

Abstract
Proteasomal stress and the accumulation of polyubiquitinated proteins are key features of numerous neurodegenerative disorders. Previously we demonstrated that stabilization of p53 and activation of its target gene, puma (p53-upregulated mediator of apoptosis), mediated proteasome inhibitor-induced apoptosis in cancer cells. Here we demonstrated that Puma also contributed to proteasome inhibitor-induced apoptosis in mouse neocortical neurons. Although protection afforded by puma gene deletion was incomplete, we found little evidence indicating contributions from other proapoptotic BH3-only proteins. Attenuation of bax expression did not further reduce Puma-independent apoptosis, suggesting that pathways other than the mitochondrial apoptosis pathway were activated. Real-time imaging experiments in wild-type and puma-deficient neurons using a fluorescence resonance energy transfer (FRET)-based caspase sensor confirmed the involvement of a second cell death pathway characterized by caspase activation prior to mitochondrial permeabilization and, more prominently, a third, caspase-independent and Puma-independent pathway characterized by rapid cell shrinkage and nuclear condensation. This pathway involved lysosomal permeabilization in the absence of autophagy activation and was sensitive to cathepsin but not autophagy inhibition. Our data demonstrate that proteasomal stress activates distinct cell death pathways in neurons, leading to both caspase-dependent and caspase-independent apoptosis, and demonstrate independent roles for Puma and lysosomal permeabilization in this model.
Accumulation of protein aggregates and buildup of polyubiquitinated proteins are characteristic features of several neurological disorders, including Parkinson's disease (30), amyotrophic lateral sclerosis (ALS) (28, 29), and ischemic stroke (25). Inhibition of the proteasome is per se sufficient to activate apoptosis in neurons and nonneuronal cells (38, 53), and targeting the proteasome is employed as therapy in cancers such as multiple myeloma (14). Recent studies on proteasome inhibitor-induced apoptosis suggest that proteasome inhibitors as therapeutics may have detrimental effects on the nervous system (8, 9). However, the molecular mechanisms of proteasome inhibition-induced cell death in neurons remain poorly understood.
The proteasome is the principal site of cellular protein degradation and recycling. It consists of a 20S barrel that facilitates protein dismantling and two 19S regulatory caps. Proteins destined for recycling are tagged with polyubiquitin chains by the E family of ligases in an ATP-dependent manner (13). Defects in ubiquitin proteasome system (UPS)-mediated protein degradation can lead to accumulation of signaling proteins and expression of transcription factors that are tightly regulated by the proteasome, such as the NF-κB inhibitor, IκB (44), and the proapoptotic transcription factor, p53 (59). Previously, we have demonstrated that stabilization of p53 and the subsequent transcriptional activation of the BH3-only protein p53-upregulated mediator of apoptosis (Puma) partially mediated proteasome inhibitor-induced apoptosis in human cancer cells (15). BH3-only proteins are proapoptotic Bcl-2 family proteins that are transcriptionally or posttranslationally activated in response to prolonged cellular stress (26). All BH3-only proteins have the capacity to neutralize antiapoptotic Bcl-2 family proteins, but some BH3-only proteins, including Bid, Bim, and Puma, are also believed to directly activate Bax and Bak in mitochondrial membranes (12). Activation of Bax and Bak is an essential step in the mitochondrial apoptosis pathway and leads to mitochondrial outer membrane permeabilization (MOMP) and the release of caspase-activating factors into the cytosol (26). Here, we sought to establish the role of Puma during proteasome inhibitor-induced apoptosis in neocortical neuron cultures, using gene deletion and silencing approaches in combination with real-time single-cell imaging of caspase-dependent and caspase-independent cell death pathways. Our data suggest the existence of at least three cell death pathways in neurons that mediate proteasome inhibitor-induced apoptosis in both puma-dependent and puma-independent manners.
MATERIALS AND METHODS
Materials.
Fetal calf serum, minimal essential medium (MEM), and tetramethylrhodamine methyl ester (TMRM) were from Invitrogen (Bio Sciences, Dublin, Ireland). Caspase substrate acetyl-DEVD-7-amido-4-methylcoumarin (Ac-DEVD-AMC) and inhibitor N-benzyloxycarbonyl-Val-Ala-Asp (O-methyl)-fluoromethyl ketone (Z-VAD-FMK) were purchased from Bachem (St. Helens, United Kingdom). Bortezomib was from Millennium Pharmaceuticals (Cambridge, MA). All other chemicals, including epoxomicin, came in analytical grade purity from Sigma-Aldrich (Tallaght, Dublin, Ireland).
Gene-targeted mice.
puma−/−, bim−/−, and bid−/− mice were generated as previously described (6, 27, 69). The puma−/− and bid−/− mice were generated on an inbred C57BL/6 background, using C57BL/6-derived ES cells. The bim−/− mice were originally generated on a mixed C57BL/6 × 129SV genetic background, using 129SV-derived embryonic stem (ES) cells, but had been backcrossed for >12 generations onto the C57BL/6 background.
Preparation of mouse primary neocortical neurons.
Cortical neurons were prepared as previously described (16). Gestation day 16-to-18 embryos (E16 to E18) were isolated by hysterectomy of the uterus, using an abdominal injection of 40 mg/kg of pentobarbital (Dolethal) as lethal anesthesia. The harvested embryos were transferred to dissection medium on ice in phosphate-buffered saline (PBS; with 0.25% glucose and 0.3% bovine serum albumin [BSA]). The cerebral cortices from each of the embryos were isolated. The surrounding meninges were removed, and the tissue was pooled in dissection medium on ice. The tissue was incubated with trypsin-EDTA (0.25%) at 37°C for 15 min. Following incubation, the trypsinization was stopped by the addition of medium containing sera. The neurons were then dissociated by gentle pipetting, and following centrifugation (400 × g for 3 min), the medium containing trypsin was aspirated. The neurons were then resuspended in fresh plating medium (minimal essential medium [MEM] containing 5% fetal calf serum, 5% horse serum, 100 U/ml penicillin/streptomycin, 0.5 mM l-glutamine, 0.6% d-glucose). Cells were plated at 2 × 105 cells per cm2 on polylysine-coated plates and incubated at 37°C in 5% CO2. The plating medium was exchanged with 50% feeding medium (NBM-embryonic containing 100 U/ml of penicillin-streptomycin [Pen/Strep], 2% B27, and 0.5 mM l-glutamine) plus 50% plating medium with additional cytosine arabinofuranoside (600 nM). Two days later, the medium was again exchanged for feeding medium and experiments were carried out on days in vitro (DIV) 5 to 6.
RT-qPCR.
Total RNA was extracted using the RNeasy minikit (Qiagen, Hilden Germany). First-strand cDNA synthesis was performed using 2 μg of total RNA as the template and reverse transcribed using Superscript II (Invitrogen) primed with 50 pmol of random hexamers. Quantitative real-time PCR (RT-qPCR) was performed using the LightCycler 4.0 (Roche Diagnostics, Basel, Switzerland) and the QuantiTech SYBR green PCR kit (Qiagen) as per the manufacturer's protocol. Specific primers for each gene analyzed were designed using Primer3 software. The sense and antisense primers were CAACACAAACCCAAGTCCT and CATTTGCAAACACCCTCCTT for bim, TCTCAGGAAAGGCTGCTGGT and CCGCCGCTCGTACTGCGCGTT for puma, ACGACAAGGCCATGCTGATA and AGGCACCCTCAGTCCATCTC for bid, TGAGCACACTCGTCCTTCAA and TCAGGAGATCGGACAAA for noxa, and GGTGTGATGGTGGGAATGG and GGTTGGCCTTAGGGTTCAGG for the β-actin gene. The PCRs were performed in 20-μl volumes with the following parameters: 95°C for 15 min, followed by 40 cycles of 94°C for 20 s, 59°C for 20 s, and 72°C for 20 s. The generation of specific PCR products was confirmed by melting curve analysis and gel electrophoresis. The data were analyzed using the Lightcycler software 4.0, with all samples normalized to β-actin.
SDS-PAGE and Western blotting.
Preparation of cell lysates and Western blotting were carried out as previously described (50a). The resulting blots were probed with either a mouse monoclonal antibody detecting mono- and polyubiquitinylated proteins (Biomol, Enzo Life Sciences, Exeter, United Kingdom) diluted 1:1,000, a mouse monoclonal anti-Hsp70 antibody (Stressgen, Victoria, Canada) diluted 1:1,000, a mouse monoclonal anti-CHOP antibody (Santa Cruz, CA) diluted 1:500, a mouse anti-caspase 8 antibody (Alexis, San Diego, CA) diluted 1:1,000, a mouse monoclonal anti-p53 antibody (Nova Castra, Leica, Microsystems, IL) diluted 1:1,000, a rabbit polyclonal anti-Bim antibody (Stressgen, Victoria, Canada) diluted 1:1,000, a goat polyclonal anti-Bid antibody (R&D Systems, Minneapolis, MN) diluted 1:1,000, a rabbit polyclonal anti-PUMA NT antibody (Prosci, Poway, CA) diluted 1:1,000, a mouse monoclonal anti-LC3 antibody (Abgent, San Diego, CA) diluted 1:1,000, a rabbit anti p62 antibody (Enzo Life Sciences, Exeter, United Kingdom) diluted 1:1,000, a rabbit polyclonal anti-Bax antibody (Upstate, Millipore, Billerica, MA) diluted 1:1,000, and a mouse monoclonal anti-β-actin antibody (clone DM 1A; Sigma, Dublin, Ireland) diluted 1:5,000. Horseradish peroxidase-conjugated secondary antibodies diluted 1:10,000 (Pierce, Northumberland, United Kingdom) were detected using SuperSignal West Pico chemiluminescent substrate (Pierce) and imaged using a FujiFilm LAS-3000 imaging system (Fuji, Sheffield, United Kingdom).
Determination of caspase-3-like protease activity.
DEVDase activity was determined fluorometrically using N-benzyloxylcarbonyl-Asp-Glu-Val-Asp-7-amino-4-methyl-coumarin (DEVD-AMC) as the substrate (10 μM). Cleavage of DEVD-AMC to liberate free AMC was monitored in live cells by measuring fluorescence after 1- and 2-h intervals. Protein content was determined using the Pierce Coomassie Plus protein assay reagent (Perbio, Northumberland, United Kingdom). Caspase activity was expressed as change in fluorescent units per hour and per microgram of protein.
Determination of neuronal injury: Hoechst and propidium iodide staining of nuclear chromatin.
Neocortical neurons were stained live with Hoechst 33258 (1 μg/ml) or propidium iodide (5 μM) in medium. Nuclear morphology was assessed with an Nikon Eclipse TE 300 inverted microscope (Nikon, Düsseldorf, Germany) with a 20× 0.43 NA phase-contrast objective using the appropriate filter set for Hoechst and a charge-coupled device (CCD) camera (SPOT RT SE 6; Diagnostic Instruments, Sterling Heights, MI). For each time point, images of nuclei were captured in three subfields and repeated in triplicate. Images were processed using Image J software (Micron-Optica; http://rsb.info.nih.gov/ij/). Condensed and/or fragmented nuclei were scored as percent nuclear condensation and expressed as a percentage of the total population.
Plasmids, transfections, and siRNA.
Neocortical neurons were transfected at DIV 5 or 6 using Lipofectamine 2000 (Invitrogen, Paisley, United Kingdom) with expression plasmids for green fluorescent protein (GFP)-LC3 (24), SCAT (i.e., sensor for activated caspases based on FRET)-DEVD-FRET (66), or Smac-yellow fluorescent protein (YFP) (49) as per the manufacturer's instructions. For inhibition of noxa expression, three synthetic small interfering RNAs (siRNAs) that target noxa were designed using the RNA Workbench software (67a) and cloned into a pFIV plasmid (System Biosciences, Cambridge Bioscience, Cambridge, United Kingdom). Neurons were transfected with a mixture of vectors expressing either a siRNA targeting noxa or a scramble sequence by electroporation using the AMAXA apparatus (program 0.05) with a mouse neuron kit (Lonza, Basel, Switzerland). The following sequences were utilized: control sense, 5′-ACUUAACCGGCAUACCGGC(dTdT)-3′; control antisense, 5′-GCCGGUAUGCCGGUUAAGU(dTdT)-3′; noxa 1 sense, 5′-TTTCTTGCGTTTCTCAGTCCGAG-3′; noxa 1 antisense, 5′-CTCGGACTGAGAAACGCAAG-3′; noxa 2 sense, 5′-TTACATCAGAAGGTTGCTTGGCC-3′; noxa 2 antisense, 5′-GGCCAAGCAACCTTCTGATGTAA-3′; noxa 3 sense, 5′-TGAGATAGTGGTTGAAGGCCTGG-3′; and noxa 3 antisense, 5′-CCAGGCCTTCAACCACTATCTCA-3′.
Silencing of bax expression was performed utilizing siRNA duplexes (sc-29213) purchased from Santa Cruz Biotechnology containing three target-specific siRNAs. Sequences were cotransfected with an enhanced green fluorescent protein (EGFP)-expressing plasmid in a ratio of 3:1 using Lipofectamine 2000.
Real-time live cell imaging.
Primary neocortical neurons transfected with either SCAT-DEVD-FRET (66) or Smac-YFP (49) probes and loaded with TMRM (20 nM) in experimental buffer (120 mM NaCl, 3.5 mM KCl, 0.4 mM KH2PO4, 20 mM HEPES, 5 mM NaHCO3, 1.2 mM Na2SO4, 1.2 mM CaCl2, 1.2 mM MgCl2, and 15 mM glucose, pH 7.4) were placed on the stage of an LSM 5Live Zeiss confocal microscope with a thermostatically regulated chamber. Following a 45-min equilibration time, drug dissolved in experimental buffer was added to the medium. TMRM was excited at 543 nm, and the emission was collected by a 560-nm long pass filter. YFP was excited at 500 ± 20 nm, and emission was collected at 535 ± 30 nm for Smac-YFP. Cyan fluorescent protein (CFP) was excited at 436 ± 10 nm, and emission was collected at 480 ± 20 nm. FRET was measured using an excitation of 436 ± 10 nm, and emission was collected at 535 ± 30 nm for the SCAT-DEVD-FRET as described previously (48). Images were captured every 5 min throughout these experiments. All microscope settings including laser intensity and scan time were kept constant. Control experiments for cytotoxicity were also carried out and determined to have negligible impact. The resulting data were processed using LSM and MetaMorph software as described previously (48, 50).
Immunocytochemistry and cell staining.
Posttreatment, cells grown on 13-mm coverslips were fixed with 4% paraformaldehyde for 15 min, permeabilized in PBS containing 0.5% Triton X-100, washed three times with PBS, and blocked for 1 h in 5% goat serum (or donkey serum-cathepsin B) in PBS. The cells were then incubated for 2 h with either a native anti-cytochrome c antibody diluted 1:250 (R&D Systems) or a goat polyclonal anti-cathepsin B antibody (Santa Cruz Biotechnology, Santa Cruz, CA) diluted 1:50 in 5% serum in PBS. Primary antibodies were detected using a 1:500 dilution of fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit secondary antibody (Jackson Immunoresearch, Plymouth, PA) for 1 h. For lysosomal staining, LysoTracker red (Molecular Probes) was incubated at 100 nM in medium 30 min prior to addition of epoxomicin. Coverslips were then transferred to glass slides with DAPI (4′,6-diamidino-2-phenylindole) mounting medium and sealed around the edges with clear varnish. Images of stained cells for quantification were captured with the Nikon Eclipse TE 300 inverted microscope with a 20× 0.43 NA phase-contrast objective and a CCD camera as described above.
Mathematical model of apoptotic cell death through caspase-3 autofeedback and imbalance of protein turnover.
Computational pathway modeling based on ordinary differential equations (ODEs) and mass action kinetics was employed to study the effect of proteasome inhibition. Initially, we induced activation of 0.1% caspase-3 caused by zymogenic activation of procaspase-3 (63). Processing of procaspase-3 and caspase-3 activation can lead to caspase-3 autoactivation (78). XIAP (X-linked inhibitor of apoptosis protein) was modeled to inhibit active caspase-3 (19). Furthermore, active caspase-3 can cleave XIAP into its BIR1-2 and BIR3-RING fragments (19), although the latter did not influence the pathway and was neglected. All proteins produced in the apoptotic cascade were subject to proteasomal degradation (39, 68), with those bound to XIAP assumed to be degraded at a higher rate. The entities involved and the reaction network with constants for mass action kinetics are described in Tables 1 and 2. Proteasome inhibition reduced the degradation constants (k−1, k−2, k+10, k+11) by the percentage rate given in Fig. 6.
TABLE 1.
Considered entities in the computational model
| Abbreviation | Definition | Initial value (nM) |
|---|---|---|
| C3 | Procaspase-3 | 100 (estimated) |
| C3a | Free active caspase-3 | 0 |
| XIAP | Free X-linked inhibitor of apoptosis protein | 100 (estimated) |
| XIAP∼C3a | XIAP in complex with caspase-3 | 0 |
| BIR12 | XIAP fragment comprising baculoviral IAP repeats 1 and 2 | 0 |
| BIR12∼C3a | XIAP Bir12 fragment in complex with caspase-3 | 0 |
TABLE 2.
The reaction network of the computational model
| Step | Substrate(s) | Reaction | Product(s) | Ki+ (μM−1 min−1)a | Ki− (min−1)a | Source or reference |
|---|---|---|---|---|---|---|
| 1 | C3 | → | Production and degradation | 0.00039 | 0.0039 | 75 (estimated) |
| 2 | XIAP | → | Production and degradation | 0.00116 | 0.0116 | 50 (estimated) |
| 3 | C3 + C3a | → | C3a + C3a | 2.4 | 0 | 78 (estimated) |
| 4 | C3a + XIAP | ↔ | [XIAP∼C3a] | 156 | 0.144 | 54 |
| 5 | C3a + XIAP | → | C3a + BIR12 | 12 | 0 | 63 |
| 6 | C3a + [XIAP∼C3a] | → | C3a + BIR12c3a | 12 | 0 | 63 |
| 7 | C3a + BIR12 | ⇆ | BIR12∼C3a | 156 | 0.144 | 54 |
| 8 | BIR12 | → | Degradation | 0.0058 | 0 | 20 |
| 9 | BIR12c3a | → | Degradation | 0.0058 | 0 | 20 |
| 10 | C3a | → | Degradation | 0.0058 | 0 | 20 |
| 11 | [XIAP∼C3a] | → | Degradation | 0.0347 | 0 | 75 |
| 12 | Substrate + C3a | → | C3a | 12 | 0 | 63 |
Kinetic constants refer to parameters for mass action kinetics for the forward (+) and backward (−) reactions.
Local stability analysis.
For a mathematically rigorous investigation of the effect of proteasome inhibitors, we performed a local stability analysis (as described by Eissing et al. [20]) of the pseudo-reaction system of Table 2 at the initial state (right column in Table 1). We therefore analytically calculated the eigenvalues λ of the Jacobian J
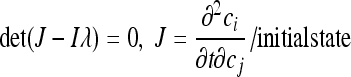 |
(1) |
with guaranteed stability, if all eigenvalues λ have a negative real part. Employing Mathematica (Wolfram Research, United Kingdom), we analytically calculated equation 1 and employed the Hurvitz criterion (20) for the solution of the seventh order polynomial. This led to a criterion for stability
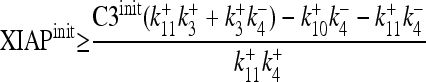 |
(2) |
with the initial concentrations of XIAP and caspase-3 (XIAPinit and C3init, respectively) given by the balance from transcription/translation and degradation
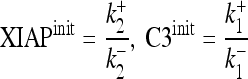 |
(3) |
Equation 2 serves as a criterion for signal depletion and relates the XIAP and procaspase-3 initial concentration (equation 3), the caspase-3 autofeedback strength, k+3, the XIAP binding to active caspase-3 (k+4 and k−4), and the degradation rate for free and XIAP-bound active caspase-3 and XIAP (k+10 and k+11) to each other with values given in Table 2. To investigate the effect of proteasomal inhibition, we replaced the degradation constant (k−1, k−2, k+10, k+11) by such that are modulated by an inhibition factor, A, indicating the range between full proteasome function (A = 1) to full inhibition (A = 0). Inserting them into equation 2 leads to stability for equation 4
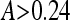 |
(4) |
or alternatively instability, and thus apoptosis execution, for proteasome inhibition higher than 90%.
Statistics.
Data are given as means ± standard errors of the means (SEM). For statistical comparison, one-way analysis of variance followed by Tukey's post hoc test was employed unless otherwise stated in the figure legends. P values smaller than 0.05 were considered to be statistically significant.
RESULTS
Prolonged proteasome inhibition results in cell death associated with release of cytochrome c and activation of effector caspases.
Proteasome inhibition has been shown to induce apoptosis in several paradigms. We characterized the morphological and biochemical features of cell death induced by prolonged proteasomal stress in neocortical neurons. Exposure to epoxomicin (50 nM) resulted in a time-dependent increase in ubiquitinated proteins (Fig. 1A), indicative of impaired proteasome-mediated protein degradation. Epoxomicin induced 60 to 80% apoptosis over 24 h, as assessed by quantification of propidium iodide (PI) inclusion (Fig. 1B and D). Hoechst staining of nuclear chromatin showed morphological changes indicative of apoptosis, including condensation and fragmentation (Fig. 1C). Using the fluorogenic probe AC-DEVD-AMC, a substrate for caspase 3 and other effector caspases, we assessed caspase activation over time (Fig. 1D). Caspase 3-like activity increased in a time-dependent manner which coincided with cell death quantified by PI uptake (Fig. 1D). Neurons with condensed nuclei also exhibited a loss of mitochondrial cytochrome c immunofluorescence, suggesting activation of the mitochondrial apoptosis pathway by epoxomicin (Fig. 1E).
FIG. 1.

Epoxomicin induces protein ubiquitination and cell death associated with cytochrome c release, caspase activation, and nuclear apoptotic morphology in neocortical neurons. (A) Cortical neurons were treated with epoxomicin (Epoxo; 50 nM) or the control (Con) (dimethyl sulfoxide [DMSO], 0.1%) for the indicated periods. Western blotting was performed using an antibody recognizing mono- and polyubiquitinated proteins. Probing for β-actin served as a loading control. (B) Bright-phase, PI-positive neurons and the merged image in control and neurons treated with epoxomicin for 24 h. Scale bar, 50 μm. (C) Hoechst-stained neurons illustrating nuclear condensation and fragmentation in DMSO- and epoxomicin-treated samples. Scale bar, 20 μm. (D) Quantification of PI-positive neurons and caspase-3-like (DEVDase) activity following epoxomicin treatment. PI-positive nuclei were expressed as a percentage of total cells per field. A minimum of 300 neurons in at least three different fields were captured per well, and at least three wells were analyzed per time point (*, P < 0.05 compared to control). Caspase 3-like activity was assessed by measuring the cleavage of the fluorogenic substrate Ac-DEVD-AMC (10 μM). DEVDase activity was expressed as fold increase over the control. The data are the results from three measurements per well and three wells per time point (#, P < 0.05 compared to control). (E) Immunocytochemistry of cytochrome c (Cyt-c) in neurons. The redistribution of cytochrome c is observed in treated samples (scale bar, 10 μm). All data are means ± SEM from three wells; experiments were repeated three times from independent cultures with similar results.
Levels of mRNA and BH3-only proteins are increased following proteasomal stress.
Previous studies have suggested the involvement of the mitochondrial apoptosis pathway (15, 35); therefore, we investigated the transcriptional and posttranslational activation of BH3-only protein family members following proteasome inhibition. The relative levels of mRNA and BH3-only proteins compared to those in control samples were measured by real-time quantitative PCR (RT-qPCR) and Western blotting. Significant increases were observed in puma, bim, and noxa mRNA expression, while bid mRNA remained unchanged over time (Fig. 2A to D). Coinciding with mRNA changes were increased Bimextra long (BimEL) and Puma (2- and 4-fold respectively, data not shown) protein levels at 24 h post-epoxomicin treatment (Fig. 2E), a time period when increased levels of cell death were evident (Fig. 1D). As with bid mRNA, no change was observed in either Bid protein levels or Bid proteolytic cleavage (Fig. 2E). Due to a lack of specific and commercially available antibodies against mouse Noxa protein, Noxa protein levels could not be explored. We next analyzed caspase-8 protein levels as it has been suggested that p53-dependent caspase activation may occur through activation of the death receptor pathway (43, 72). We did however detect only a transient activation of caspase-8 activation (1.5-fold; data not shown) in response to proteasome inhibition (Fig. 2E).
FIG. 2.

Determination of the transcriptional and posttranslational activation of BH3-only proteins. (A to D) Real-time quantitative PCR of BH3-only genes puma, bim, noxa, and bid. Cortical neurons were treated with epoxomicin (50 nM) or the control (DMSO; 0.1%) for the indicated time periods. The relative mRNA expression levels were assessed by RT-qPCR and normalized to β-actin mRNA levels. Expression levels were normalized to control-treated cells, and data are represented as means ± SEM from three wells. *, P < 0.05 compared to control-treated controls (ANOVA and Tukey's post hoc test). (E) Cortical neurons were treated with epoxomicin or DMSO for the indicated time periods. The expression of Puma, Bim, Bid, and caspase-8 was analyzed by Western blotting. Probing for β-actin served as the loading control. Similar results were observed in two other independent experiments. t-Bid, truncated Bid.
Loss of puma protects cortical neurons from proteasome inhibitor-induced apoptosis.
Having established that specific BH3-only proteins were increased following proteasomal stress in cortical neurons, we next investigated if and to what extent such changes contributed to cell death in our paradigm. We quantified the number of neurons with apoptotic nuclei from BH3-only knockout mice compared to their wild-type (WT) counterparts following epoxomicin treatment. In spite of increased bim mRNA and protein, neurons that lacked bim were found to be equally susceptible to cell death compared to WT neurons (Fig. 3A). This finding coincided with previously obtained results in HCT116 colon cancer cells and mouse embryonic fibroblasts deficient for bim (15). Similarly cell death levels in bid−/− neurons did not differ from WT neurons (Fig. 3B), suggesting that neither transcriptional induction nor posttranslational activation of Bid was required for epoxomicin-induced apoptosis. We did, however, observe a robust reduction in apoptotic neurons at both 16 and 24 h post-epoxomicin treatment in neuronal cultures derived from puma-deficient mice (Fig. 3C), confirming a prominent role for Puma in this model.
FIG. 3.

Cortical neurons deficient in puma are protected from proteasomal stress-induced apoptosis. (A to C) Cortical neurons from WT or bim−/− (A), bid−/− (B), or puma−/− (C) mice were treated with epoxomicin (50 nM) or the control (DMSO; 0.1%), and cell death was assessed by quantifying apoptotic nuclei by Hoechst staining. Nuclear apoptosis was expressed as a percentage of total neurons in the field. Each field contained approximately 300 to 400 neurons, three fields were captured per well, and at least three wells were analyzed per time point. Data are means ± SEM from three wells per condition. *, P < 0.05 compared to similarly treated WT cultures (ANOVA and Tukey's post hoc test). (D and E) DEVDase activity of WT and puma−/− cortical neurons was assessed following treatment with epoxomicin (D) or 10 μM colchicine (E). Data are expressed relative to control-treated cultures. (F) WT and puma−/− neurons were treated with bortezomib (Bort; 100 nM), and cell death was assessed by quantifying the percentage of nuclear condensation. Data are means ± SEM from four cultures. *, P < 0.05 compared to WT-treated cultures (ANOVA and Tukey's post hoc test). (G and H) WT and puma−/− cortical neurons were treated with epoxomicin (50 nM) for the indicated time periods. Protein levels of mono- and polyubiquitinylated proteins (G) or Chop, Hsp70, and p53 (H) were assessed by Western blotting. The experiments were repeated three times from different preparations with similar results.
To further characterize the protection afforded in the puma-deficient neurons, we monitored cleavage of the caspase-3 substrate Ac-DEVD-AMC over time. Neurons from puma−/− mice had significantly less caspase-3-like protease activity at 16 and 24 h post-epoxomicin treatment (Fig. 3D). As a control, we assessed Ac-DEVD-AMC cleavage following treatment with colchicine (10 μM). Colchicine inhibits microtubule polymerization and has been shown to induce apoptosis in cerebral granular neurons (5) and organotypic slices (33). We observed no significant difference in caspase 3-like protease activities between WT and puma−/− neuron cultures following colchicine treatment (Fig. 3E). Furthermore, to assess if the protection observed was a general response to proteasomal stress, we treated WT and puma−/− neuronal cultures with 100 nM bortezomib (Velcade), a proteasomal inhibitor used clinically in the treatment of multiple myeloma (77). Similar to epoxomicin-treated neurons, bortezomib-induced cell death was significantly attenuated in puma−/− cultures (Fig. 3F).
Next, we wanted to ensure that the protection observed in puma−/− neurons was specific to the puma gene deletion and not due to altered stress levels between genotypes. The levels of ubiquitinated proteins observed were found to be similar in WT and puma-deficient neurons following treatment (Fig. 3G), indicating comparable inhibition of the proteasome. In addition, we assessed the levels of stress-associated proteins induced by proteasome dysfunction and observed similar levels of Chop and heat shock protein 70 (Hsp70) induction in both genotypes (Fig. 3H). Furthermore, levels of induction of the tumor suppressor p53, an important protein regulated by the proteasome and central to the regulation of Puma, as previously demonstrated (15), were also comparable between genotypes (Fig. 3H). Taken together, these results demonstrate that the observed protection was specific to proteasome inhibitor-induced cell death and not a result of a generally reduced susceptibility of puma−/− neocortical neurons to caspase activation and apoptosis.
Attenuation of noxa or bax expression in puma−/− neurons does not confer additional protection.
The protection afforded to puma−/− neurons, although significant in replicate experiments, was incomplete (Fig. 3C, D, and F). Real-time qPCR analysis revealed a significant increase in another BH3-only protein gene, noxa (Fig. 2C), also a p53 target gene (42). In some systems, Puma is more dominant than Noxa in contributing to p53-dependent cell death (58). However, at least in certain cell types, such as thymocytes, responding to gamma irradiation, Puma and Noxa can cooperate (40). To investigate if Noxa played a prominent role in epoxomicin-induced cell death, either by acting in conjunction with Puma or by compensating in part for its absence, we used siRNA to knock down noxa gene expression. Expression of three different sequences was found to attenuate noxa gene expression following epoxomicin treatment (Fig. 4A and B). A mixture of all three siRNA plasmids was then transfected into WT and puma−/− cortical neurons. The plasmid coexpressed GFP to allow for identification of Noxa siRNA-expressing neurons. Apoptotic nuclei of neurons that were GFP positive, had noxa attenuated, or had been transfected with scrambled sequence were quantified in control and epoxomicin-treated samples. Attenuation of noxa expression in WT neurons did not significantly alter cell death induced by epoxomicin. More importantly, attenuation of noxa expression in puma−/− cortical neurons did not result in additional protection (Fig. 4C).
FIG. 4.

Knockdown of proapoptotic Bcl-2 family genes noxa and bax, does not confer additional protection to puma−/− neocortical neurons. Three noxa siRNAs were tested for their ability to attenuate noxa gene expression in neurons. (A) The siRNAs were transfected into neurons during culture preparation (DIV 0) using Amaxa (mouse neofection kit). Neurons were subsequently (DIV 5) treated with epoxomicin for 24 h or vehicle (DMSO, 0.1%), and samples were prepared for RT-qPCR. noxa gene expression was given as n-fold expression over control and normalized to β-actin. (B) The transfection efficiency of Amaxa-transfected neurons at DIV 5 was assessed by quantifying GFP-positive neurons within the cultures. KD, knockdown. (C) The number of apoptotic neurons from GFP-expressing cells in WT and puma−/− in scramble or noxa siRNA-transfected neurons was quantified in control- or epoxomicin (50 nM)-treated samples after 24 h (n = 123 to 150 cells/time point quantified). *, P < 0.05 compared to epoxomicin-treated control siRNA (ANOVA and Tukey's post hoc test). ns, not significant. (D) Western blotting of Bax expression 24 h posttransfection with either control or Bax siRNA sequences. β-Actin served as a loading control. (E) The number of apoptotic nuclei in WT and puma−/− neurons transfected with either Bax siRNA or scramble siRNA was quantified in control- or epoxomicin-treated cultures. siRNA was cotransfected with a plasmid expressing GFP to allow for identification of transfected neurons. (F) WT and puma−/− cortical neurons were transfected with scramble or Bax siRNA and subsequently treated with colchicine (10 μM) or vehicle, and apoptosis was assessed as in panel E. *, P < 0.05 compared drug-treated control siRNA (ANOVA and Tukey's post hoc test). ns, not significant. The experiments were repeated three times with independent culture preparations with similar results.
The possibility remained that other BH3-only proteins not tested in this study or identified to date contributed to epoxomicin-induced apoptosis. We therefore decided to test whether bax gene silencing afforded additional protection in puma-deficient neurons. Cortical neurons, as for most central and peripheral neurons, exclusively express an alternatively spliced, antiapoptotic variant of Bak, N-Bak (65), with bax deletion being sufficient to inhibit the activation of the mitochondrial apoptosis pathway (17). bax knockdown was achieved by cotransfecting three bax siRNA duplexes along with a GFP-expressing plasmid (Fig. 4D). Apoptotic nuclei of GFP-expressing bax siRNA- or control siRNA-transfected neurons were quantified in control and epoxomicin-treated samples. As expected, transfection of bax siRNA decreased apoptosis in response to epoxomicin (Fig. 4E). We did not, however, observe a difference in the degree of protection achieved by bax gene silencing when compared to the protection observed in puma−/− cells. Moreover, puma−/− cells transfected with bax siRNA did not offer significant protection over puma−/− neurons transfected with the scrambled sequence (Fig. 4E). In contrast, WT and puma−/− neurons transfected with bax siRNA were protected from colchicine-induced cell death compared to cells transfected with the scrambled sequence (Fig. 4F). Taken together, these results suggest that Puma and Bax constitute the major proteins responsible for the activation of the mitochondrial apoptosis pathway and suggest the existence of alternative cell death pathways leading to proteasome inhibition-induced apoptosis.
Proteasome inhibition induces caspase-dependent as well as caspase-independent cell death in puma-deficient neurons.
To explore potential alternative cell death pathways, we took an alternative approach and analyzed cell death dynamics at the single-cell level. We analyzed caspase-3-like activity by measuring cleavage of the caspase-3 substrate DEVD in single cells using a fluorescence resonance energy transfer (FRET)-based probe, SCAT3. Cleavage of the SCAT3-DEVD-FRET probe by caspase-3 or caspase-3-like proteases results in FRET disruption and is detected by a reduced fluorescence in the FRET channel and a coinciding increase in CFP fluorescence with a decrease in YFP fluorescence (Fig. 5A and B), as shown previously (50). Therefore, monitoring for increases in the ratio of CFP/YFP fluorescence serves as an indicator for caspase-3 like activity. In parallel, we measured changes in mitochondrial membrane potential (Δψm) using the fluorometric probe tetramethylrhodamine methyl ester (TMRM). As a control, WT cortical neurons were treated with 300 nM staurosporine (STS), a kinase inhibitor which is not associated with the activation of proteasomal stress and which predominantly activates the mitochondrial apoptosis pathway in neurons (34). Experiments in STS-treated neurons demonstrated that the loss of TMRM fluorescence coincided with the release of the mitochondrial intermembrane protein Smac-YFP, an indicator of MOMP (data not shown), similar to previous findings by our group and others (23, 49).
FIG. 5.

FRET-based single-cell analysis of caspase-3-like activity and mitochondrial membrane potential in WT and puma−/− neurons reveals caspase-dependent and caspase-independent cell death. (A and B) The SCAT3 FRET probe used consisted of CFP and YFP fluorophores linked together by a region containing the caspase-3 substrate sequence DEVD. Upon cleavage of the DEVD linker, the fluorescent resonance energy transfer (FRET) excitation between CFP and YFP is disrupted and detected by a decrease in FRET fluorescent intensity. This results in an increased energy transfer to CFP, as detected by an increase in CFP fluorescence. YFP excitation was used as a control for changes in fluorescence not directly related to probe cleavage, such as changes in cell volume, and therefore the data are expressed as a ratio of CFP to YFP. TMRM is used as a Δψm indicator in the nonquenched mode and measured in parallel. Here, WT neurons were treated with STS (300 nM, 8 h), or WT and puma−/− neurons were treated with epoxomicin for 24 h on the stage of a Zeiss 5Live confocal microscope. Fluorescent measurements were captured for TMRM, FRET, CFP, and YFP in real-time. All neurons that lost Δψm were categorized into those where DEVD cleavage occurred prior to Δψm loss or post-Δψm loss or those in which no DEVD cleavage was detected. Sigmoidal fits were applied to traces and the point initiation of onset (indicated by arrows) or endpoints determined as previously described (48). (C, E, and G) Representative traces of cells which undergo FRET disruption post-Δψm loss (C) or prior to Δψm loss (E) or which undergo Δψm loss in the absence of FRET disruption (G). t-onset, time of onset. (D, F, and H) Quantification of the number of cells in STS-treated WT neurons (n = 11) or in epoxomicin (50 nM, 24 h)-treated WT (n = 75) and puma−/− (n = 96) neurons with FRET disruption either prior to Δψm loss (D) or post-Δψm loss (F) or cells which undergo Δψm loss in the absence of FRET disruption (H). Data were obtained from 6 (WT-STS), 20 (WT-epoxo), and 28 (puma−/−-epoxo) separate experiments from 5 to 25 independent cultures. All data are means ± SEM. *, P < 0.05 compared to WT (D to H) as assessed by Fisher's exact t test.
Using loss of Δψm or cell shrinkage as an indicator of cell death, we noted that puma-deficient neurons did not lose Δψm and did not undergo cell shrinkage (data not shown) as frequently as WT cells in response to epoxomicin, reflecting the protection observed in our population-based cell death assays (Fig. 3C and D). We then focused on the kinetics of cell death signaling in individual dying neurons. In the case of STS treatment, all dying neurons monitored displayed a loss in Δψm, followed by an increase in CFP/YFP ratio indicative of FRET probe cleavage within a period of 15 ± 3 min, as previously described in other systems (49, 50). In response to epoxomicin, however, we noted that the individual responses were much more diverse. Only 75% of WT and 53% of puma−/− neurons displayed FRET probe cleavage following Δψm loss (Fig. 5C and D). Interestingly, a small percentage of both WT and puma−/− neurons showed FRET probe cleavage prior to loss of Δψm (11% of neurons in WT versus 8% in puma−/−) (Fig. 5E and F). As active caspases are subject to enforced proteasomal degradation (10), it was possible that this submaximal caspase activity was a result of autoactivation of caspase-3 (or potentially of initiator caspases) upon inhibition of proteasomal degradation with epoxomicin treatment. Using our recently developed and experimentally validated mathematical model of effector caspase activation (50), we could indeed successfully remodel submaximal caspase-3 activity upon 90 to 99% inhibition of the proteasome, which would occur with a delay of several hours following epoxomicin treatment (Fig. 6A and B). However, due to the relatively small number of neurons showing this behavior, this cell death pathway was not further experimentally explored. More interestingly, we also noted that a subset of neurons showed a significant loss of Δψm in the absence of FRET disruption prior to or following the loss of Δψm (Fig. 5G and H). This was more frequently observed in puma−/− neurons than in WT neurons (39% in puma−/− neurons compared to 14% in WT neurons).
FIG. 6.

Proteasome inhibition and subsequent reduced degradation of active caspases can lead to autoactivation. Shown is a mathematical model of apoptotic cell death through spontaneous (spont.) activation and imbalance of protein turnover. (A) Model schematic assuming a 0.1% initial, spontaneous caspase-3 activity which gets amplified by caspase-3 autofeedback. Cleavage of the cellular substrate is prevented through heterodimerization and inhibition of caspase-3 by XIAP (indicated by “XIAP ∼ Caspase-3”). The model further considered caspase-3 cleavage (solid arrows) of XIAP to its fragments BIR12 and BIR3R (neglected). Dashed arrows indicate the constant turnover of endogenous proteins XIAP and procaspase-3, as well as the decay of proteins that are activated in the signaling cascade. (B) Cellular substrate cleavage as a consequence of the deregulation of protein turnover balance induced by proteasome (prot.) inhibition. With inhibition higher than 90%, a robust cleavage of cellular substrate was predicted with onset of 5% substrate cleavage at approximately 10 to 30 h. Inhibitions less than 80% led to a complete abolishment of robust caspase-3 activation and therefore no substrate cleavage. admin, administration.
Characterization of puma- and caspase-independent cell death.
To investigate this cell death pathway further, we determined whether these cells underwent MOMP. Using single-cell imaging of neurons transfected with the MOMP indicator Smac-YFP, we determined that a minority of WT neurons lost Δψm without a coinciding Smac release (Fig. 7A and C) and that this occurred more frequently in puma−/− neurons (Fig. 7B and C). Approximately 16% of WT neurons and 40% of puma−/− neurons lost Δψm in the absence of Smac-YFP release (Fig. 7C). This distribution was similar to the distribution of WT and puma−/− neurons displaying Δψm depolarization in the absence of caspase activation (Fig. 5H).
FIG. 7.

Characterization of puma- and caspase-independent cell death. WT (A) and puma−/− (B) neurons were transfected with Smac-YFP and 24 h posttransfection loaded with TMRM (20 nM) in experimental buffer and mounted on the thermostatically regulated stage of a Zeiss 5Live confocal microscope. Neurons were treated with epoxomicin (50 nM), and images were acquired every 5 min. (C) Quantification of the percentage of WT and puma−/− neurons where Δψm loss occurred in the absence of Smac release. Smac release was taken as reduction in the standard deviation (S.D.) of Smac-YFP fluorescence (n = 26 and n = 15 cells for WT and puma−/− neurons, respectively, from at least five independent experiments; *, P < 0.05 for comparisons as assessed by Fisher's exact t test). (D) Duration of Δψm loss was measured in WT neurons undergoing FRET disruption and puma-deficient neurons in which disruption was absent (n = 15 for WT and n = 16 for puma−/− cells from at least 10 independent experiments; *, P < 0.05). (E) Images illustrating type I and type II apoptosis. puma−/− neurons were transfected with Smac-YFP and 24 h posttransfection loaded with TMRM (20 nM) and Hoechst (1 μg/ml) in experimental buffer. Neurons were treated with epoxomicin (50 nM), and images were acquired in real time every 5 min. Scale bar, 10 μm. (F) Images illustrating the lack of nuclear fragmentation in puma−/− neurons. WT and puma−/− neurons were treated with epoxomicin (50 nM) and stained with Hoechst 24 h posttreatment. Scale bar, 15 μm.
Neurons displaying caspase-independent apoptosis underwent a more delayed loss of Δψm, which occurred over 78 ± 12 min compared to 34 ± 6 min in neurons exhibiting caspase activity (Fig. 7D). In addition, these neurons underwent rapid nuclear condensation and cell shrinkage in the absence of Smac-YFP release (Fig. 7E). Moreover, nuclear fragmentation indicative of caspase activation was virtually absent in puma-deficient cells compared to WT cells (Fig. 7F). This piece of data suggested the activation of a type II apoptosis/apoptosis-like programmed cell death (21, 22) that could substitute for puma deficiency.
Autophagy does not contribute to proteasome inhibitor-induced cell death.
Proteasome inhibition has been associated with the activation of macroautophagy in cancer cells and neurons (57, 62). Macroautophagy, referred to hereafter as “autophagy,” is primarily a survival response whereby catabolic processes such as degradation of cellular components result in recycling of constituents to facilitate essential processes during stress or starvation, but it has also been implicated as a cell death pathway (73).
To investigate the contribution of autophagy, we performed Western blot analysis for autophagy-associated proteins LC3-II and p62 on WT and puma−/− neocortical lysates from control or epoxomicin-treated cultures. LC3-II and p62 are incorporated into complete autophagosomes and have been previously used as markers for autophagy (31). Cultures were also treated in the presence or absence of E64 D and pepstatin A (E/P) to inhibit lysosomal degradation of autophagocytosed proteins, enabling the monitoring of LC3-II accumulation as a measure of autophagic flux (31). In WT and puma−/− cultures treated with epoxomicin, LC3-II protein levels did not increase at time points when cell death occurred (Fig. 8A). We also could not detect alterations in p62 levels during treatment, suggesting that autophagic pathways were not prominently activated by epoxomicin.
FIG. 8.

Autophagy is not active at time points associated with epoxomicin-induced cell death. (A) WT and puma−/− cortical neurons were treated with epoxomicin in the presence or absence of E64-D/pepstatin A (E/P; 10 μg/ml). Western blotting was performed for LC3 and p62. β-Actin was used as a loading control. (B) WT and puma−/− neurons were transfected with GFP-LC3, and 24 h posttransfection, the neurons were treated with epoxomicin (50 nM) or bortezomib (100 nM) for 24 h or serum starved (Starv.) in Hanks' balanced salt solution (HBSS) for 4 h in the presence and absence of 1 mM autophagy inhibitor 3-methyl adenine (3-MA). Scale bar, 10 μm. (C) Quantification of the percentage of GFP-LC3 cells positive for puncta in neurons treated as described in panel B above (n = 200 to 300 cells/treatment). Similar results were observed in two independent experiments. (D and E) WT and puma−/− cortical neurons were treated with epoxomicin (D) or bortezomib (E) in the presence or absence of 3-MA. Cells with condensed nuclei were expressed as a percentage of total neurons in a field. All data are means ± SEM. *, P < 0.05 compared to the WT control (Con) (ANOVA and Tukey's post hoc test). Experiments were repeated three times from independent cultures with similar results.
Furthermore, cells transfected with GFP-LC3 and subsequently treated with epoxomicin or bortezomib did not show increased GFP puncta, indicative of autophagosome formation (Fig. 8B and C). GFP puncta were formed in serum-starved neurons, a positive control for autophagy induction, but were absent when starvation was induced in the presence of 3-methyl adenine (3-MA) (Fig. 8B and C). Moreover, WT and puma−/− neurons exposed to either epoxomicin or bortezomib were not rescued from cell death by treatment with 3-MA (Fig. 8D and E). Taken together, these data demonstrated that autophagy was not prominent at time points associated with cell death in our paradigm and was unlikely to contribute to caspase-independent, proteasome inhibitor-induced cell death in neurons.
Proteasome inhibition induces lysosomal permeabilization in WT and puma−/− neurons, and induction of puma-independent cell death is cathepsin-dependent.
Cathepsins released into the cytosol through lysosomal membrane permeabilization (LMP) may result in proteolysis and progression of cell death (22). LMP has been suggested to be induced by p53 (76) and has recently been linked to proteasome inhibition (2). To investigate whether proteasome inhibition could induce LMP, we stained WT and puma−/− neurons with the lysosomotropic dye LysoTracker Red and quantified the percentage of cells with diffuse LysoTracker Red staining, indicative of lysosomal acidification and rupture (7, 70). Treatment with epoxomicin or bortezomib reduced the number of neurons with intact LysoTracker Red staining in WT and puma−/− neurons (Fig. 9A). Cathepsin B, one of the main proteases released from lysosomes, has previously been shown to induce caspase-independent cell death associated with nuclear condensation (21), similar to that observed in our paradigm (Fig. 7E and F). Cathepsin B immunostaining of WT and puma−/− neurons treated with epoxomicin or bortezomib resulted in redistribution of cathepsin B staining with a loss of vesicular localization (Fig. 9B), suggesting that lysosomal rupture occurred. Moreover, staining with the lysosomal marker LAMP1 revealed no significant loss of staining following epoxomicin or bortezomib treatment, suggesting that the decrease in LysoTracker Red staining was not related to decreased lysosomal number (Fig. 9C).
FIG. 9.

Proteasomal stress induces lysosomal leakage. (A) WT and puma−/− neurons were treated with epoxomicin (50 nM), bortezomib (100 nM), or vehicle (0.1% DMSO) for 24 h in the presence of Lysotracker Red (0.25 μM). Determination of the reduction in lysosomal membrane integrity was assessed by quantification of the percentage of cells with reduced Lysotracker puncta/staining. Images were captured from random fields (200 to 300 neurons/field) with three fields captured per well under identical camera settings, and three wells were analyzed per time point. (B) Neurons were treated with epoxomicin (50 nM) or bortezomib (100 nM) for 24 h and stained for cathepsin B. Lysosomal disruption was assessed by quantification of the percentage of cells with redistributed cathepsin B staining. Images were captured from random fields, with each field assessed containing approximately 100 neurons, three fields were captured per well, and three wells were analyzed per time point. (C) WT and puma−/− cortical neurons were treated with epoxomicin (50 nM) or bortezomib (100 nM) for 24 h and stained for LAMP1, a membrane-associated lysosomal marker (scale bar, 10 μm). Experiments were performed three times from independent cultures with similar results obtained.
Interestingly, real-time single-cell imaging experiments with puma−/− neurons pretreated with CA-074 methyl ester (CA-074-ME), a specific cathepsin B inhibitor, and subsequently treated with epoxomicin revealed a significant reduction in the percentage of neurons showing caspase-independent cell death (Fig. 10A). Furthermore, apoptotic nuclei in cultures pretreated with CA-074-ME prior to treatment with epoxomicin or bortezomib were significantly reduced in both WT and puma−/− neurons (Fig. 10B and C). In addition, the protection afforded by caspase inhibition using Z-VAD-FMK was exacerbated to complete protection when CA-074-ME was present too (Fig. 10D and E). Taken together, these data suggest that lysosomally mediated cell death proceeded independently of caspase activity and that both caspase-dependent and lysosomally mediated caspase-independent pathways mediate cell death during prolonged proteasome inhibition in neurons.
FIG. 10.

Proteasomal stress induces cathepsin-mediated caspase-independent cell death. (A) puma−/− neurons were protected from cell death in the presence of CA-074-ME, as indicated by reduced loss of Δψm and FRET disruption. Shown is quantification of the number of cells undergoing FRET disruption in Puma-deficient neurons in the presence or absence of CA074-ME (n = 15 and n = 18 cells, respectively). AU, arbitrary units. *, P < 0.05 as assessed by Fisher's exact t test. (B and C) WT and puma−/− cortical neurons were treated with epoxomicin (B) or bortezomib (C) in the presence or absence of CA-074-ME (10 μM). The number of neurons with apoptotic nuclei was expressed as a percentage of total neurons in a field. *, P < 0.05 compared to WT treated; #, P < 0.05, as indicated (ANOVA and Tukey's post hoc test). (D and E) WT and puma−/− cortical neurons were treated with epoxomicin (D) or bortezomib (E) in the presence or absence of CA074-ME (10 μM) and/or Z-VAD-FMK (100 μM). The number of neurons with condensed nuclei was expressed as a percentage of total neurons in a field. Data are shown as means ± SEM. *, P < 0.05 compared to WT treated cultures; #, P < 0.05, as indicated (ANOVA and Tukey's post hoc test). Experiments were carried out at least three times from independent cultures with similar results.
DISCUSSION
Similar to dysregulation in other protein quality control systems such as the endoplasmic reticulum (ER) and autophagy, disturbances in the UPS can have severe consequences for the cell. Proteasomal stress initially activates conversed, cytoprotective stress responses, such as the expression of molecular chaperones facilitating protein degradation and preventing the accumulation of protein aggregates. However, excessive or prolonged proteasome inhibition results in apoptosis (38). We demonstrate here that in neurons, proteasome inhibitor-induced cell death is partially mediated by a Bcl-2 family-controlled mitochondrial apoptosis pathway. This occurs via induction of the p53 target gene puma, coding for a proapoptotic BH3-only protein, with little evidence for a contribution of other BH3-only proteins. Moreover, using a single-cell imaging approach, we also demonstrate the importance of alternative cell death pathways that bypass a requirement for puma, compensating for its deficiency. These involve MOMP-independent caspase activation and, more prominently, lysosomal permeabilization and the activation of cathepsin-mediated, caspase-independent cell death.
Previous studies have demonstrated a key role for the p53 target gene puma in proteasome inhibiton (15). The tumor suppressor p53 is constitutively degraded by the proteasome and is stabilized following proteasome inhibition (59), allowing increased expression of its target genes puma and noxa. Concurrent with our previous results in human cancer cells (15), we identified Puma as a central mediator of proteasome inhibition-induced apoptosis. Noxa has previously been reported to contribute to bortezomib-induced apoptosis in myeloma and melanoma cells (47). However, we found little evidence implicating Noxa as an instigator of cell death in neurons. Noxa, unlike Puma, is believed to be an enabler BH3-only protein which may not have the functional capacity to activate Bax directly and induce MOMP independently of other BH3-only proteins (36, 40). In support of this hypothesis, we found no evidence of a role for Noxa in the absence of puma. Moreover, despite induction at both mRNA and protein levels, loss of bim expression did not afford protection either. We previously obtained similar results in HCT116 colon cancer cells silenced for bim gene expression (in the presence and absence of puma), as well as mouse embryonic fibroblasts deficient for bim (15), and similar findings have been reported in hepatocytes (1). A lack of protection in bim-deficient cells, despite induction in WT cells, was also observed in cortical neurons treated with arsenite (71) or exposed to oxidative stress (61) as well as PC12 cells exposed to 6-hydroxydopamine (3). It therefore appears that Bim is subject to further posttranslational control steps or is possibly maintained in an inactive state through the action of cytoprotective proteins.
In spite of significant evidence for the activation of the Puma/Bax-dependent mitochondrial apoptosis pathway in our model, we found that puma or bax deficiency only partially protected. Clearly, we cannot fully exclude that the bax gene-silencing approach did not provide a complete gene knockout and that alternative BH3-only proteins such as Nbk/Bik, Bmf, or Bad may have contributed to cell death. However, using a single-cell imaging approach, we demonstrate that the remaining cell death observed was rather due to the activation of alternative caspase-dependent and -independent cell death pathways. Indeed, to date the majority of studies concerning signal transduction pathways following proteasome inhibition have been performed in cell populations. Time-lapse analysis of mitochondrial function and caspase activation in single neurons identified two additional cell populations in which caspase activity occurred prior to mitochondrial engagement or in which cell death occurred in the absence of caspase activity. Importantly, this was specific to proteasomal stress, as a similar diversity was not seen after treatment with staurosporine.
A very small fraction of neurons underwent caspase activation prior to mitochondrial depolarization. It is possible that this caspase activity resulted from ligand-induced activation of a death receptor pathway triggered by the activation of p53 (43, 72) or by a caspase-2-dependent pathway. Minimal activation of caspase-8 as detected by Western blotting could be responsible for apoptosis in these cells as caspase-8 may also cleave the DEVD probe. Since procaspases exhibit a low zymogenic activity (56), which can lead to their autoactivation, and activated caspases are readily degraded by the proteasome (64), in instances of proteasome inhibition the ability of activate caspases to further cleave and activate their pro-forms may induce a feedback amplification loop resulting in a slow but detectable caspase activity. Indeed, computational modeling based on our previously established APOPTO-CELL model of caspase activation (50) demonstrated that proteasome inhibition of 90 to 99% can lead to caspase autoactivation following an initial lag time of approximately 10 to 30 h. The model accounted for direct autoactivation of caspase-3, as suggested previously (67), yet similar principles may apply for indirect caspase-3 feedback loops via caspase-8 or caspase-6 and caspase-8 (60, 67). Upon sufficient caspase-3 or -8 activation, the initially mitochondrion-independent pathway may also be amplified through caspase-dependent posttranslational activation of other Bcl-2 proteins such as Bid, Bcl-2, and Bcl-xL (11, 37). We did not, however, observe any protection against epoxomicin-induced apoptosis in bid-deficient neurons. It is possible that the delayed loss of Δψm detected in these cells was due to a caspase-mediated mitochondrial dysfunction (51).
While the fraction of cells exhibiting puma-independent caspase activation was relatively small, a more significant proportion of neurons underwent a puma- and caspase-independent apoptosis. We first hypothesized that this cell death was due to increased autophagy. Catabolic degradation of bulky long-lived proteins, aggregates, and even whole organelles is facilitated by autophagy and subsequent lysosomal degradation. Recently, proteasomal degradation and lysosomal degradation have been linked with the view that impaired proteasomal degradation may be compensated for by increased autophagic flux (45). It seems, however, that autophagy induction as a compensatory mechanism for proteasomal dysfunction is not universal and may be cell type and/or time-dependent specific (32). Interestingly, we found that epoxomicin treatment did not result in increased autophagic flux in WT or puma−/− neurons but did result in an apparent decrease in autophagy indicated by reduced LC3-II accumulation independent of puma expression. It has been suggested that detection of autophagy in cortical neurons may be difficult due to the highly efficient clearance of autophagosomes by lysosomes (4). However, we showed robust autophagic flux in untreated neurons by measuring LC3-II accumulation under conditions that prevent lysosomal protein turnover (E63 D and pepstatin A). In contrast, epoxomicin or bortezomib treatment caused a complete inhibition of LC3-II accumulation, suggesting that LC3-mediated autophagy was inhibited. Experiments in which autophagy was prevented at the sequestration phase with 3-MA did not alter cell death dynamics. Therefore, we conclude that autophagy or associated cell death is not induced in our paradigm or contributing to puma-independent cell death.
In contrast, we provide evidence that LMP and cathepsins are major contributors of puma-independent cell death in response to proteasome inhibition. Proteasome inhibition has previously been shown to result in increased lysosomal enzyme activity in neurons (52). Moreover, cytoprotective chaperones induced following proteasomal stress such as Hsp70 have been demonstrated to exert their protective activity via a stabilization of lysosomal membrane integrity (41). Evidence has also been provided that LMP can inhibit autophagy in a cathepsin-dependent manner following proteasomal inhibition in breast cancer cells (46), a finding that could explain the inhibition of autophagic activation in our study. Release of cathepsins has been shown to mediate cell death in vitro in cytokine- or bile salt-induced apoptosis in cancer cells (18, 55) and in vivo following global cerebral ischemia in primates (74). In our study, the protection offered by inhibition of cathepsins was additive to the protection observed with the puma gene knockout, suggesting that cathepsin-mediated cell death was puma independent. The increased cathepsin-dependent cell death observed in puma-deficient cells suggested that lysosomal/cathepsin-mediated cell death can function as a backup mechanism when puma expression is absent or delayed. Finally, it should be noted that inhibition of cathepsins and caspases led to a complete protection from epoxomicin or bortezomib-induced cell death. Our data therefore suggest that caspases and cathepsins are the major executioners of puma-dependent and puma-independent cell death following proteasomal inhibition in neurons (Fig. 11).
FIG. 11.
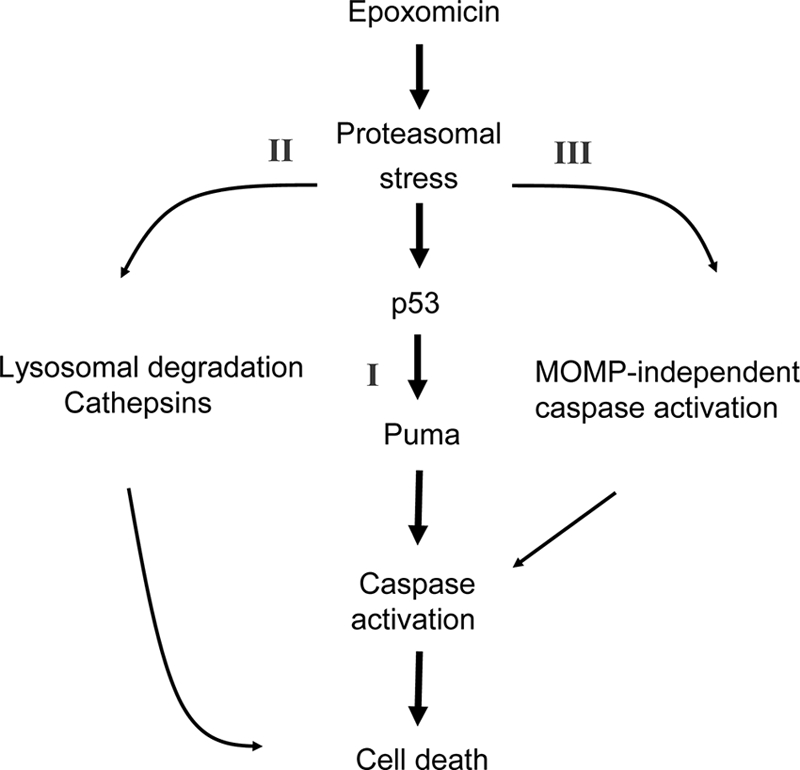
Proposed pathways of proteasomal stress-induced cell death. In neocortical neurons, proteasome inhibition induces three distinct cell death pathways. Increased levels of p53 and the p53 target gene puma partially mediate caspase-dependent cell death. In addition, in a small number of neurons, mitochondrion-independent activation of caspases may occur. Moreover, lysosomal degradation and cathepsin-mediated cell death constitute another distinct cell death pathway, which may substitute for puma deficiency following proteotoxic stress.
Acknowledgments
This study was supported by grants from Science Foundation Ireland (08/IN1/1949) and the Health Research Board (RP/2006/333) to J. H. M. Prehn. L. P. Tuffy was a recipient of an IRCSET scholarship (325/2005-2009).
We thank T. Nagai for the gift of cyan fluorescent protein-DEVD-Venus plasmid used for construction of the SCAT-DEVD-FRET probe; N. Mizushima for the gift of the LC3 plasmid; A. Strasser for puma-, bim-, and brd-deficient mice; H. Düssmann and T. Bernas for imaging expertise; and Sarah Cannon for excellent technical assistance.
Footnotes
Published ahead of print on 4 October 2010.
REFERENCES
- 1.Anan, A., E. S. Baskin-Bey, S. F. Bronk, N. W. Werneburg, V. H. Shah, and G. J. Gores. 2006. Proteasome inhibition induces hepatic stellate cell apoptosis. Hepatology 43:335-344. [DOI] [PubMed] [Google Scholar]
- 2.Berndtsson, M., M. Beaujouin, L. Rickardson, A. M. Havelka, R. Larsson, J. Westman, E. Liaudet-Coopman, and S. Linder. 2009. Induction of the lysosomal apoptosis pathway by inhibitors of the ubiquitin-proteasome system. Int. J. Cancer 124:1463-1469. [DOI] [PubMed] [Google Scholar]
- 3.Biswas, S. C., E. Ryu, C. Park, C. Malagelada, and L. A. Greene. 2005. Puma and p53 play required roles in death evoked in a cellular model of Parkinson disease. Neurochem. Res. 30:839-845. [DOI] [PubMed] [Google Scholar]
- 4.Boland, B., A. Kumar, S. Lee, F. M. Platt, J. Wegiel, W. H. Yu, and R. A. Nixon. 2008. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J. Neurosci. 28:6926-6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonfoco, E., S. Ceccatelli, L. Manzo, and P. Nicotera. 1995. Colchicine induces apoptosis in cerebellar granule cells. Exp. Cell Res. 218:189-200. [DOI] [PubMed] [Google Scholar]
- 6.Bouillet, P., D. Metcalf, D. C. Huang, D. M. Tarlinton, T. W. Kay, F. Kontgen, J. M. Adams, and A. Strasser. 1999. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science 286:1735-1738. [DOI] [PubMed] [Google Scholar]
- 7.Boya, P., K. Andreau, D. Poncet, N. Zamzami, J. L. Perfettini, D. Metivier, D. M. Ojcius, M. Jaattela, and G. Kroemer. 2003. Lysosomal membrane permeabilization induces cell death in a mitochondrion-dependent fashion. J. Exp. Med. 197:1323-1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bruna, J., E. Udina, A. Ale, J. J. Vilches, A. Vynckier, J. Monbaliu, L. Silverman, and X. Navarro. 2010. Neurophysiological, histological and immunohistochemical characterization of bortezomib-induced neuropathy in mice. Exp. Neurol. 223:599-608. [DOI] [PubMed] [Google Scholar]
- 9.Cavaletti, G., A. Gilardini, A. Canta, L. Rigamonti, V. Rodriguez-Menendez, C. Ceresa, P. Marmiroli, M. Bossi, N. Oggioni, M. D'Incalci, and R. De Coster. 2007. Bortezomib-induced peripheral neurotoxicity: a neurophysiological and pathological study in the rat. Exp. Neurol. 204:317-325. [DOI] [PubMed] [Google Scholar]
- 10.Chen, L., L. Smith, Z. Wang, and J. B. Smith. 2003. Preservation of caspase-3 subunits from degradation contributes to apoptosis evoked by lactacystin: any single lysine or lysine pair of the small subunit is sufficient for ubiquitination. Mol. Pharmacol. 64:334-345. [DOI] [PubMed] [Google Scholar]
- 11.Cheng, E. H., D. G. Kirsch, R. J. Clem, R. Ravi, M. B. Kastan, A. Bedi, K. Ueno, and J. M. Hardwick. 1997. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science 278:1966-1968. [DOI] [PubMed] [Google Scholar]
- 12.Chipuk, J. E., T. Moldoveanu, F. Llambi, M. J. Parsons, and D. R. Green. 2010. The BCL-2 family reunion. Mol. Cell 37:299-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ciechanover, A., D. Finley, and A. Varshavsky. 1984. The ubiquitin-mediated proteolytic pathway and mechanisms of energy-dependent intracellular protein degradation. J. Cell Biochem. 24:27-53. [DOI] [PubMed] [Google Scholar]
- 14.Colland, F. 2010. The therapeutic potential of deubiquitinating enzyme inhibitors. Biochem. Soc. Trans. 38:137-143. [DOI] [PubMed] [Google Scholar]
- 15.Concannon, C. G., B. F. Koehler, C. Reimertz, B. M. Murphy, C. Bonner, N. Thurow, M. W. Ward, A. Villunger, A. Strasser, D. Kogel, and J. H. Prehn. 2007. Apoptosis induced by proteasome inhibition in cancer cells: predominant role of the p53/PUMA pathway. Oncogene 26:1681-1692. [DOI] [PubMed] [Google Scholar]
- 16.Concannon, C. G., L. P. Tuffy, P. Weisova, H. P. Bonner, D. Davila, C. Bonner, M. C. Devocelle, A. Strasser, M. W. Ward, and J. H. Prehn. 2010. AMP kinase-mediated activation of the BH3-only protein Bim couples energy depletion to stress-induced apoptosis. J. Cell Biol. 189:83-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deckwerth, T. L., J. L. Elliott, C. M. Knudson, E. M. Johnson, Jr., W. D. Snider, and S. J. Korsmeyer. 1996. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron 17:401-411. [DOI] [PubMed] [Google Scholar]
- 18.Deiss, L. P., H. Galinka, H. Berissi, O. Cohen, and A. Kimchi. 1996. Cathepsin D protease mediates programmed cell death induced by interferon-gamma, Fas/APO-1 and TNF-alpha. EMBO J. 15:3861-3870. [PMC free article] [PubMed] [Google Scholar]
- 19.Deveraux, Q. L., E. Leo, H. R. Stennicke, K. Welsh, G. S. Salvesen, and J. C. Reed. 1999. Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases. EMBO J. 18:5242-5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eissing, T., H. Conzelmann, E. D. Gilles, F. Allgower, E. Bullinger, and P. Scheurich. 2004. Bistability analyses of a caspase activation model for receptor-induced apoptosis. J. Biol. Chem. 279:36892-36897. [DOI] [PubMed] [Google Scholar]
- 21.Foghsgaard, L., D. Wissing, D. Mauch, U. Lademann, L. Bastholm, M. Boes, F. Elling, M. Leist, and M. Jaattela. 2001. Cathepsin B acts as a dominant execution protease in tumor cell apoptosis induced by tumor necrosis factor. J. Cell Biol. 153:999-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guicciardi, M. E., M. Leist, and G. J. Gores. 2004. Lysosomes in cell death. Oncogene 23:2881-2890. [DOI] [PubMed] [Google Scholar]
- 23.Heiskanen, K. M., M. B. Bhat, H. W. Wang, J. Ma, and A. L. Nieminen. 1999. Mitochondrial depolarization accompanies cytochrome c release during apoptosis in PC6 cells. J. Biol. Chem. 274:5654-5658. [DOI] [PubMed] [Google Scholar]
- 24.Hetschko, H., V. Voss, C. Senft, V. Seifert, J. H. Prehn, and D. Kogel. 2008. BH3 mimetics reactivate autophagic cell death in anoxia-resistant malignant glioma cells. Neoplasia 10:873-885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu, B. R., M. E. Martone, Y. Z. Jones, and C. L. Liu. 2000. Protein aggregation after transient cerebral ischemia. J. Neurosci. 20:3191-3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang, D. C., and A. Strasser. 2000. BH3-only proteins—essential initiators of apoptotic cell death. Cell 103:839-842. [DOI] [PubMed] [Google Scholar]
- 27.Kaufmann, T., L. Tai, P. G. Ekert, D. C. Huang, F. Norris, R. K. Lindemann, R. W. Johnstone, V. M. Dixit, and A. Strasser. 2007. The BH3-only protein bid is dispensable for DNA damage- and replicative stress-induced apoptosis or cell-cycle arrest. Cell 129:423-433. [DOI] [PubMed] [Google Scholar]
- 28.Kieran, D., I. Woods, A. Villunger, A. Strasser, and J. H. Prehn. 2007. Deletion of the BH3-only protein puma protects motoneurons from ER stress-induced apoptosis and delays motoneuron loss in ALS mice. Proc. Natl. Acad. Sci. U. S. A. 104:20606-20611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kikuchi, H., G. Almer, S. Yamashita, C. Guegan, M. Nagai, Z. Xu, A. A. Sosunov, G. M. McKhann II, and S. Przedborski. 2006. Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc. Natl. Acad. Sci. U. S. A. 103:6025-6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kitada, T., S. Asakawa, N. Hattori, H. Matsumine, Y. Yamamura, S. Minoshima, M. Yokochi, Y. Mizuno, and N. Shimizu. 1998. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605-608. [DOI] [PubMed] [Google Scholar]
- 31.Klionsky, D. J., et al. 2008. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4:151-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Korolchuk, V. I., F. M. Menzies, and D. C. Rubinsztein. 2010. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 584:1393-1398. [DOI] [PubMed] [Google Scholar]
- 33.Kristensen, B. W., H. Noer, J. B. Gramsbergen, J. Zimmer, and J. Noraberg. 2003. Colchicine induces apoptosis in organotypic hippocampal slice cultures. Brain Res. 964:264-278. [DOI] [PubMed] [Google Scholar]
- 34.Krohn, A. J., E. Preis, and J. H. Prehn. 1998. Staurosporine-induced apoptosis of cultured rat hippocampal neurons involves caspase-1-like proteases as upstream initiators and increased production of superoxide as a main downstream effector. J. Neurosci. 18:8186-8197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lang-Rollin, I., K. Vekrellis, Q. Wang, H. J. Rideout, and L. Stefanis. 2004. Application of proteasomal inhibitors to mouse sympathetic neurons activates the intrinsic apoptotic pathway. J. Neurochem. 90:1511-1520. [DOI] [PubMed] [Google Scholar]
- 36.Letai, A., M. C. Bassik, L. D. Walensky, M. D. Sorcinelli, S. Weiler, and S. J. Korsmeyer. 2002. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2:183-192. [DOI] [PubMed] [Google Scholar]
- 37.Li, H., H. Zhu, C. J. Xu, and J. Yuan. 1998. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94:491-501. [DOI] [PubMed] [Google Scholar]
- 38.Lopes, U. G., P. Erhardt, R. Yao, and G. M. Cooper. 1997. p53-dependent induction of apoptosis by proteasome inhibitors. J. Biol. Chem. 272:12893-12896. [DOI] [PubMed] [Google Scholar]
- 39.MacFarlane, M., W. Merrison, S. B. Bratton, and G. M. Cohen. 2002. Proteasome-mediated degradation of Smac during apoptosis: XIAP promotes Smac ubiquitination in vitro. J. Biol. Chem. 277:36611-36616. [DOI] [PubMed] [Google Scholar]
- 40.Michalak, E. M., A. Villunger, J. M. Adams, and A. Strasser. 2008. In several cell types tumour suppressor p53 induces apoptosis largely via Puma but Noxa can contribute. Cell Death Differ. 15:1019-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nylandsted, J., M. Gyrd-Hansen, A. Danielewicz, N. Fehrenbacher, U. Lademann, M. Hoyer-Hansen, E. Weber, G. Multhoff, M. Rohde, and M. Jaattela. 2004. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J. Exp. Med. 200:425-435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oda, E., R. Ohki, H. Murasawa, J. Nemoto, T. Shibue, T. Yamashita, T. Tokino, T. Taniguchi, and N. Tanaka. 2000. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288:1053-1058. [DOI] [PubMed] [Google Scholar]
- 43.Owen-Schaub, L. B., W. Zhang, J. C. Cusack, L. S. Angelo, S. M. Santee, T. Fujiwara, J. A. Roth, A. B. Deisseroth, W.-W. Zhang, E. Kruzel, and R. Radinsky. 1995. Wild-type human p53 and a temperature-sensitive mutant induce Fas/APO-1 expression. Mol. Cell. Biol. 15:3032-3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Palombella, V. J., O. J. Rando, A. L. Goldberg, and T. Maniatis. 1994. The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell 78:773-785. [DOI] [PubMed] [Google Scholar]
- 45.Pandey, U. B., Z. Nie, Y. Batlevi, B. A. McCray, G. P. Ritson, N. B. Nedelsky, S. L. Schwartz, N. A. DiProspero, M. A. Knight, O. Schuldiner, R. Padmanabhan, M. Hild, D. L. Berry, D. Garza, C. C. Hubbert, T. P. Yao, E. H. Baehrecke, and J. P. Taylor. 2007. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447:859-863. [DOI] [PubMed] [Google Scholar]
- 46.Periyasamy-Thandavan, S., W. H. Jackson, J. S. Samaddar, B. Erickson, J. R. Barrett, L. Raney, E. Gopal, V. Ganapathy, W. D. Hill, K. N. Bhalla, and P. V. Schoenlein. 2010. Bortezomib blocks the catabolic process of autophagy via a cathepsin-dependent mechanism, affects endoplasmic reticulum stress and induces caspase-dependent cell death in antiestrogen-sensitive and resistant ER+ breast cancer cells. Autophagy 6:19-35. [DOI] [PubMed] [Google Scholar]
- 47.Qin, J. Z., J. Ziffra, L. Stennett, B. Bodner, B. K. Bonish, V. Chaturvedi, F. Bennett, P. M. Pollock, J. M. Trent, M. J. Hendrix, P. Rizzo, L. Miele, and B. J. Nickoloff. 2005. Proteasome inhibitors trigger NOXA-mediated apoptosis in melanoma and myeloma cells. Cancer Res. 65:6282-6293. [DOI] [PubMed] [Google Scholar]
- 48.Rehm, M., H. Dussmann, R. U. Janicke, J. M. Tavare, D. Kogel, and J. H. Prehn. 2002. Single-cell fluorescence resonance energy transfer analysis demonstrates that caspase activation during apoptosis is a rapid process. Role of caspase-3. J. Biol. Chem. 277:24506-24514. [DOI] [PubMed] [Google Scholar]
- 49.Rehm, M., H. Dussmann, and J. H. Prehn. 2003. Real-time single cell analysis of Smac/DIABLO release during apoptosis. J. Cell Biol. 162:1031-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rehm, M., H. J. Huber, H. Dussmann, and J. H. Prehn. 2006. Systems analysis of effector caspase activation and its control by X-linked inhibitor of apoptosis protein. EMBO J. 25:4338-4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50a.Reimertz, C., D. Kögel, S. Lankiewiez, M. Poppe, and J. H. Prehn. 2001. Ca2+-induced inhibition of apoptosis in human SH-SY5Y neuroblastoma cells: degradation of apoptotic protease activating factor-I (APAF-1). J. Neurochem. 78:1256-1266. [DOI] [PubMed] [Google Scholar]
- 51.Ricci, J. E., C. Munoz-Pinedo, P. Fitzgerald, B. Bailly-Maitre, G. A. Perkins, N. Yadava, I. E. Scheffler, M. H. Ellisman, and D. R. Green. 2004. Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell 117:773-786. [DOI] [PubMed] [Google Scholar]
- 52.Rideout, H. J., I. Lang-Rollin, and L. Stefanis. 2004. Involvement of macroautophagy in the dissolution of neuronal inclusions. Int. J. Biochem. Cell Biol. 36:2551-2562. [DOI] [PubMed] [Google Scholar]
- 53.Rideout, H. J., and L. Stefanis. 2002. Proteasomal inhibition-induced inclusion formation and death in cortical neurons require transcription and ubiquitination. Mol. Cell Neurosci. 21:223-238. [DOI] [PubMed] [Google Scholar]
- 54.Riedl, S. J., M. Renatus, R. Schwarzenbacher, Q. Zhou, C. Sun, S. W. Fesik, R. C. Liddington, and G. S. Salvesen. 2001. Structural basis for the inhibition of caspase-3 by XIAP. Cell 104:791-800. [DOI] [PubMed] [Google Scholar]
- 55.Roberts, L. R., P. N. Adjei, and G. J. Gores. 1999. Cathepsins as effector proteases in hepatocyte apoptosis. Cell Biochem. Biophys. 30:71-88. [DOI] [PubMed] [Google Scholar]
- 56.Salvesen, G. S., and V. M. Dixit. 1999. Caspase activation: the induced-proximity model. Proc. Natl. Acad. Sci. U. S. A. 96:10964-10967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schwartz, A. L., R. A. Brandt, H. Geuze, and A. Ciechanover. 1992. Stress-induced alterations in autophagic pathway: relationship to ubiquitin system. Am. J. Physiol. 262:C1031-C1038. [DOI] [PubMed] [Google Scholar]
- 58.Shibue, T., S. Suzuki, H. Okamoto, H. Yoshida, Y. Ohba, A. Takaoka, and T. Taniguchi. 2006. Differential contribution of Puma and Noxa in dual regulation of p53-mediated apoptotic pathways. EMBO J. 25:4952-4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shkedy, D., H. Gonen, B. Bercovich, and A. Ciechanover. 1994. Complete reconstitution of conjugation and subsequent degradation of the tumor suppressor protein p53 by purified components of the ubiquitin proteolytic system. FEBS Lett. 348:126-130. [DOI] [PubMed] [Google Scholar]
- 60.Slee, E. A., M. T. Harte, R. M. Kluck, B. B. Wolf, C. A. Casiano, D. D. Newmeyer, H. G. Wang, J. C. Reed, D. W. Nicholson, E. S. Alnemri, D. R. Green, and S. J. Martin. 1999. Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J. Cell Biol. 144:281-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Steckley, D., M. Karajgikar, L. B. Dale, B. Fuerth, P. Swan, C. Drummond-Main, M. O. Poulter, S. S. Ferguson, A. Strasser, and S. P. Cregan. 2007. Puma is a dominant regulator of oxidative stress induced Bax activation and neuronal apoptosis. J. Neurosci. 27:12989-12999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stefanis, L., K. E. Larsen, H. J. Rideout, D. Sulzer, and L. A. Greene. 2001. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J. Neurosci. 21:9549-9560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stennicke, H. R., M. Renatus, M. Meldal, and G. S. Salvesen. 2000. Internally quenched fluorescent peptide substrates disclose the subsite preferences of human caspases 1, 3, 6, 7 and 8. Biochem. J. 350:563-568. [PMC free article] [PubMed] [Google Scholar]
- 64.Stennicke, H. R., and G. S. Salvesen. 2000. Caspases—controlling intracellular signals by protease zymogen activation. Biochim. Biophys. Acta 1477:299-306. [DOI] [PubMed] [Google Scholar]
- 65.Sun, Y. F., L. Y. Yu, M. Saarma, T. Timmusk, and U. Arumae. 2001. Neuron-specific Bcl-2 homology 3 domain-only splice variant of Bak is anti-apoptotic in neurons, but pro-apoptotic in non-neuronal cells. J. Biol. Chem. 276:16240-16247. [DOI] [PubMed] [Google Scholar]
- 66.Takemoto, K., T. Nagai, A. Miyawaki, and M. Miura. 2003. Spatio-temporal activation of caspase revealed by indicator that is insensitive to environmental effects. J. Cell Biol. 160:235-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Van de Craen, M., W. Declercq, I. Van den brande, W. Fiers, and P. Vandenabeele. 1999. The proteolytic procaspase activation network: an in vitro analysis. Cell Death Differ. 6:1117-1124. [DOI] [PubMed] [Google Scholar]
- 67a.Vareková, R. S., I. Bradác, M. Plchút, M. Skrdla, M. Wacenovsky, H. Mahr, G. Mayer, H. Tanner, H. Brugger, J. Withalm, P. Lederer, H. Huber, G. Gierlinger, R. Graf, H. Tafer, I. Hofacker, P. Schuster, and M. Polcík. 2008. www.rnaworkbench.com: a new program for analyzing RNA interference. Comput. Methods Programs Biomed. 90:89-94. [DOI] [PubMed] [Google Scholar]
- 68.Vaux, D. L., and J. Silke. 2005. IAPs, RINGs and ubiquitylation. Nat. Rev. Mol. Cell Biol. 6:287-297. [DOI] [PubMed] [Google Scholar]
- 69.Villunger, A., E. M. Michalak, L. Coultas, F. Mullauer, G. Bock, M. J. Ausserlechner, J. M. Adams, and A. Strasser. 2003. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 302:1036-1038. [DOI] [PubMed] [Google Scholar]
- 70.Werneburg, N. W., M. E. Guicciardi, S. F. Bronk, and G. J. Gores. 2002. Tumor necrosis factor-alpha-associated lysosomal permeabilization is cathepsin B dependent. Am. J. Physiol. Gastrointest. Liver Physiol. 283:G947-G956. [DOI] [PubMed] [Google Scholar]
- 71.Wong, H. K., M. Fricker, A. Wyttenbach, A. Villunger, E. M. Michalak, A. Strasser, and A. M. Tolkovsky. 2005. Mutually exclusive subsets of BH3-only proteins are activated by the p53 and c-Jun N-terminal kinase/c-Jun signaling pathways during cortical neuron apoptosis induced by arsenite. Mol. Cell. Biol. 25:8732-8747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu, G. S., T. F. Burns, E. R. McDonald III, W. Jiang, R. Meng, I. D. Krantz, G. Kao, D. D. Gan, J. Y. Zhou, R. Muschel, S. R. Hamilton, N. B. Spinner, S. Markowitz, G. Wu, and W. S. el-Deiry. 1997. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat. Genet. 17:141-143. [DOI] [PubMed] [Google Scholar]
- 73.Xue, L., G. C. Fletcher, and A. M. Tolkovsky. 1999. Autophagy is activated by apoptotic signalling in sympathetic neurons: an alternative mechanism of death execution. Mol. Cell Neurosci. 14:180-198. [DOI] [PubMed] [Google Scholar]
- 74.Yamashima, T., Y. Kohda, K. Tsuchiya, T. Ueno, J. Yamashita, T. Yoshioka, and E. Kominami. 1998. Inhibition of ischaemic hippocampal neuronal death in primates with cathepsin B inhibitor CA-074: a novel strategy for neuroprotection based on ‘calpain-cathepsin hypothesis. ’ Eur. J. Neurosci. 10:1723-1733. [DOI] [PubMed] [Google Scholar]
- 75.Yoo, S. J., J. R. Huh, I. Muro, H. Yu, L. Wang, S. L. Wang, R. M. Feldman, R. J. Clem, H. A. Muller, and B. A. Hay. 2002. Hid, Rpr and Grim negatively regulate DIAP1 levels through distinct mechanisms. Nat. Cell Biol. 4:416-424. [DOI] [PubMed] [Google Scholar]
- 76.Yuan, X. M., W. Li, H. Dalen, J. Lotem, R. Kama, L. Sachs, and U. T. Brunk. 2002. Lysosomal destabilization in p53-induced apoptosis. Proc. Natl. Acad. Sci. U. S. A. 99:6286-6291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhu, H., L. Zhang, F. Dong, W. Guo, S. Wu, F. Teraishi, J. J. Davis, P. J. Chiao, and B. Fang. 2005. Bik/NBK accumulation correlates with apoptosis-induction by bortezomib (PS-341, Velcade) and other proteasome inhibitors. Oncogene 24:4993-4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zou, H., R. Yang, J. Hao, J. Wang, C. Sun, S. W. Fesik, J. C. Wu, K. J. Tomaselli, and R. C. Armstrong. 2003. Regulation of the Apaf-1/caspase-9 apoptosome by caspase-3 and XIAP. J. Biol. Chem. 278:8091-8098. [DOI] [PubMed] [Google Scholar]