

Abstract
The sepsis syndrome represents an improper immune response to infection and is associated with unacceptably high rates of mortality and morbidity. The interactions between T cells and the innate immune system while combating sepsis are poorly understood. In this report, we observed that treatment with the potent, antiapoptotic cytokine interleukin-7 (IL-7) accelerated neutrophil recruitment and improved bacterial clearance. We first determined that T cells were necessary for the previously observed IL-7-mediated enhanced survival. Next, IL-7 increased Bcl-2 expression in T cells isolated from septic mice as early as 3 h following treatment. This treatment resulted in increased gamma interferon (IFN-γ) and IP-10 production within the septic peritoneum together with local and systemic increases of IL-17 in IL-7-treated mice. We further demonstrate that the increase in IL-17 was largely due to increased recruitment and production by γδ T cells, which express CXCR3. Consistent with increased IL-17 production, IL-7 treatment increased CXCL1/KC production, neutrophil recruitment, and bacterial clearance. Significantly, end-organ tissue injury was not significantly different between vehicle- and IL-7-treated mice. Collectively, these data illustrate that IL-7 can mediate the cross talk between Th1 and Th17 lymphocytes during sepsis such that neutrophil recruitment and bacterial clearance is improved while early tissue injury is not increased. All together, these observations may underlay novel potential therapeutic targets to improve the host immune response to sepsis.
Sepsis entails a complex immune process involving both the innate and adaptive immune systems, which can effect concurrent changes on the host (39, 56). The immune response to infection is typically modeled by phagocytic cells as first responders for the early removal of pathogenic bodies, while the adaptive system becomes involved later for mop-up duty and maintenance of a long-term immune response against the current pathogens. Recent literature suggests an early role for the adaptive immune system in mediating sepsis (30); however, a paucity of effective sepsis therapies suggests a gap in our knowledge concerning cross talk and the respective roles of the adaptive and innate immune during the host response to sepsis. An increased understanding of the complex interplay of these immune response arms is critical, given the annual toll this diagnosis places on both human life and health care dollars (2, 41).
γδ T cells represent a small subset of cells with a restricted T-cell receptor (TCR) repertoire of ligands. These cells are widespread in epithelium-rich tissues such as the skin and intestine, with cytokine production dictated by the tissues in which they reside (8, 12, 20, 40, 54). Contradictory results have been published on the role of γδ T cells during sepsis, suggesting that these cells become more pathogenic as the severity of the sepsis model used increases (6, 10, 13). Our previous report demonstrates that γδ T cells play a key role in neutrophil recruitment to the site of infection and control of bacterial load following cecal ligation and puncture (CLP). Recently, γδ T cells have been implicated as the major producer of the cytokine interleukin-17 (IL-17) during sepsis (13). IL-17A is part of the IL-17 family of cytokines, produced by diverse cell populations such as conventional CD4 and CD8 T cells as well as γδ T cells (58). While the protective or pathogenic role of IL-17 in sepsis is not yet clear (13, 16), it is known that IL-17 mediates recruitment of myeloid cells through downstream production of CXCL1/KC and CXCL2/MIP-2, presumably through the stabilization of mRNA (18). Additionally, the IL-17 receptor (IL-17R) is necessary for neutrophil recruitment to the site of infection and control of bacterial load following CLP (16). The functions of IL-17 on myeloid cell recruitment, along with its production by T cells during sepsis, make this cytokine an interesting target in mediating the cross talk that occurs between the innate and adaptive responses to sepsis.
The suggestion of early cross talk between the innate and adaptive immune responses extends from recent reports, indicating the necessity of T cells in bacterial clearance following infection (29, 37, 54, 55). Additionally, T-cell activation was newly linked to bacterial clearance through increased neutrophil oxidative burst and phagocytosis (29). These findings provide a bridge to the literature, demonstrating worsened outcomes in septic T-cell-deficient mice (26) that are reversed with adoptive transfer of T cells (47). Further, it is known that T cells undergo apoptosis within the first 24 h after onset of sepsis (25, 27, 57), and the degree of apoptosis correlates with the sepsis severity (24, 33). Additionally, remaining T cells demonstrate reduced functionality illustrated by decreased production of gamma interferon (IFN-γ) (28). Taken together, these results provide mechanistic insight into how prevention of T-cell apoptosis improves survival in a murine model of sepsis (26) and illustrate a therapeutic avenue for reduction of morbidity and mortality associated with this disease process.
The cytokines IL-2, IL-7, and IL-15 are ligands for the cytokine receptor common gamma chain. The IL-7 receptor is expressed mainly on T lymphocytes, with IL-7 playing a key role in T-cell differentiation, survival, and proliferation (36). Recently, we observed that IL-7 treatment significantly reduced T-cell apoptosis in the spleen and mesenteric lymph nodes following sepsis (55). Additionally, we reported that treatment with recombinant human IL-7 (rhIL-7) improved the delayed-type hypersensitivity (DTH) response while partially preventing a sepsis-induced decrease in T-cell IFN-γ production (55). The attenuation of T-cell apoptosis and improved functionality following rhIL-7 treatment were associated with improved survival (55). To build upon our previous study, we undertook investigations to further determine how IL-7 stabilization of T-cell numbers and function mediated tissue injury, bacterial load, and the neutrophil response during sepsis. Here, we found that IL-7 treatment significantly accelerated γδ T-cell recruitment to the site of infection, such that the bacterial load was decreased. Significantly, we postulate that the early IL-7 stabilization of the adaptive immune response allows for a more beneficial innate immune response during sepsis.
MATERIALS AND METHODS
Cecal ligation and puncture.
Male C57BL/6 (wild-type [WT]) mice between 6 and 8 weeks of age (20 to 28 g), TCR αβ/γδ-deficient mice (B6.129P2-Tcrbtm1MOM Tcrdtm1MOM/J; Jackson Laboratory, Bar Harbor, ME), and TCR γδ-deficient mice (B6.129P2-Tcrdtm1Mom/J; Jackson Laboratory) were utilized. All experiments involving animals were performed under protocols approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Cincinnati. Polymicrobial sepsis was induced as previously described (9). Briefly, cecal ligation and puncture (CLP) operations were always performed between 6 a.m. and 10 a.m. Normal fed mice were anesthetized to effect by 2.5% isoflurane in oxygen via face masks. After laparotomy, the latter 80% of the cecum was ligated and punctured once on the antimesenteric side with a 23-gauge needle. A small amount of bowel content was extruded through the puncture hole to ensure full thickness of the perforation. The cecum was replaced in its original location, and the midline incision was closed by a two-layer suture. Prior to closure of the peritoneum with one figure-of-eight stitch, 5 μg human recombinant IL-7 (Cytheris, Issy-les-Moulineaux, France) or an equivalent volume of vehicle was injected via pipette. Sham-operated animals received midline laparotomies, exteriorization of the cecum with prompt replacement, and closure of incisions in two layers. The animals were resuscitated with 1 ml of sterile saline subcutaneously and kept on a heating blanket, with additional oxygen supply for 1 h. In survival studies, animals were given ad libitum access to food and water and followed until death or humane sacrifice per protocol. Animals were evaluated every 12 h following CLP.
Flow cytometry for surface and intracellular staining.
Analyses of cell surface antigen expression and in situ cytokine and Bcl-2 expression were performed as previously described (5) on spleen and peritoneal lavage samples. Flow cytometry data acquisition and analysis were performed on an LSR II using FACS Diva software (BD Biosciences, Mountain View, CA). The antibodies used were CD4 (clone RM4-5; BioLegend, San Diego, CA), CD8 (clone 53-6.7; BD Biosciences, San Diego, CA), CD69 (clone H1.2F3; BD Biosciences), CD44 (clones Pgp-1 and Ly-24; BD Biosciences), CD62L (clone MEL-14; Invitrogen, Carlsbad, CA), αβ T cell (clone H57-597), γδ T cell (clone GL3; BioLegend), CXCR3 (clone 49801; R&D, Minneapolis, MN), Bcl-2 (clone 3F11; BD Biosciences), IL-17A (clone TC11-18H10.1; BioLegend), CD11b (clone M1/70; BD Biosciences), and Gr-1/Ly6G (clone 1A8; BD Biosciences). Leukocyte subsets were defined as follows: CD4+ naive (CD62Lhi CD44lo), CD4+ central memory (CD62Lhi CD44hi), CD4+ effector memory (CD62Llo CD44hi), CD8+ naive (CD62Lhi CD44lo), CD8+ central memory (CD62Lhi CD44hi), CD8+ effector memory (CD62Llo CD44hi), and neutrophils (Gr-1hi CD11b+).
Bacterial load determination.
Bacterial load determination was performed with blood samples harvested aseptically by cardiac puncture as previously described (54). Samples were serially diluted in sterile saline and cultured on soy agar pour plates. Plates were incubated at 37°C for 24 h, and colony enumeration was performed.
Cytokine and chemokine measurements by ELISA.
Whole-blood samples were harvested by sterile cardiac punching, and sera were isolated using serum separator tubes. Peritoneal lavage fluid and cells were obtained by injection of 9 ml 0.9% normal saline intraperitoneally and removal via syringe. IL-6 (Invitrogen), IFN-γ (Invitrogen), CXCL1/KC (R&D), IL-17 (eBioscience, San Diego, CA), and CXCL10/IP-10 (Peprotech, Rocky Hill, NJ) levels were analyzed by enzyme-linked immunosorbent assay (ELISA), according to the manufacturers' protocol.
Phagocytosis determination.
Cells from peritoneal lavage fluid were harvested 24 h after CLP and mixed with opsonized Alexa Fluor 488-labeled Escherichia coli (K-12 strain; Invitrogen). This suspension was incubated at 37°C for 5 min. This time point was chosen based upon previous optimization experiments. Following the incubation period, unattached extracellular bacteria were removed by being washed three times. Samples were run on a Becton Dickinson LSR II to determine the mean fluorescence intensity (MFI), a measure of the number of bacteria taken up per cell. To quench the fluorescence of adherent bacteria, trypan blue was added 1 min before the second acquisition. Quenching with trypan blue reduced the fluorescein isothiocyanate (FITC) fluorescence of adherent bacteria by excitation energy transfer (21).
Neutrophil oxidative burst assay.
The oxidative burst was determined as previously described (48). Briefly, polystyrene tissue culture dishes were coated with 1 μg/well fibronectin for 2 h at 37°C and 5% CO2 and then washed. A total of 100 μl of reaction mixture (10 mM scopoletin, 1 mg/ml horseradish peroxidase, 4 mM NaN3 in Krebs-Ringer phosphate buffer [KRPG]) and 20 μl neutrophils resuspended in KRPG at 7.5 × 105 cells/ml were allowed to incubate for 10 min at 37°C in the wells prior to stimulation with either buffer or N-formyl-methionyl-leucyl-phenylalanine (fMLP; 100 nM). Fluorescence was measured immediately and at 10-min intervals for 60 min. H2O2 production was calculated from the decrease in fluorescence due to oxidation of scopoletin. The data are expressed as nanomoles of H2O2 produced per 1.5 × 104 polymorphonuclear leukocytes (PMNs).
Tissue injury determination.
For general tissue damage assessment, serum samples were obtained as described above, diluted 1:4, and run for the aspartate transaminase (AST) level on the Siemens Dimension Xpand Plus integrated chemistry system (Siemens Healthcare Diagnostics, Deerfield, IL). Lung tissue (100 mg) was homogenized in 2 ml buffer A (3.4 mM KH2HPO4 and 16 mM Na2HPO4, pH 7.4). After centrifugation for 20 min at 10,000 × g, the pellet was resuspended in 10 vol buffer B (43.2 mM KH2HPO4, 6.5 mM Na2HPO4, 10 mM EDTA, and 0.5% hexadecyltrimethylammonium, pH 6.0) and was sonicated for 10 s. After being heated for 2 h at 60°C, the supernatant was reacted with 3,3′,5,5′-tetramethylbenzidine (Sigma Chemical Co., St. Louis, MO), and the optical density (OD) at 655 nm was determined. Two-centimeter sections of intestine were harvested and placed on previously weighed trays. Samples were incubated at 55°C for 72 h and reweighed. They were incubated for another 24 h at 55°C and reweighed to ensure complete fluid evaporation. The wet weight-to-dry weight ratio was then calculated.
Statistical analyses.
Statistical comparisons were performed using the Kaplan-Meier log rank test (survival), Student t test (two groups), or one-way analysis of variance (ANOVA) with the Holm-Sidak post hoc test (more than two groups), using StatView 3.5 (SAS Institute, Cary, NC) and GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA). The mean and standard error of the mean were calculated in experiments containing multiple data points. A P value of ≤0.05 was considered statistically significant.
RESULTS
Previously, we demonstrated enhanced survival in septic wild-type mice treated with recombinant human IL-7 at the time of CLP (54). To confirm the requirement for αβ and γδ T cells for the rhIL-7-mediated survival advantage during sepsis, we subjected mice deficient in these cells treated with either rhIL-7 or vehicle and then monitored them for survival. Here, we observed no differences in survival or median survival times (Fig. 1). All together, these data implicate the necessity of αβ and/or γδ T cells for the IL-7-induced increase in survival of septic wild-type mice.
FIG. 1.
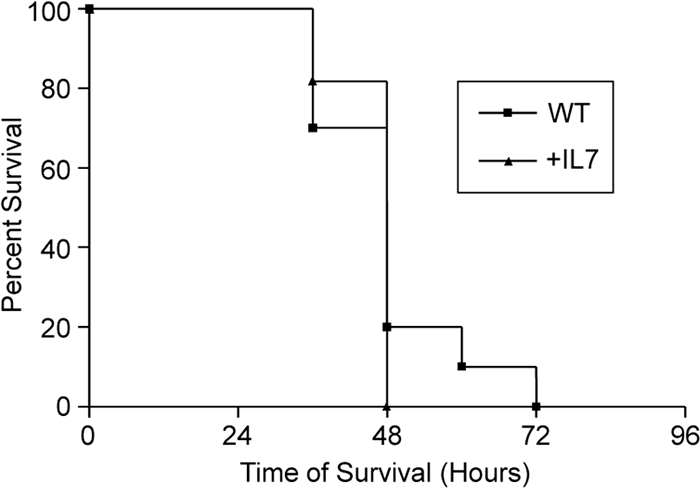
Effects of IL-7 on survival following sepsis are mediated by TCR αβ or TCR γδ cells. Combined TCR αβ/γδ-deficient mice were treated with 5 μg human recombinant IL-7 or vehicle at the time of CLP and then monitored for survival. The sample size was 10 to 11 mice/group. Data are presented as a Kaplan-Meier survival curve.
A signature hallmark of sepsis is the profound and rapid depletion of lymphocytes (23). T-cell apoptosis has been reported as the underlying cause of decreased lymphocyte numbers following sepsis (27, 60). Previously, we observed that splenic T cells isolated from rhIL-7-treated mice had increased Bcl-2 expression as well as reduced T-cell apoptosis at 24 h after CLP. As increased expression of Bcl-2 at 24 h after CLP may represent a selection bias of surviving lymphocytes, we sought to elucidate the effect of rhIL-7 on T-cell Bcl-2 expression at 3 h after sepsis induction, when cells have not yet undergone significant depletion. Here, we determined that IL-7 treatment resulted in increased Bcl-2 expression in naive CD4 and CD8 T cells and effector memory CD4 T cells compared to that in T cells isolated from vehicle-treated septic mice (Fig. 2 A to D). Thus, rhIL-7 treatment can begin to enhance T-cell survival quite early after treatment.
FIG. 2.

IL-7 treatment elevates CD4 and CD8 lymphocyte Bcl-2 expression as early as 3 h after CLP. Sham- and CLP-operated WT mice were treated with 5 μg human recombinant IL-7 or vehicle (VEH) at the time of CLP. Splenocytes obtained 3 h after CLP were directly analyzed by flow cytometry for Bcl-2 expression on naive CD4+ (A), effector memory (EM) CD4+ (B), naive CD8+ (C), and EM CD8+ (D) T-cell subsets. Naive T cells had CD62Lhi CD44lo expression, while EM T cells had CD62Llo CD44hi expression. The sample size was 4 mice/group. Data are expressed as the mean values ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
IL-7 treatment enhances IFN-γ and IP-10 production during sepsis.
IFN-γ is a key cytokine in the host immune response to sepsis, as evidenced by decreased survival of IFN-γ knockout mice subjected to CLP compared to that of WT mice (37). Here, we observed increased peritoneal IFN-γ concentrations in rhIL-7-treated mice at almost double the level found in vehicle-treated animals (Fig. 3). Sham levels were undetectable, as were serum levels of IFN-γ at this time point (data not shown). Of the many roles IFN-γ can play during the immune response, one involves stimulating the release of CXCL10/IP-10 by macrophages (52). CXCL10/IP-10 is one ligand for CXCR3. It has been previously reported that γδ T cells can express CXCR3 and that this receptor can mediate recruitment (1). Therefore, we evaluated the CXCR3 ligand in WT mice following CLP and observed its expression upon 89% of peritoneal γδ T cells (Fig. 4 A). Additionally, we demonstrated an approximately 2-fold increase in CXCL10/IP-10 in the rhIL-7-treated mice versus that in the vehicle-treated mice, while sham mice had no appreciable levels of this ligand (Fig. 4B). Thus, rhIL-7 treatment, possibly through prevention of T-cell apoptosis, enhances peritoneal levels of IFN-γ with a subsequent downstream increase in CXCL10/IP-10 production.
FIG. 3.
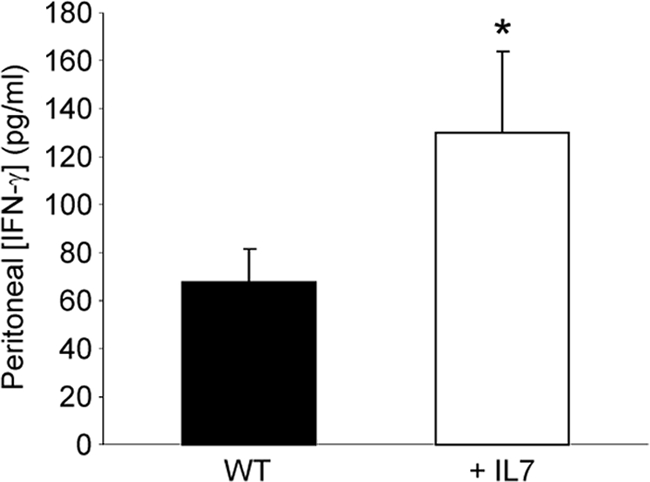
IL-7 treatment enhances IFN-γ levels in the septic peritoneum. CLP-operated WT mice were treated with 5 μg human recombinant IL-7 or vehicle at the time of CLP. IFN-γ was measured by ELISA in peritoneal samples obtained 3 h following CLP. The sample size was 8 mice/group. Data are expressed as the mean values ± SEM. *, P < 0.05.
FIG. 4.

CXCR3 expression on peritoneal TCR γδ cells. (A) Flow cytometric analysis of WT peritoneal TCR γδ cells obtained 3 h following CLP-evaluated CXCR3 expression. The sample size was 4 mice/group. (B) Sham- and CLP-operated WT mice were treated with 5 μg human recombinant IL-7 or vehicle at the time of CLP. Peritoneal lavage fluid obtained 3 h after CLP was analyzed by ELISA for CXCL10/IP-10. The sample size was 4 mice/group. Data are expressed as the mean values ± SEM. ^, P < 0.01 compared to sham groups; &, P < 0.01 compared to all groups; PE-A, phycoerythrin; APC-A, allophycocyanin.
Neutrophil recruitment, but not activation or functionality, is accelerated by rhIL-7 treatment.
Neutrophils clear bacterial pathogens during sepsis as part of a complex process involving recruitment to the site of infection, activation, cytokine secretion, phagocytosis, and oxidative burst killing of bacteria. Here, we evaluated the enumeration, activation, and functionality of neutrophils found at the site of infection at multiple time points following CLP. Treatment of mice with rhIL-7 caused an accelerated recruitment to the peritoneum at 6 h following CLP compared to that of vehicle-treated mice, with no difference between groups seen at 24 h (Fig. 5 A). CD11b expression was used as a surrogate marker for neutrophil activation and showed no difference between WT and rhIL-7-treated mice at 6 or 24 h following CLP (Fig. 5B) or between sham animals treated with rhIL-7 and phosphate-buffered saline (PBS) (data not shown). Lastly, we evaluated the phagocytosis and oxidative burst potential of neutrophils obtained from the site of infection at 6 h and 24 h following CLP in WT and rhIL-7-treated mice. Interestingly, we found no differences in either phagocytosis (Fig. 5C) or oxidative burst (Fig. 5D) between the two groups at either time point. These findings suggest that rhIL-7 plays a singular role in neutrophil recruitment to the site of infection during sepsis.
FIG. 5.

IL-7 treatment enhances neutrophil recruitment but not activation or functionality during sepsis. CLP-operated WT mice were treated with 5 μg human recombinant IL-7 or vehicle at the time of CLP. Neutrophils at the site of infection were analyzed by flow cytometry for enumeration (A) and determination of CD11b expression (B) at the indicated times following CLP. The sample size was 9 to 13 mice/group at 6 h and 12 to 13 mice/group at 24 h. Neutrophil functionality was evaluated using phagocytosis (C) and oxidative burst (D) at the indicated time points. The sample size was 6 mice/group at 3 h and 4 mice/group at 6 h. Data are expressed as the mean values ± SEM. **, P < 0.01.
rhIL-7 treatment increased γδ T-cell numbers, IL-17 production, and CXCL1/KC production.
Neutrophil recruitment can be mediated by IL-17 and subsequent production of CXCL1/KC. During sepsis, γδ T cells have been reported to be significant producers of IL-17 (13) and can play a critical role in increasing peritoneal neutrophil accumulation (54). In pilot experiments, we were unable to observe significant numbers of peritoneal γδ T cells in sham animals. Following CLP, we determined that rhIL-7 treatment resulted in both increasing the numbers of IL-17-producing γδ T cells and enhancing IL-17 production (Fig. 6 A and B). To confirm the necessity of γδ T cells for IL-7-induced enhancement of IL-17 during sepsis, we subjected wild-type and γδ T-cell-deficient mice to CLP with and without rhIL-7 treatment. Evaluation of the peritoneal lavage fluid at 6 h following the injury demonstrated significantly elevated IL-17 only in WT mice treated with rhIL-7 compared to that in all other treatment groups (Fig. 6C). Lastly, a positive correlation between neutrophil recruitment and CXCL1/KC expression has been reported (17, 34, 44, 51, 59). As IL-7 treatment increased neutrophil numbers at the site of infection, we next evaluated local levels of CXCL1/KC and observed significantly increased CXCL1/KC in the peritoneal lavage fluid of rhIL-7-treated mice compared to that in the peritoneal lavage fluid of WT mice (Fig. 6D). Thus, treatment with rhIL-7 rapidly increases the number of peritoneal γδ T cells present following sepsis, the production of IL-17 by these cells, and increased CXCL1/KC accumulation.
FIG. 6.

IL-7 treatment increases the number of, and IL-17 production by, peritoneal γδ T cells following CLP. CLP-operated WT mice were treated with 5 μg human recombinant IL-7 or vehicle at the time of CLP. Flow cytometric analysis was used to enumerate the number of peritoneal γδ T cells producing IL-17 (A) and the intensity of IL-17 expression by peritoneal γδ T cells (B) 3 h after CLP. The sample size was 4 mice/group. (C) Peritoneal lavage fluid obtained 6 h after CLP from WT and TCR γδ-deficient mice treated with IL-7 or vehicle was analyzed by ELISA for IL-17. The sample size was 5 mice/group. (D) Peritoneal lavage fluid obtained 6 h after CLP was analyzed by ELISA for CXCL1/KC. The sample size was 11 to 14 mice/group. Data are expressed as the mean values ± SEM. *, P < 0.05; **, P < 0.01.
rhIL-7 treatment increases inflammation and bacterial clearance without changing end-organ tissue injury.
Properly regulated neutrophils play an important role in bacterial clearance (32); however, decreased apoptosis and increased half-life may result in increased tissue injury during sepsis (22). During sepsis, an optimal balance among early pathogen clearance, associated inflammation, and end-organ tissue damage will likely allow for a reduction of host morbidity and mortality. As IL-6 has previously been utilized as a surrogate marker for inflammation (42), we evaluated the serum levels of this cytokine at 6 and 24 h. Mice treated with rhIL-7 had significantly higher serum IL-6 levels at 6 h than WT mice, with no difference existing at 24 h (Fig. 7 A). Evaluation of bacteremia at 24 h following CLP demonstrated a significantly lower bacterial load in those animals treated with rhIL-7 rather than vehicle (Fig. 7B), consistent with previous findings. We next evaluated general, lung, and intestine tissue injury. We observed that serum aspartate aminotransferase (AST) levels (Fig. 7C), lung myeloperoxidase (MPO) levels (Fig. 7D), bronchoalveolar lavage (BAL) fluid protein content (data not shown), and intestinal wet/dry ratios (Fig. 7E) were not significantly different between vehicle-treated WT mice and those treated with rhIL-7. Experiments conducted with sham-operated mice did not demonstrate detectable levels of IL-6, bacteremia, or tissue injury. All together, mice treated with rhIL-7 had diminished bacteremia and early increased, but not sustained, inflammation, but not worsened tissue damage, compared to those of vehicle-treated mice.
FIG. 7.

IL-7 treatment mediates serum IL-6 levels, decreases bacteremia, and does not alter tissue injury after CLP. WT mice were subjected to CLP, as indicated in Materials and Methods, and treated with 5 μg human recombinant IL-7 or vehicle at the time of CLP. (A) Sera obtained at the indicated time points were analyzed by ELISA for IL-6. (B) Serum bacterial loads were measured 24 h after induction of sepsis. (C) Serum AST levels were measured 24 h after induction of sepsis. (D) Lungs were harvested at the indicated times after CLP, and MPO was measured. (E) Sections (2 cm) of terminal ileum were obtained at the indicated times following CLP, weighed, and dried as described in Materials and Methods. The sample size was 8 to 11 mice/group. Data are expressed as the mean values ± SEM. *, P < 0.05; ***, P < 0.001.
DISCUSSION
In this study, we tested the hypothesis that IL-7, a potent T-cell antiapoptotic cytokine, would mediate the early innate immune response to sepsis. First, we demonstrated the necessity of αβ and/or γδ T cells for the IL-7-induced increase in survival of septic wild-type mice. Additionally, we show the upregulation of Bcl-2 expression on CD4 T cells as early as 3 h following CLP in mice treated with rhIL-7. Here, mice treated with rhIL-7 were shown to have increased neutrophil recruitment to the peritoneum without increased end-organ tissue damage compared to that of WT controls. Elevated neutrophil recruitment at the site of infection was correlated with elevated local and systemic production of CXCL1/KC and IL-17. We determined that following rhIL-7 treatment, septic mice had increased numbers of γδ T cells in the peritoneum, producing elevated amounts of IL-17. Additionally, γδ T cells were necessary for an elevated peritoneal IL-17 concentration following IL-7 treatment. Previously, we demonstrated that T cells isolated from IL-7-treated mice had a reversal of the sepsis-induced defective production of IFN-γ (55). Here, we built upon these findings by demonstrating an IL-7-induced increase in peritoneal IFN-γ levels. Increased IFN-γ production has been shown to promote macrophage or endothelial CXCL10/IP-10 expression (3, 19, 31), a ligand for the CXCR3 receptor. We show that this receptor is expressed upon γδ T cells. These results suggest that IL-7 treatment improves the host response to sepsis through the rapid recruitment of γδ T cells to the site of infection, coupled with accelerated γδ T cell-mediated recruitment of neutrophils to the site of infection. All together, these data are summarized in Fig. 8.
FIG. 8.
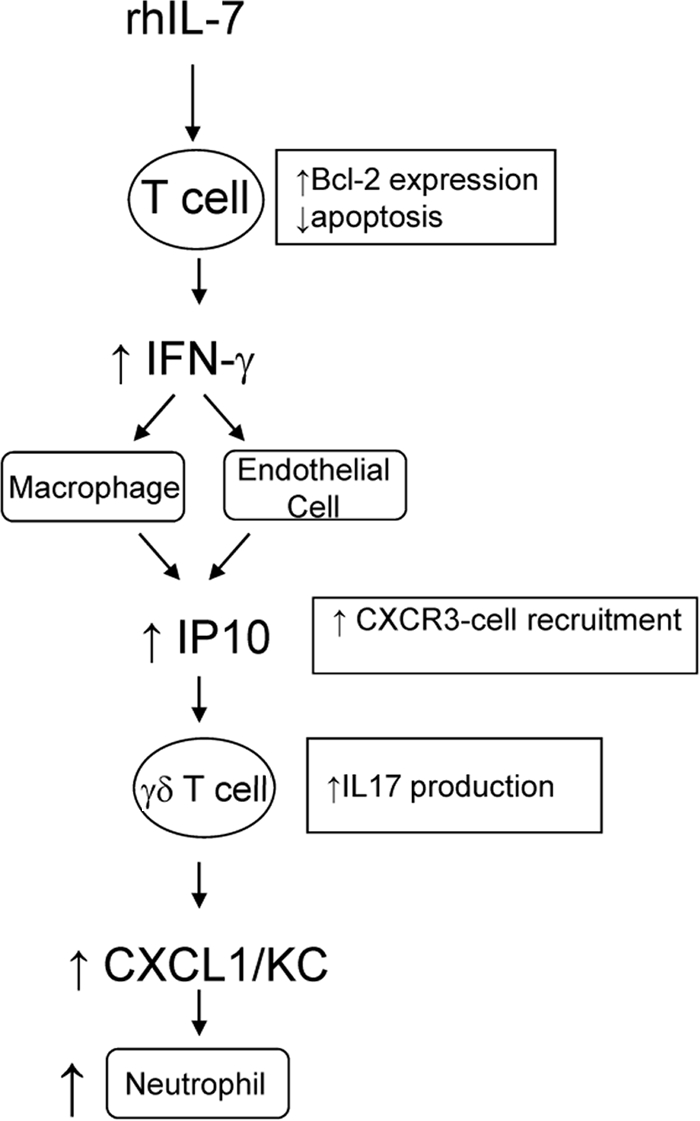
Model for IL-7's role in modulating neutrophil recruitment in sepsis. This figure represents a schematic hypothesis, demonstrating how rhIL-7 treatment, through maintenance of appropriate T-cell immunocompetence, results in enhanced neutrophil recruitment to the site of infection.
The work detailing that rhIL-7 treatment improves the murine host response to sepsis has recently been reported (55), although its effects on lymphocyte apoptosis and proliferation in human diseases such as hepatitis C and cancer stem from ongoing clinical trials (46, 49). At 24 h following CLP, IL-7 treatment significantly increased Bcl-2 expression in splenic CD4 and CD8 T-cell subsets compared to that in wild-type animals and produced a relative change of mRNA abundance in CD4 T cells (55). Additionally, rhIL-7 blunted both splenic and mesenteric lymph node T-cell apoptosis at 24 h following CLP, resulting in significantly elevated numbers of CD4 and CD8 T cells in treated versus WT mice at this time point (55). Clinical findings of decreased Bcl-2 gene expression in septic human patients at 12 and 24 h following diagnosis were previously noted to correlate highly with decreased absolute numbers of CD4 lymphocytes (4, 24). Furthermore, prevention of apoptosis with overexpression of Bcl-2 is known to prevent T-cell apoptosis, providing protection from sepsis-related mortality (26). Here, we show that naive CD4, CD8, and effector memory CD4 cells obtained from rhIL-7-treated animals have significantly elevated Bcl-2 protein expression at 3 h following CLP, suggesting that apoptotic signaling in T cells takes place soon after induction of sepsis. One key effect of early T-cell apoptosis in sepsis is reduced immunocompetence through decreased IFN-γ production. The production of IFN-γ by CD4 T cells during sepsis is associated with improved survival, decreased bacterial load, and increased neutrophil functionality (37, 53). Interestingly, we show that IL-7 treatment significantly increases the peritoneal concentration of IFN-γ as early as 3 h following CLP. These data suggest that rhIL-7 treatment upregulates Bcl-2 expression very soon after treatment, such that T-cell apoptosis can be reduced and IFN-γ production can be preserved.
The enhanced production of IFN-γ in rhIL-7-treated mice relative to WT controls exerts downstream effects on both adaptive and innate cells. Gamma interferon-inducible protein 10 (CXCL10/IP-10) is a chemokine secreted by endothelial cells, macrophages, and neutrophils upon stimulation with IFN-γ and serves as a chemoattractant for T lymphocytes (7, 35, 61). It has been suggested that CXCL10/IP-10 plays a distinct role in the recruitment of T cells expressing CXCR3 to sites of inflammation (7), consistent with findings that TCR γδ T cells express CXCR3 and transmigrate upon stimulation with IP-10 (11, 45). Here, we demonstrate that a large percentage of γδ T cells and neutrophils present at the site of infection soon following CLP express CXCR3. Further, septic mice treated with IL-7 had significantly larger amounts of CXCL10/IP-10 at the peritoneum. Consistent with these findings, we observed that rhIL-7 treatment resulted in an elevated number of γδ T cells at the site of infection. All together, we postulate that IL-7 treatment enhanced T-cell IFN-γ production such that CXCL10/IP-10 was increased and γδ T cells entered the site of infection.
After exposure to CXCL10/IP-10, γδ T cells can transmigrate and mediate inflammatory processes in tissue compartments such as the lung and peritoneum. The inflammatory effects of γδ T cells result, in part, from production of the cytokine IL-17 in response to early and sustained stimulation by IL-1β, TLR-2, and IL-23 (1, 38, 43, 50). IL-17 acts upon mesothelial cells to produce CXCL1/KC and CXCL2/MIP-2 (15) and has been shown to be necessary for the promotion of granulopoiesis and neutrophil recruitment in peripheral tissues (14, 62). Here, we show that local and systemic levels of IL-17 are elevated with rhIL-7 treatment following CLP, with a subsequent and complementary increase in CXCL1/KC. Further, elevated IL-17 following rhIL-7 treatment in sepsis is dependent upon the presence of γδ T cells. This demonstrates a sequential and mechanistic pathway through which rhIL-7-mediated attenuation of T-cell dysfunction results in increased neutrophil recruitment to the site of infection following CLP. The elevated number of neutrophils early in the septic course is likely responsible for the decreased bacterial load in rhIL-7-treated mice, even in the absence of neutrophil functionality changes. The rapid return of neutrophil numbers to WT levels at 24 h may explain the comparable levels of end-organ tissue damage seen in these groups, consistent with previous findings in this model assessing the role of neutrophils in sepsis (22). The lack of a functional effect on neutrophils following IL-7 treatment may further explain the consonant end-organ results in these studies. All together, we speculated that the ability of rhIL-7 to maintain numbers and functionality of CD4 T cells following sepsis resulted in accelerated γδ T-cell recruitment and functionality at the site of infection, ultimately aiding bacterial clearance without worsening tissue damage.
Our data reported here demonstrate that rhIL-7 treatment increased peritoneal IFN-γ levels, IP-10 levels, and CXCR3-expressing γδ T-cell accumulation. However, one limitation to this report is a lack of data verifying that IP-10, through CXCR3, recruits γδ T cells to the peritoneum. While trying to address this, we subjected CXCR3-deficient mice to CLP and found that these mice had significantly increased peritoneal bacterial loads compared to those of WT mice. In addition, neutrophils isolated from these mice demonstrated a decreased phagocytic capacity. Finally, the CXCR3 knockout (KO) had increased numbers of neutrophils and macrophages at the site of infection compared to those of controls. We speculate that the CXCR3-mediated defect in phagocytosis resulted in sustained leukocyte chemotaxis that ultimately confounded the experimental results. Thus, the use of more specific reagents will be needed to fully address this potentially important mechanism.
Sepsis involves a complex interplay between the innate and adaptive immune systems that occurs very early after the onset of disease and not in a delayed fashion, as previously accepted. γδ T cells, through production of IL-17, appear to play a central role in immune response cross talk between the two arms of the immune response. In addition, the existence of cross talk between these two responses suggests that multiple targets for future therapeutic intervention may be available. One such therapeutic target is the cytokine IL-7, shown to play a direct role in prevention of T-cell apoptosis, with subsequent downstream effects resulting in an enhanced innate response to sepsis. Going forward, these insights may play a role in the ultimate production of a viable sepsis therapeutic target that reduces the health care burden imposed by this disease process.
Acknowledgments
This work was supported in part by National Institutes of Health grants GM72760 (to C.C.C.), GM44118 and GM55194 (to R.S.H.), and AI057753 (to D.A.H.) and by the Alan A. and Edith L. Wolff Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Michel Morre is the CEO of Cytheris Corporation, a company that makes IL-7 for clinical trials. The other authors have no financial conflicts of interest.
Editor: J. N. Weiser
Footnotes
Published ahead of print on 7 September 2010.
REFERENCES
- 1.Ajuebor, M. N., Y. Jin, G. L. Gremillion, R. M. Strieter, Q. Chen, and P. A. Adegboyega. 2008. GammadeltaT cells initiate acute inflammation and injury in adenovirus-infected liver via cytokine-chemokine cross talk. J. Virol. 82:9564-9576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Angus, D. C., W. T. Linde-Zwirble, J. Lidicker, G. Clermont, J. Carcillo, and M. R. Pinsky. 2001. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit. Care Med. 29:1303-1310. [DOI] [PubMed] [Google Scholar]
- 3.Bacon, K. B., and J. J. Oppenheim. 1998. Chemokines in disease models and pathogenesis. Cytokine Growth Factor Rev. 9:167-173. [DOI] [PubMed] [Google Scholar]
- 4.Bilbault, P., T. Lavaux, A. Lahlou, B. Uring-Lambert, M. P. Gaub, C. Ratomponirina, N. Meyer, P. Oudet, and F. Schneider. 2004. Transient Bcl-2 gene down-expression in circulating mononuclear cells of severe sepsis patients who died despite appropriate intensive care. Intensive Care Med. 30:408-415. [DOI] [PubMed] [Google Scholar]
- 5.Caldwell, C. C., H. Kojima, D. Lukashev, J. Armstrong, M. Farber, S. G. Apasov, and M. V. Sitkovsky. 2001. Differential effects of physiologically relevant hypoxic conditions on T lymphocyte development and effector functions. J. Immunol. 167:6140-6149. [DOI] [PubMed] [Google Scholar]
- 6.Chung, C. S., L. Watkins, A. Funches, J. Lomas-Neira, W. G. Cioffi, and A. Ayala. 2006. Deficiency of gammadelta T lymphocytes contributes to mortality and immunosuppression in sepsis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 291:R1338-R1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dufour, J. H., M. Dziejman, M. T. Liu, J. H. Leung, T. E. Lane, and A. D. Luster. 2002. IFN-gamma-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J. Immunol. 168:3195-3204. [DOI] [PubMed] [Google Scholar]
- 8.Duhindan, N., A. J. Farley, S. Humphreys, C. Parker, B. Rossiter, and C. G. Brooks. 1997. Patterns of lymphokine secretion amongst mouse gamma delta T cell clones. Eur. J. Immunol. 27:1704-1712. [DOI] [PubMed] [Google Scholar]
- 9.Ebong, S. J., D. R. Call, G. Bolgos, D. E. Newcomb, J. I. Granger, M. O'Reilly, and D. G. Remick. 1999. Immunopathologic responses to non-lethal sepsis. Shock 12:118-126. [DOI] [PubMed] [Google Scholar]
- 10.Enoh, V. T., S. H. Lin, C. Y. Lin, T. Toliver-Kinsky, E. D. Murphey, T. K. Varma, and E. R. Sherwood. 2007. Mice depleted of alphabeta but not gammadelta T cells are resistant to mortality caused by cecal ligation and puncture. Shock 27:507-519. [DOI] [PubMed] [Google Scholar]
- 11.Ferrero, E., P. Biswas, K. Vettoretto, M. Ferrarini, M. Uguccioni, L. Piali, B. E. Leone, B. Moser, C. Rugarli, and R. Pardi. 2003. Macrophages exposed to Mycobacterium tuberculosis release chemokines able to recruit selected leucocyte subpopulations: focus on gammadelta cells. Immunology 108:365-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferrick, D. A., M. D. Schrenzel, T. Mulvania, B. Hsieh, W. G. Ferlin, and H. Lepper. 1995. Differential production of interferon-gamma and interleukin-4 in response to Th1- and Th2-stimulating pathogens by gamma delta T cells in vivo. Nature 373:255-257. [DOI] [PubMed] [Google Scholar]
- 13.Flierl, M. A., D. Rittirsch, H. Gao, L. M. Hoesel, B. A. Nadeau, D. E. Day, F. S. Zetoune, J. V. Sarma, M. S. Huber-Lang, J. L. Ferrara, and P. A. Ward. 2008. Adverse functions of IL-17A in experimental sepsis. FASEB J. 22:2198-2205. [DOI] [PubMed] [Google Scholar]
- 14.Forlow, S. B., J. R. Schurr, J. K. Kolls, G. J. Bagby, P. O. Schwarzenberger, and K. Ley. 2001. Increased granulopoiesis through interleukin-17 and granulocyte colony-stimulating factor in leukocyte adhesion molecule-deficient mice. Blood 98:3309-3314. [DOI] [PubMed] [Google Scholar]
- 15.Fossiez, F., O. Djossou, P. Chomarat, L. Flores-Romo, S. Ait-Yahia, C. Maat, J. J. Pin, P. Garrone, E. Garcia, S. Saeland, D. Blanchard, C. Gaillard, B. Das Mahapatra, E. Rouvier, P. Golstein, J. Banchereau, and S. Lebecque. 1996. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J. Exp. Med. 183:2593-2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freitas, A., J. C. Alves-Filho, T. Victoni, T. Secher, H. P. Lemos, F. Sonego, F. Q. Cunha, and B. Ryffel. 2009. IL-17 receptor signaling is required to control polymicrobial sepsis. J. Immunol. 182:7846-7854. [DOI] [PubMed] [Google Scholar]
- 17.Frink, M., Y. C. Hsieh, C. H. Hsieh, H. C. Pape, M. A. Choudhry, M. G. Schwacha, and I. H. Chaudry. 2007. Keratinocyte-derived chemokine plays a critical role in the induction of systemic inflammation and tissue damage after trauma-hemorrhage. Shock 28:576-581. [DOI] [PubMed] [Google Scholar]
- 18.Gaffen, S. L. 2009. Structure and signalling in the IL-17 receptor family. Nat. Rev. Immunol. 9:556-567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han, C., J. Fu, Z. Liu, H. Huang, L. Luo, and Z. Yin. 2010. Dipyrithione inhibits IFN-gamma-induced JAK/STAT1 signaling pathway activation and IP-10/CXCL10 expression in RAW264.7 cells. Inflamm. Res. 59:809-816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hayday, A., and R. Tigelaar. 2003. Immunoregulation in the tissues by gammadelta T cells. Nat. Rev. Immunol. 3:233-242. [DOI] [PubMed] [Google Scholar]
- 21.Hed, J. 1986. Methods for distinguishing ingested from adhering particles. Methods Enzymol. 132:198-204. [DOI] [PubMed] [Google Scholar]
- 22.Hoesel, L. M., T. A. Neff, S. B. Neff, J. G. Younger, E. W. Olle, H. Gao, M. J. Pianko, K. D. Bernacki, J. V. Sarma, and P. A. Ward. 2005. Harmful and protective roles of neutrophils in sepsis. Shock 24:40-47. [DOI] [PubMed] [Google Scholar]
- 23.Hotchkiss, R. S., and D. W. Nicholson. 2006. Apoptosis and caspases regulate death and inflammation in sepsis. Nat. Rev. Immunol. 6:813-822. [DOI] [PubMed] [Google Scholar]
- 24.Hotchkiss, R. S., S. B. Osmon, K. C. Chang, T. H. Wagner, C. M. Coopersmith, and I. E. Karl. 2005. Accelerated lymphocyte death in sepsis occurs by both the death receptor and mitochondrial pathways. J. Immunol. 174:5110-5118. [DOI] [PubMed] [Google Scholar]
- 25.Hotchkiss, R. S., P. E. Swanson, B. D. Freeman, K. W. Tinsley, J. P. Cobb, G. M. Matuschak, T. G. Buchman, and I. E. Karl. 1999. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit. Care Med. 27:1230-1251. [DOI] [PubMed] [Google Scholar]
- 26.Hotchkiss, R. S., P. E. Swanson, C. M. Knudson, K. C. Chang, J. P. Cobb, D. F. Osborne, K. M. Zollner, T. G. Buchman, S. J. Korsmeyer, and I. E. Karl. 1999. Overexpression of Bcl-2 in transgenic mice decreases apoptosis and improves survival in sepsis. J. Immunol. 162:4148-4156. [PubMed] [Google Scholar]
- 27.Hotchkiss, R. S., K. W. Tinsley, P. E. Swanson, R. E. Schmieg, Jr., J. J. Hui, K. C. Chang, D. F. Osborne, B. D. Freeman, J. P. Cobb, T. G. Buchman, and I. E. Karl. 2001. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J. Immunol. 166:6952-6963. [DOI] [PubMed] [Google Scholar]
- 28.Kasten, K. R., H. S. Goetzman, M. R. Reid, A. M. Rasper, S. G. Adediran, C. T. Robinson, C. M. Cave, J. S. Solomkin, A. B. Lentsch, J. A. Johannigman, and C. C. Caldwell. 2010. Divergent adaptive and innate immunological responses are observed in humans following blunt trauma. BMC Immunol. 11:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kasten, K. R., J. Tschoep, H. S. Goetzman, L. G. England, J. R. Dattilo, C. M. Cave, A. P. Seitz, D. A. Hildeman, and C. C. Caldwell. T cell activation differentially mediates the host response to sepsis. Shock, in press. [DOI] [PubMed]
- 30.Kasten, K. R., J. Tschop, S. G. Adediran, D. A. Hildeman, and C. C. Caldwell. T cells are potent early mediators of the host response to sepsis. Shock, in press. [DOI] [PubMed]
- 31.Kopydlowski, K. M., C. A. Salkowski, M. J. Cody, N. van Rooijen, J. Major, T. A. Hamilton, and S. N. Vogel. 1999. Regulation of macrophage chemokine expression by lipopolysaccharide in vitro and in vivo. J. Immunol. 163:1537-1544. [PubMed] [Google Scholar]
- 32.Leendertse, M., R. J. Willems, I. A. Giebelen, J. J. Roelofs, M. J. Bonten, and T. van der Poll. 2009. Neutrophils are essential for rapid clearance of Enterococcus faecium in mice. Infect. Immun. 77:485-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Le Tulzo, Y., C. Pangault, A. Gacouin, V. Guilloux, O. Tribut, L. Amiot, P. Tattevin, R. Thomas, R. Fauchet, and B. Drenou. 2002. Early circulating lymphocyte apoptosis in human septic shock is associated with poor outcome. Shock 18:487-494. [DOI] [PubMed] [Google Scholar]
- 34.Li, X., D. Klintman, Q. Liu, T. Sato, B. Jeppsson, and H. Thorlacius. 2004. Critical role of CXC chemokines in endotoxemic liver injury in mice. J. Leukoc. Biol. 75:443-452. [DOI] [PubMed] [Google Scholar]
- 35.Lin, A. A., P. K. Tripathi, A. Sholl, M. B. Jordan, and D. A. Hildeman. 2009. Gamma interferon signaling in macrophage lineage cells regulates central nervous system inflammation and chemokine production. J. Virol. 83:8604-8615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu, Y. J., V. Soumelis, N. Watanabe, T. Ito, Y. H. Wang, W. Malefyt Rde, M. Omori, B. Zhou, and S. F. Ziegler. 2007. TSLP: an epithelial cell cytokine that regulates T cell differentiation by conditioning dendritic cell maturation. Annu. Rev. Immunol. 25:193-219. [DOI] [PubMed] [Google Scholar]
- 37.Martignoni, A., J. Tschop, H. S. Goetzman, L. G. Choi, M. D. Reid, J. A. Johannigman, A. B. Lentsch, and C. C. Caldwell. 2008. CD4-expressing cells are early mediators of the innate immune system during sepsis. Shock 29:591-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin, B., K. Hirota, D. J. Cua, B. Stockinger, and M. Veldhoen. 2009. Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity 31:321-330. [DOI] [PubMed] [Google Scholar]
- 39.Monneret, G., F. Venet, A. Pachot, and A. Lepape. 2008. Monitoring immune dysfunctions in the septic patient: a new skin for the old ceremony. Mol. Med. 14:64-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pennington, D. J., D. Vermijlen, E. L. Wise, S. L. Clarke, R. E. Tigelaar, and A. C. Hayday. 2005. The integration of conventional and unconventional T cells that characterizes cell-mediated responses. Adv. Immunol. 87:27-59. [DOI] [PubMed] [Google Scholar]
- 41.Polderman, K. H., and A. R. Girbes. 2004. Drug intervention trials in sepsis: divergent results. Lancet 363:1721-1723. [DOI] [PubMed] [Google Scholar]
- 42.Remick, D. G., G. R. Bolgos, J. Siddiqui, J. Shin, and J. A. Nemzek. 2002. Six at six: interleukin-6 measured 6 h after the initiation of sepsis predicts mortality over 3 days. Shock 17:463-467. [DOI] [PubMed] [Google Scholar]
- 43.Roark, C. L., P. L. Simonian, A. P. Fontenot, W. K. Born, and R. L. O'Brien. 2008. Gammadelta T cells: an important source of IL-17. Curr. Opin. Immunol. 20:353-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rollins, B. J. 1997. Chemokines. Blood 90:909-928. [PubMed] [Google Scholar]
- 45.Romagnani, P., F. Annunziato, E. Lazzeri, L. Cosmi, C. Beltrame, L. Lasagni, G. Galli, M. Francalanci, R. Manetti, F. Marra, V. Vanini, E. Maggi, and S. Romagnani. 2001. Interferon-inducible protein 10, monokine induced by interferon gamma, and interferon-inducible T-cell alpha chemoattractant are produced by thymic epithelial cells and attract T-cell receptor (TCR) alphabeta+ CD8+ single-positive T cells, TCRgammadelta+ T cells, and natural killer-type cells in human thymus. Blood 97:601-607. [DOI] [PubMed] [Google Scholar]
- 46.Rosenberg, S. A., C. Sportes, M. Ahmadzadeh, T. J. Fry, L. T. Ngo, S. L. Schwarz, M. Stetler-Stevenson, K. E. Morton, S. A. Mavroukakis, M. Morre, R. Buffet, C. L. Mackall, and R. E. Gress. 2006. IL-7 administration to humans leads to expansion of CD8+ and CD4+ cells but a relative decrease of CD4+ T-regulatory cells. J. Immunother. 29:313-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shelley, O., T. Murphy, H. Paterson, J. A. Mannick, and J. A. Lederer. 2003. Interaction between the innate and adaptive immune systems is required to survive sepsis and control inflammation after injury. Shock 20:123-129. [DOI] [PubMed] [Google Scholar]
- 48.Solomkin, J. S., C. T. Robinson, C. M. Cave, B. Ehmer, and A. B. Lentsch. 2007. Alterations in membrane cholesterol cause mobilization of lipid rafts from specific granules and prime human neutrophils for enhanced adherence-dependent oxidant production. Shock 28:334-338. [DOI] [PubMed] [Google Scholar]
- 49.Sportes, C., F. T. Hakim, S. A. Memon, H. Zhang, K. S. Chua, M. R. Brown, T. A. Fleisher, M. C. Krumlauf, R. R. Babb, C. K. Chow, T. J. Fry, J. Engels, R. Buffet, M. Morre, R. J. Amato, D. J. Venzon, R. Korngold, A. Pecora, R. E. Gress, and C. L. Mackall. 2008. Administration of rhIL-7 in humans increases in vivo TCR repertoire diversity by preferential expansion of naive T cell subsets. J. Exp. Med. 205:1701-1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sutton, C. E., S. J. Lalor, C. M. Sweeney, C. F. Brereton, E. C. Lavelle, and K. H. Mills. 2009. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity 31:331-341. [DOI] [PubMed] [Google Scholar]
- 51.Tanimoto, N., M. Terasawa, M. Nakamura, D. Kegai, N. Aoshima, Y. Kobayashi, and K. Nagata. 2007. Involvement of KC, MIP-2, and MCP-1 in leukocyte infiltration following injection of necrotic cells into the peritoneal cavity. Biochem. Biophys. Res. Commun. 361:533-536. [DOI] [PubMed] [Google Scholar]
- 52.Tebo, J. M., H. S. Kim, J. Gao, D. A. Armstrong, and T. A. Hamilton. 1998. Interleukin-10 suppresses IP-10 gene transcription by inhibiting the production of class I interferon. Blood 92:4742-4749. [PubMed] [Google Scholar]
- 53.Thiel, M., C. C. Caldwell, S. Kreth, S. Kuboki, P. Chen, P. Smith, A. Ohta, A. B. Lentsch, D. Lukashev, and M. V. Sitkovsky. 2007. Targeted deletion of HIF-1alpha gene in T cells prevents their inhibition in hypoxic inflamed tissues and improves septic mice survival. PLoS One 2:e853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tschop, J., A. Martignoni, H. S. Goetzman, L. G. Choi, Q. Wang, J. G. Noel, C. K. Ogle, T. A. Pritts, J. A. Johannigman, A. B. Lentsch, and C. C. Caldwell. 2008. Gammadelta T cells mitigate the organ injury and mortality of sepsis. J. Leukoc. Biol. 83:581-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Unsinger, J., M. McGlynn, K. R. Kasten, A. S. Hoekzema, E. Watanabe, J. T. Muenzer, J. S. McDonough, J. Tschoep, T. A. Ferguson, J. E. McDunn, M. Morre, D. A. Hildeman, C. C. Caldwell, and R. S. Hotchkiss. 2010. IL-7 promotes T cell viability, trafficking, and functionality and improves survival in sepsis. J. Immunol. 184:3768-3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Venet, F., F. Davin, C. Guignant, A. Larue, M. A. Cazalis, R. Darbon, C. Allombert, B. Mougin, C. Malcus, F. Poitevin-Later, A. Lepape, and G. Monneret. Early assessment of leukocyte alterations at diagnosis of septic shock. Shock, in press. [DOI] [PubMed]
- 57.Wang, S. D., K. J. Huang, Y. S. Lin, and H. Y. Lei. 1994. Sepsis-induced apoptosis of the thymocytes in mice. J. Immunol. 152:5014-5021. [PubMed] [Google Scholar]
- 58.Weaver, C. T., R. D. Hatton, P. R. Mangan, and L. E. Harrington. 2007. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu. Rev. Immunol. 25:821-852. [DOI] [PubMed] [Google Scholar]
- 59.Wengner, A. M., S. C. Pitchford, R. C. Furze, and S. M. Rankin. 2008. The coordinated action of G-CSF and ELR + CXC chemokines in neutrophil mobilization during acute inflammation. Blood 111:42-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wesche, D. E., J. L. Lomas-Neira, M. Perl, C. S. Chung, and A. Ayala. 2005. Leukocyte apoptosis and its significance in sepsis and shock. J. Leukoc. Biol. 78:325-337. [DOI] [PubMed] [Google Scholar]
- 61.Widney, D. P., Y. R. Xia, A. J. Lusis, and J. B. Smith. 2000. The murine chemokine CXCL11 (IFN-inducible T-cell alpha chemoattractant) is an IFN-gamma- and lipopolysaccharide-inducible glucocorticoid-attenuated response gene expressed in lung and other tissues during endotoxemia. J. Immunol. 164:6322-6331. [DOI] [PubMed] [Google Scholar]
- 62.Ye, P., F. H. Rodriguez, S. Kanaly, K. L. Stocking, J. Schurr, P. Schwarzenberger, P. Oliver, W. Huang, P. Zhang, J. Zhang, J. E. Shellito, G. J. Bagby, S. Nelson, K. Charrier, J. J. Peschon, and J. K. Kolls. 2001. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J. Exp. Med. 194:519-527. [DOI] [PMC free article] [PubMed] [Google Scholar]