gram staining further confirmed the presence of MGAS5005 Δsrv ΔspeB microcolonies in the infected samples (Fig. 7C). Finally, viable MGAS5005 Δsrv ΔspeB was recovered from both the effusion and macroscopic structures (Fig. 8).
FIG. 8.
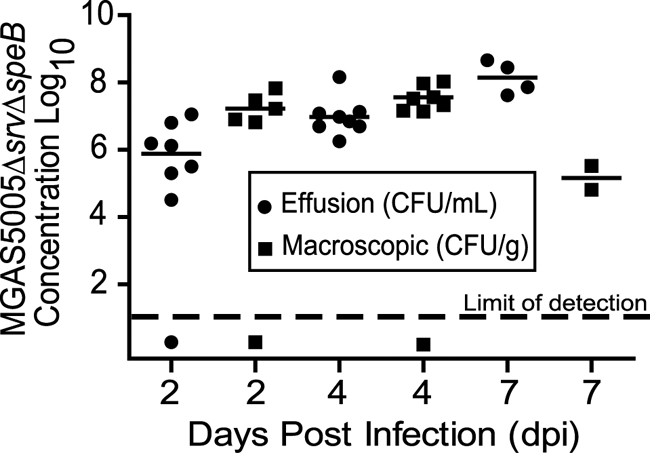
MGAS5005 Δsrv ΔspeB was maintained within middle ear effusion and macroscopic structures over the course of infection. Animal euthanasia reduced the number of samples recovered at 7 dpi.
DISCUSSION
In this study, we sought to test the hypothesis that GAS colonizes the middle ear in a biofilm. The chinchilla animal model has been a useful tool for studying in vivo biofilm formation (2, 3, 15, 22, 38). Recently, the chinchilla model was used to investigate Streptococcus pneumoniae biofilm formation in vivo (38). In that study, middle ear effusion and inflammation characteristic of OM were observed along with macroscopic structures that were indistinguishable by the naked eye from the structures observed in this study (38). Similar effusion, inflammation, and structure characteristics were also observed following infection with Haemophilus influenzae (22). Thus, the formation of these structures appears to often accompany the onset of otitis media in this model; however, our data indicate that the presence of these macroscopic structures does not determine the distribution of the infecting organism within the structure. That is, the pathogen may be aggregated into microcolonies or biofilms or may be more randomly distributed throughout the structure and the accompanying effusion.
In the S. pneumoniae study, biofilm formation correlated with resistance to clearance by the host immune system (38). Given that MGAS5005 Δsrv does not form biofilms in vitro (14), we hypothesized that we would see decreased biofilm formation by MGAS5005 Δsrv and an increased rate of clearance in vivo. While we did note that biofilm formation was decreased or absent, animals infected with MGAS5005 Δsrv had higher bacterial loads in middle ear effusions until 7 dpi and significantly higher bacterial loads in the effusion at 2 and 4 dpi (Fig. 3A). Furthermore, animals infected with MGAS5005 Δsrv had a lower rate of clearance of the organism from the effusion than those infected with MGAS5005 (Fig. 3A). While the data did not reach statistical significance, the rate of mortality for those animals infected with MGAS5005 Δsrv was also greater (Fig. 3C).
In contrast, a different phenotype was observed in the MGAS5005-infected samples. Plate counts of the homogenized macroscopic structures revealed the presence of MGAS5005 in all 8 of the experimentally infected ears at 7 dpi (Fig. 3B). A combination of SEM with LIVE/DEAD and Gram staining revealed the presence of three-dimensional aggregates of cells or microcolonies indicative of biofilms within the macroscopic structures removed from the middle ears of MGAS5005-infected samples. For GAS, evidence of biofilm formation as dense clusters of bacteria or microcolonies in zebrafish muscle tissue has been previously presented (6). Microcolonies have also been previously presented as evidence of GAS biofilms on human cells in vitro (30). These microcolonies were largely absent in the analyzed MGAS5005 Δsrv-infected samples. Rather, the MGAS5005 Δsrv organisms were randomly distributed throughout the macroscopic structure and dispersed into the effusion. This is perhaps best shown in the LIVE/DEAD stained samples (Fig. 5), in which living MGAS5005 cells were clustered into a densely packed three-dimensional core compared to the randomly distributed viable MGAS5005 Δsrv organisms. This is not to say that some clustering of MGAS5005 Δsrv was not observed, but it was found in dispersed form far more frequently than MGAS5005. Thus, the MGAS5005 Δsrv structures are less adherent, less organized, and less stable. These results do not contrast with our in vitro MGAS5005 Δsrv data (14); rather, the in vivo data suggest that host components may partially rescue a biofilm-deficient mutant and likely add to the overall biofilm structural stability of a biofilm-proficient strain (21, 22, 38). We conclude that the increased number of CFU of MGAS5005 Δsrv found in the effusion were due to the decreased biofilm phenotype exhibited by this strain. Furthermore, the increase in CFU of MGAS5005 found in the effusion at 7 dpi may reflect a population density cue that signals dispersal into the effusion. We are continuing to investigate this hypothesis.
In our in vitro work examining GAS biofilm formation, we presented evidence that the significant deficiency in the ability of MGAS5005 Δsrv to form biofilms was due to constitutive production of SpeB. SpeB has previously been shown to degrade GAS proteins such as M protein and DNase (5, 34, 44) as well as host proteins, including complement, antibodies, and extracellular matrix components (7, 16, 23, 24). Further work has supported a role for SpeB in dissemination, tissue damage, and prevention of phagocytosis by polymorphonuclear leukocytes (25, 27). In addition, patients suffering from severe, invasive GAS disease have decreased levels of antibodies to SpeB compared to healthy counterparts, suggesting that individuals with low anti-SpeB antibody titers may be at a higher risk for developing severe disease (20, 31). Thus, if our hypothesis is correct, allelic replacement of speB in the MGAS5005 Δsrv background should restore biofilm formation. SEM and Gram staining revealed the presence of biofilms within the macroscopic structures recovered from MGAS5005 Δsrv ΔspeB-infected animals (Fig. 7B and C).
Taken together, we believe this work is significant on three counts. First, we have demonstrated a model for the study of GAS otitis media. While the chinchilla model itself is not new, we are unaware of GAS being used in this model previously. Transbullar infection with GAS resulted in inflammation, including vessel dilation, fluid accumulation, and opacity of the tympanic membrane associated with otitis media and similar to what has been previously described for other organisms (2, 3, 15, 22, 38). It is important to continue to develop this model to determine whether intranasal infection can lead to GAS OM. Alternatively, we are currently developing this model to study the advancement of GAS OM to the more severe disease stage of mastoiditis in an effort to understand how GAS biofilms or biofilm dispersal may be involved in this process.
Second, we have provided evidence that GAS naturally forms biofilms during otitis media infection and that these biofilms may contribute to persistence within the macroscopic structures that form as a result of the disease and immune response. However, through the MGAS5005 Δsrv data, we have also demonstrated that biofilms are not required for infection or overt disease following transbullar inoculation. Thus, it appears that GAS can exist at the site of infection in either a biofilm or nonbiofilm state. Longer time courses of infection are needed to determine whether or not the GAS biofilm does provide a resistance to overall clearance from the middle ear, but it appears that, based upon increased bacterial loads in the effusion and increased mortality, the nonbiofilm state, as seen in the MGAS5005 Δsrv-infected animals, is more virulent.
Finally, the ability of GAS to exist in a biofilm or nonbiofilm state during an active infection strongly suggests that there is a mechanism for the dispersal of the biofilm. Our previous in vitro work and the genetic evidence presented here support one hypothetical mechanism by which Srv-mediated regulation of SpeB contributes to biofilm stability or dispersal and dissemination (14; Roberts, submitted). During colonization and biofilm maturation, Srv-mediated control, whether direct or indirect, of speB/SpeB is tightly regulated, allowing little to no production of SpeB. Upon the sensing of some environmental signal(s), control is relaxed, SpeB is produced, and the biofilm is dispersed, allowing dissemination and possible disease transmission. We are continuing to explore this hypothesis.
Acknowledgments
This work was supported by Wake Forest Venture Funds and Public Health Service grant R01AI063453 from the National Institutes of Health to S.D.R.
We thank the Swords laboratory for expert technical assistance. We thank Rajendar Deora, Karen Haas, Steve Richardson, and Ed Swords (Wake Forest University School of Medicine) for helpful discussions and a detailed critique of the manuscript. Special thanks go to Dan Wozniak (Ohio State University) for his mentorship. We thank Nancy Kock for assistance with histopathology.
We declare no conflicts of interest.
Editor: J. N. Weiser
Footnotes
Published ahead of print on 30 August 2010.
REFERENCES
- 1.Akiyama, H., S. Morizane, O. Yamasaki, T. Oono, and K. Iwatsuki. 2003. Assessment of Streptococcus pyogenes microcolony formation in infected skin by confocal laser scanning microscopy. J. Dermatol. Sci. 32:193-199. [DOI] [PubMed] [Google Scholar]
- 2.Allegrucci, M., and K. Sauer. 2007. Characterization of colony morphology variants isolated from Streptococcus pneumoniae biofilms. J. Bacteriol. 189:2030-2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bakaletz, L. O., R. L. Daniels, and D. J. Lim. 1993. Modeling adenovirus type 1-induced otitis media in the chinchilla: effect on ciliary activity and fluid transport function of eustachian tube mucosal epithelium. J. Infect. Dis. 168:865-872. [DOI] [PubMed] [Google Scholar]
- 4.Baldassarri, L., R. Creti, S. Recchia, M. Imperi, B. Facinelli, E. Giovanetti, M. Pataracchia, G. Alfarone, and G. Orefici. 2006. Therapeutic failures of antibiotics used to treat macrolide-susceptible Streptococcus pyogenes infections may be due to biofilm formation. J. Clin. Microbiol. 44:2721-2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berge, A., and L. Bjorck. 1995. Streptococcal cysteine proteinase releases biologically active fragments of streptococcal surface proteins. J. Biol. Chem. 270:9862-9867. [DOI] [PubMed] [Google Scholar]
- 6.Cho, K. H., and M. G. Caparon. 2005. Patterns of virulence gene expression differ between biofilm and tissue communities of Streptococcus pyogenes. Mol. Microbiol. 57:1545-1556. [DOI] [PubMed] [Google Scholar]
- 7.Collin, M., M. D. Svensson, A. G. Sjoholm, J. C. Jensenius, U. Sjobring, and A. Olsen. 2002. EndoS and SpeB from Streptococcus pyogenes inhibit immunoglobulin-mediated opsonophagocytosis. Infect. Immun. 70:6646-6651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conley, J., M. E. Olson, L. S. Cook, H. Ceri, V. Phan, and H. D. Davies. 2003. Biofilm formation by group A streptococci: is there a relationship with treatment failure? J. Clin. Microbiol. 41:4043-4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Costerton, J. W., K. J. Cheng, G. G. Geesey, T. I. Ladd, J. C. Nickel, M. Dasgupta, and T. J. Marrie. 1987. Bacterial biofilms in nature and disease. Annu. Rev. Microbiol. 41:435-464. [DOI] [PubMed] [Google Scholar]
- 10.Costerton, J. W., Z. Lewandowski, D. E. Caldwell, D. R. Korber, and H. M. Lappin-Scott. 1995. Microbial biofilms. Annu. Rev. Microbiol. 49:711-745. [DOI] [PubMed] [Google Scholar]
- 11.Cunningham, M. W. 2000. Pathogenesis of group A streptococcal infections. Clin. Microbiol. Rev. 13:470-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dale, J. B. 1999. Multivalent group A streptococcal vaccine designed to optimize the immunogenicity of six tandem M protein fragments. Vaccine 17:193-200. [DOI] [PubMed] [Google Scholar]
- 13.Doern, C. D., R. C. Holder, and S. D. Reid. 2008. Point mutations within the streptococcal regulator of virulence (Srv) alter protein-DNA interactions and Srv function. Microbiology 154:1998-2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doern, C. D., A. L. Roberts, W. Hong, J. Nelson, S. Lukomski, W. E. Swords, and S. D. Reid. 2009. Biofilm formation by group A Streptococcus: a role for the streptococcal regulator of virulence (Srv) and streptococcal cysteine protease (SpeB). Microbiology 155:46-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Donlan, R. M., and J. W. Costerton. 2002. Biofilms: survival mechanisms of clinically relevant microorganisms. Clin. Microbiol. Rev. 15:167-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eriksson, A., and M. Norgren. 2003. Cleavage of antigen-bound immunoglobulin G by SpeB contributes to streptococcal persistence in opsonizing blood. Infect. Immun. 71:211-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fux, C. A., P. Stoodley, L. Hall-Stoodley, and J. W. Costerton. 2003. Bacterial biofilms: a diagnostic and therapeutic challenge. Expert Rev. Anti Infect. Ther. 1:667-683. [DOI] [PubMed] [Google Scholar]
- 18.Gilbert, P., J. Das, and I. Foley. 1997. Biofilm susceptibility to antimicrobials. Adv. Dent. Res. 11:160-167. [DOI] [PubMed] [Google Scholar]
- 19.Heslop, A., and T. Ovesen. 2006. Severe acute middle ear infections: microbiology and treatment. Int. J. Pediatr. Otorhinolaryngol. 70:1811-1816. [DOI] [PubMed] [Google Scholar]
- 20.Holm, S. E., A. Norrby, A. M. Bergholm, and M. Norgren. 1992. Aspects of pathogenesis of serious group A streptococcal infections in Sweden, 1988-1989. J. Infect. Dis. 166:31-37. [DOI] [PubMed] [Google Scholar]
- 21.Hong, W., R. A. Juneau, B. Pang, and W. E. Swords. 2009. Survival of bacterial biofilms within neutrophil extracellular traps promotes nontypeable Haemophilus influenzae persistence in the chinchilla model for otitis media. J. Innate Immun. 1:215-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hong, W., K. Mason, J. Jurcisek, L. Novotny, L. O. Bakaletz, and W. E. Swords. 2007. Phosphorylcholine decreases early inflammation and promotes the establishment of stable biofilm communities of nontypeable Haemophilus influenzae strain 86-028NP in a chinchilla model of otitis media. Infect. Immun. 75:958-965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kapur, V., S. Topouzis, M. W. Majesky, L. L. Li, M. R. Hamrick, R. J. Hamill, J. M. Patti, and J. M. Musser. 1993. A conserved Streptococcus pyogenes extracellular cysteine protease cleaves human fibronectin and degrades vitronectin. Microb. Pathog. 15:327-346. [DOI] [PubMed] [Google Scholar]
- 24.Kuo, C. F., Y. S. Lin, W. J. Chuang, J. J. Wu, and N. Tsao. 2008. Degradation of complement 3 by streptococcal pyrogenic exotoxin B inhibits complement activation and neutrophil opsonophagocytosis. Infect. Immun. 76:1163-1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuo, C. F., J. J. Wu, K. Y. Lin, P. J. Tsai, S. C. Lee, Y. T. Jin, H. Y. Lei, and Y. S. Lin. 1998. Role of streptococcal pyrogenic exotoxin B in the mouse model of group A streptococcal infection. Infect. Immun. 66:3931-3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lembke, C., A. Podbielski, C. Hidalgo-Grass, L. Jonas, E. Hanski, and B. Kreikemeyer. 2006. Characterization of biofilm formation by clinically relevant serotypes of group A streptococci. Appl. Environ. Microbiol. 72:2864-2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lukomski, S., E. H. Burns, Jr., P. R. Wyde, A. Podbielski, J. Rurangirwa, D. K. Moore-Poveda, and J. M. Musser. 1998. Genetic inactivation of an extracellular cysteine protease (SpeB) expressed by Streptococcus pyogenes decreases resistance to phagocytosis and dissemination to organs. Infect. Immun. 66:771-776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luntz, M., A. Brodsky, S. Nusem, J. Kronenberg, G. Keren, L. Migirov, D. Cohen, S. Zohar, A. Shapira, D. Ophir, G. Fishman, G. Rosen, V. Kisilevsky, I. Magamse, S. Zaaroura, H. Z. Joachims, and D. Goldenberg. 2001. Acute mastoiditis—the antibiotic era: a multicenter study. Int. J. Pediatr. Otorhinolaryngol. 57:1-9. [DOI] [PubMed] [Google Scholar]
- 29.Luo, F., S. Lizano, S. Banik, H. Zhang, and D. E. Bessen. 2008. Role of Mga in group A streptococcal infection at the skin epithelium. Microb. Pathog. 45:217-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manetti, A. G., C. Zingaretti, F. Falugi, S. Capo, M. Bombaci, F. Bagnoli, G. Gambellini, G. Bensi, M. Mora, A. M. Edwards, J. M. Musser, E. A. Graviss, J. L. Telford, G. Grandi, and I. Margarit. 2007. Streptococcus pyogenes pili promote pharyngeal cell adhesion and biofilm formation. Mol. Microbiol. 64:968-983. [DOI] [PubMed] [Google Scholar]
- 31.Mascini, E. M., M. Jansze, J. F. Schellekens, J. M. Musser, J. A. Faber, L. A. Verhoef-Verhage, L. Schouls, W. J. van Leeuwen, J. Verhoef, and H. van Dijk. 2000. Invasive group A streptococcal disease in the Netherlands: evidence for a protective role of anti-exotoxin A antibodies. J. Infect. Dis. 181:631-638. [DOI] [PubMed] [Google Scholar]
- 32.Musser, J. M., and F. R. DeLeo. 2005. Toward a genome-wide systems biology analysis of host-pathogen interactions in group A Streptococcus. Am. J. Pathol. 167:1461-1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Musser, J. M., and R. M. Krause. The revival of group A streptococcal diseases, with a commentary on staphylococcal toxic shock syndrome, p. 185-218. In R. M. Krause (ed.), Emerging infections. Academic Press, New York, NY.
- 34.Nyberg, P., M. Rasmussen, U. Von Pawel-Rammingen, and L. Bjorck. 2004. SpeB modulates fibronectin-dependent internalization of Streptococcus pyogenes by efficient proteolysis of cell-wall-anchored protein F1. Microbiology 150:1559-1569. [DOI] [PubMed] [Google Scholar]
- 35.Pichichero, M. E. 2000. Recurrent and persistent otitis media. Pediatr. Infect. Dis. J. 19:911-916. [DOI] [PubMed] [Google Scholar]
- 36.Reid, S. D., M. S. Chaussee, C. D. Doern, M. A. Chaussee, A. G. Montgomery, D. E. Sturdevant, and J. M. Musser. 2006. Inactivation of the group A Streptococcus regulator srv results in chromosome wide reduction of transcript levels, and changes in extracellular levels of Sic and SpeB. FEMS Immunol. Med. Microbiol. 48:283-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reid, S. D., N. P. Hoe, L. M. Smoot, and J. M. Musser. 2001. Group A Streptococcus: allelic variation, population genetics, and host-pathogen interactions. J. Clin. Invest. 107:393-399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reid, S. D., W. Hong, K. E. Dew, D. R. Winn, B. Pang, J. Watt, D. T. Glover, S. K. Hollingshead, and W. E. Swords. 2009. Streptococcus pneumoniae forms surface-attached communities in the middle ear of experimentally infected chinchillas. J. Infect. Dis. 199:786-794. [DOI] [PubMed] [Google Scholar]
- 39.Reid, S. D., A. G. Montgomery, and J. M. Musser. 2004. Identification of srv, a PrfA-like regulator of group A streptococcus that influences virulence. Infect. Immun. 72:1799-1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roddy, M. G., S. S. Glazier, and D. Agrawal. 2007. Pediatric mastoiditis in the pneumococcal conjugate vaccine era: symptom duration guides empiric antimicrobial therapy. Pediatr. Emerg. Care 23:779-784. [DOI] [PubMed] [Google Scholar]
- 41.Segal, N., N. Givon-Lavi, E. Leibovitz, P. Yagupsky, A. Leiberman, and R. Dagan. 2005. Acute otitis media caused by Streptococcus pyogenes in children. Clin. Infect. Dis. 41:35-41. [DOI] [PubMed] [Google Scholar]
- 42.Spratley, J., H. Silveira, I. Alvarez, and M. Pais-Clemente. 2000. Acute mastoiditis in children: review of the current status. Int. J. Pediatr. Otorhinolaryngol. 56:33-40. [DOI] [PubMed] [Google Scholar]
- 43.Stähelin-Massik, J., M. Podvinec, J. Jakscha, O. N. Rust, J. Greisser, M. Moschopulos, and H. E. Gnehm. 2008. Mastoiditis in children: a prospective, observational study comparing clinical presentation, microbiology, computed tomography, surgical findings and histology. Eur. J. Pediatr. 167:541-548. [DOI] [PubMed] [Google Scholar]
- 44.Walker, M. J., A. Hollands, M. L. Sanderson-Smith, J. N. Cole, J. K. Kirk, A. Henningham, J. D. McArthur, K. Dinkla, R. K. Aziz, R. G. Kansal, A. J. Simpson, J. T. Buchanan, G. S. Chhatwal, M. Kotb, and V. Nizet. 2007. DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat. Med. 13:981-985. [DOI] [PubMed] [Google Scholar]
- 45.Watnick, P., and R. Kolter. 2000. Biofilm, city of microbes. J. Bacteriol. 182:2675-2679. [DOI] [PMC free article] [PubMed] [Google Scholar]