

Abstract
Severe combined immunodeficient (SCID) mice carry a germ-line mutation in DNA-PK, associated with deficiency in recognition and repair DNA double strand breaks. Thus, SCID cells and tissues display increased sensitivity to radiation-induced post-mitotic (clonogenic) cell death. Nonetheless, the single radiation doses required for 50% permanent local control (TCD50) of tumors implanted in SCID mice are not significantly different from the TCD50 values of the same tumors in wild-type hosts. Whereas the tumor stroma is derived from the host, the observation that tumors implanted in SCID mice do not exhibit hypersensitivity to radiation might imply that stromal endothelial elements do not contribute substantially to tumor cure by ionizing radiation. Here we challenge this notion, testing the hypothesis that acid sphingomyelinase (ASMase)-mediated endothelial apoptosis, which results from plasma membrane alterations, not DNA damage, is a crucial element in the cure of tumors in SCID mice by single dose radiotherapy (SDRT). We show that endothelium in MCA/129 fibrosarcomas and B16 melanomas exhibit a wild-type apoptotic phenotype in SCID hosts, abrogated in tumors in SCIDasmase−/− littermates, which also acquire resistance to SDRT. Conversion into a radioresistant tumor phenotype when implanted in SCIDasmase−/− hosts provides compelling evidence that cell membrane ASMase-mediated microvascular dysfunction, rather than DNA damage-mediated endothelial clonogenic lethality, plays a mandatory role in the complex pathophysiologic mechanism of tumor cure by SDRT, and provides an explanation for the wild-type SDRT responses reported in tumors implanted in SCID mice.
Keywords: ceramide, apoptosis, radiation, SCID endothelium
Introduction
Engagement of a vascular component in tumor response to ionizing radiation (IR) has been a topic of debate in recent literature (1–7). In 1993, Budach et al. (2) reported that the TCD50 values of tumors transplanted in SCID mice were not significantly different from the corresponding TCD50 values of tumors growing in wild-type C3H/Sed or NCr/Sed (nu/nu) hosts. SCID mice carry a germ line mutation in the DNA-dependent protein kinase (DNA-PK) gene, associated with deficiency in repair of DNA double strand breaks (DSBs) (8). SCID cells and tissues exhibit a 2.5–3.0 fold increased sensitivity to IR (9, 10), consistent with a basic tenet of classical radiobiology implying that the cell type-specific blueprint of DSB repair genes determines inherent radiosensitivity, and that its impairment enhances lethality via the reproductive (also termed clonogenic or post-mitotic) cell death mechanism [reviewed in (11)]. Whereas the TCD50 is generally accepted as a surrogate of tumor stem cell lethality in vivo (2, 12–14), and since tumor stroma elements, such as the microvascular endothelium, are derived from the host and express the host DNA-PK phenotype, the Budach observations were interpreted as indicating that tumor cure by radiation is dominated by the inherent radiosensivity of tumor stem cell clonogens (termed here SCCs), and that stromal endothelial elements do not contribute substantially to tumor cure by ionizing radiation (IR) (2). This notion was reiterated by Gerwick et al. (5, 6) who studied the radiation response of isogenic tumor xenografts in NCr(nu/nu) hosts differing only in genetic inactivation of DNA-PKcs.
Recent genetic pharmacologic studies have, however, challenged this notion, demonstrating that tumor SCC lethality after single high-dose radiation is linked to an early and transient wave of acid sphingomyelinase (ASMase)-mediated endothelial apoptosis, also induced by radiation exposure (3, 4). Engagement of the endothelial component was mandatory for tumor cure, as MCA/129 fibrosarcomas and B16 melanomas grown in apoptosis-resistant asmase−/− or Bax−/− hosts were resistant to the curative effects SDRT, observed in wild-type littermates (4). The endothelial apoptotic component displays a threshold at 8–10 Gy, and is thus not observed in classical fractionated radiation schemes employing repeated radiation exposures of 1.8–5 Gy. Conversion into a tumor radioresistant phenotype when implanted in apoptosis-resistant hosts, implied that the stromal endothelial component of SDRT may serve as an epigenetic regulator of the tumor SCC blueprint of DSB repair, thus determining the observed tumor response phenotype (3). The endothelial apoptotic response is triggered by IR-induced activation of ASMase trafficking from cytosolic vesicles to the plasma membrane, where it operates an apoptogenic rheostat via formation of ceramide-rich macrodomains on the plasma membrane outer leaflet (15). Hence, ASMase-mediated apoptosis involves radiation lesions at the plasma membrane, not IR-induced DNA damage, and is independent of the host-specific blueprint of DSB repair genes. Consistent with this notion, ASMase can be activated in isolated membranes devoid of DNA damage by IR treatment (16). Furthermore, endothelial cells derived from asmase−/− tumors are completely resistant to IR-induced apoptosis (4).
Since the endothelial component of the tumor response is inert to DNA damage and its repair, we tested here the hypothesis that SCID-derived endothelium might exhibit a normal wild-type apoptotic phenotype upon radiation exposure. To address this notion, we backcrossed the asmase−/− mutation into SCID mice and tested the endothelial apoptotic phenotypes in tumors implanted in SCIDasmase+/+ and SCIDasmase−/− littermates, and their impact on tumor response to SDRT. The data provide an insight into the mechanism of the radiation response of tumor xenografts in SCID mice, and re-define the impact of host DNA repair deficiency on the radiation response of tumors implanted in DNA repair-deficient hosts.
Material and Methods
Mice
C57BL/6J and Prkdcscid (termed here SCID) mice, 8–12 weeks old, were purchased from The Jackson Laboratory (Bar Harbor, ME). SV129/C57BL/6asmase−/− and C57BL/6asmase−/− mice were bred in our colony and genotyped as previously described (17). Backcrossing of the asmase−/− genotype into SCID mice was performed by mating SCID females with C57BL/6asmase−/− male mice. Male SCIDasmase+/− mice were subsequently mated with SCIDasmase+/− female mice for ten generations to obtain the SCIDasmase−/− and its SCIDasmase+/+ littermate genotypes. Once backcrossing was established, SCIDasmase+/− mice were interbred to obtain experimental animals. Mice were housed at the Research Animal Resource Center of Memorial Sloan-Kettering Cancer Center. This facility is approved by the American Association for Accreditation of Laboratory Animal Care and is maintained in accordance with the regulations and standards of the United States Department of Agriculture and the Department of Health and Human Services, National Institutes of Health.
Tumor growth and irradiation
MCA/129 fibrosarcoma and B16F1 (termed here B16) melanoma cells were maintained and transplanted subcutaneously into the right hind limb, as described (4). Tumors were irradiated when reaching a volume of 100–150 mm3. Mice were lightly sedated with ketamine (0.1 mg/g) and xylazine (0.02 mg/g) and irradiated using a Pantak Seifert Systems X-Ray 320 at 117 cGy/min (50 cm source to skin distance), using a specialized lead jig. Tumor volume was evaluated daily using caliper measurements and calculated according to the formula of Kim et al. (18). Tumor cure was defined as no clinical evidence of tumor re-growth at 90 days after SDRT, confirmed by histologic examination of the tumor bed. Duration of tumor growth delay was defined as the time required for tumor to reach 125% of its volume at irradiation.
Quantitative evaluation of endothelial apoptosis in tissue specimens
Tumor specimens were obtained at the indicated time points after radiation, fixed in 4% fresh formaldehyde, embedded in paraffin, and 5µm sections were immunostained for apoptosis by terminal deoxytransferase-mediated deoxyuridine triphosphate nick end labeling (TUNEL), as described (4), and for endothelial cell identification using an antibody specific for the endothelial cell surface marker MECA-32 (Developmental Studies Hybridoma Bank, developed under the auspices of the NICHD and maintained by The University of Iowa, IA). Briefly, TUNEL stained sections were incubated overnight with the MECA-32 monoclonal antibody (DSHB; 1:10 dilution) and developed using biotinylated secondary anti-rat antibodies (1:100 dilution), avidin-biotin peroxidase complex (1:25 dilution; Vector Laboratories) and True Blue Peroxidase substrate as the final chromogen (KPL Laboratories). Apoptotic endothelial cells were identified as a red-brown TUNEL-positive nuclear signal surrounded by dark-blue plasma membrane signal indicative of MECA-32 staining.
Endothelial cell isolation
Endothelial cell isolation was performed as described (4). Briefly, tumors were dissected from the hind limbs and disaggregated in a collagenase A cocktail at 37°C, and separated from tissue debris by density on a 30% Percoll gradient (Amersham Pharmacia Biotech). Hematopoietic cells were removed using MACS microbeads (Miltenyi Biotec Inc.) conjugated to antibodies directed against hematopoietic cell surface markers, including anti-CD45 (Catalog # 523-01) for mouse leukocytes, anti-CD90 (# 491-01) for mouse T cells, anti-MHC Class II(Ia) (# 524-01) for mouse B-cells, monocytes, macrophages, dendritic cells and hematopoietic progenitor cells, as indicated by the manufacturer. The total Percoll gradient fraction-antibody-conjugated MACS microbeads incubation was applied to the column. The fluent fraction was then incubated with mouse P1H12 anti-human CD146 antibody (Chemicon, # MAB 16985) for positive selection of tumor endothelial cells (at 4°C, 1:100 dilution). After 60 minutes, rat anti-mouse IgG1 microbeads (Miltenyi Biotec Inc.) at 1:50 dilution were added and applied to the MACS LS Separation columns. Endothelial cells bound to microbeads were eluted as indicated by the manufacturer. Analysis by FACS showed that the final eluate contained 95% pure endothelial cells based on the binding of Griffonia (Bandeiraea) Simplicifolia Lectin I, Isolectin B4 (Vector Laboratories), a specific endothelial cell marker.
Statistical analysis
Values are expressed as mean±SD, unless otherwise noted. Paired/unpaired, two-tailed student’s t-tests were calculated using Prism v4 software.
Results
asmase genotype but not SCID mutation impacts tumor radiosensitivity
We first defined the baseline radiation response patterns of the two tumor models employed here (MCA/129 fibrosarcoma and B16 melanoma) when implanted in wild-type and asmase−/− littermates, and in SCID hosts. Fig. 1A depicts individual MCA/129 fibrosarcoma tumors growing in SV129/C57BL/6asmase+/+ and SV129/C57BL/6asmase−/− littermates. Of 26 MCA/129 fibrosarcomas exposed to a single dose of 15 Gy in asmase wild type hosts, 12 (46%) displayed a sustained complete response at 30 days, while the rest showed a partial response with tumor growth delay of 18.8±1.7 days (Fig. 1A left panel), reducing the mean growth rate from 23.5±4.5 to 3.4±3.3 mm3/day (not shown; p<0.005). In contrast, there were no significant tumor responses among 15 fibrosarcomas growing in asmase−/− littermates (Fig. 1A, right panel), displaying a mean growth rate of 19.0±2.75mm3/day compared to 26.2±2.8mm3/day in unirradiated littermates (not shown; p=0.1). B16 melanoma tumors growing in asmase wild-type C57Bl/6 hosts were significantly more radioresistant (Fig. 1B, left panel), with only 1/10 (10%) exposed to 15 Gy exhibiting a sustained complete response and the rest showing a growth delay of 11.1±1.7 days, reducing the mean growth rate from the 38±3.4mm3/day to 7.5±3.4mm3/day (not shown; p<0.001). As with the sarcoma, B16 melanomas in asmase−/− littermates were minimally affected by 15 Gy (Fig. 1B, right panel), showing no responses and a mean growth rate of 48.5±5.3mm3/day, compared to 54.8±7.5mm3/day in control unirradiated hosts (not shown; p=0.6). Similar responses were observed with B16 melanomas exposed to 20 Gy (not shown), with a tumor growth delay of 14.4±1.5 days in wild-type hosts, compared with no significant tumor growth delay in asmase−/− littermates. These data confirm our previous report on the response of MCA/129 fibrosarcomas and B16 melanomas to SDRT (4).
Figure 1. Tumor radiosensitivity depends on host asmase genotype.


A, B. Effect of radiation on MCA/129 fibrosarcomas (A) or B16 melanomas (B) implanted in the right hind limb of asmase+/+ and asmase−/− hosts and irradiated at the post-implantation day indicated by the arrows. Lines indicate change in tumor volume over time for individual tumors measured daily. n indicates number of tumors treated per group. C, D. Effect of radiation on MCA/129 fibrosarcoma (C) or B16 melanoma (D) implanted in the right hind limb of wild-type strains (as indicated) or SCID hosts and irradiated at the post-implantation day indicated by the arrows. Value at each data point is the mean±SEM. Number of mice in each group is in parentheses.
Host SCID mutation did not affect the radiation response of the tumors models used here, consistent with the observations reported by Budach et al. (2). Fig. 1C shows that the mean growth rate of control unirradiated fibrosarcomas in SCID hosts was not different from tumors implanted in SV129/C57BL/6 wild-type mice (39.7± 3.1 vs. 40.1±5.6 mm3/day; p=0.9). When exposed to 15 Gy, tumors in SCID and SV129/C57BL/6 wild-type mice displayed a similar growth delay (15.4±1.6 vs. 13.2±1.6 days; p=0.3). A similar pattern was observed in B16 melanomas in SCID and wild type C57BL/6 mice (Fig. 1D, right panel) with regards to growth rates of unirradiated control tumors (79.1±17.0 mm3/day vs. 87.2±18.1 mm3/day, respectively; p=0.9) and 20 Gy irradiated tumors (growth delay of 14.4±2.3 vs. 11.4 days; p=0.25).
The SCID mutation does not impact radiation-induced endothelial apoptosis
Our prior studies demonstrated that high dose exposure induces a rapid wave of endothelial cell apoptosis in the tumors studied here when implanted in asmase+/+ but not asmase−/− hosts, peaking at 4–6 hours after radiation (4). We reported that the B16 melanoma tumors contain an average of 34.0±0.5 endothelial cells and 1216±64 tumor cells per 400X high power field (hpf). Fig. 2A shows that baseline endothelial apoptosis in unirradiated B16 melanomas was not significantly different for tumors grown in wild-type C57BL/6 or SCID hosts (2.8±0.2 vs. 2.6±0.2 cells/400X hpf, respectively). Apoptosis increased similarly in the tumor endothelium in both hosts types after exposure to 20 Gy, and the differences remained insignificant at all time points up to 8 hours after irradiation (15.9±1.5 vs. 17.3±1.1 endothelial cells per hpf at 6 hours for tumors in C57BL/6 and SCID, respectively), decreasing to background levels by 12 hours (not shown). In contrast less than 1% tumor cell apoptosis was observed at all times up to 48 hours post-irradiation (4).
Figure 2. The SCID mutation does not impact radiation-induced apoptosis of B16 melanoma endothelium.

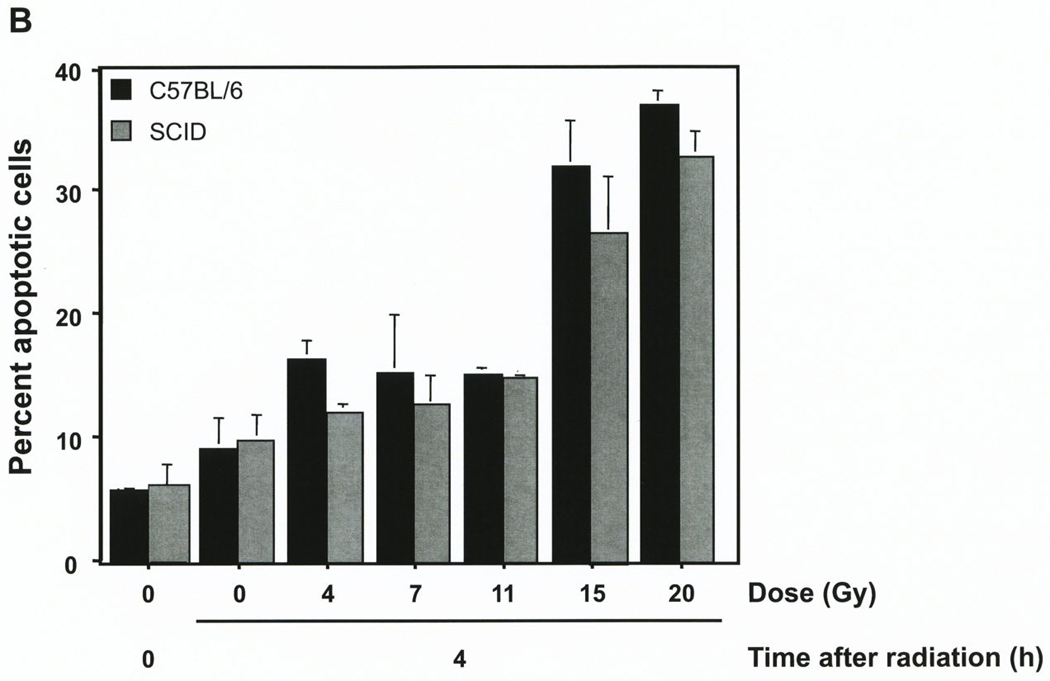
A. B16 melanomas were removed before and at the indicated times after exposure to 20 Gy, and 5 µm histologic sections were immunostained for apoptosis by TUNEL and for the endothelial cell surface marker MECA-32. Blinded scoring of apoptotic endothelial (TUNEL-MECA-32 double stained) cells was carried out manually. Data (means±SD) were compiled from 20 random hpfs (400X magnification) for each time point and collated from three independent experiments. B. A homogenous population of endothelial cells (~95% purity) was isolated by negative and positive selection from B16 melanoma tumors growing in either SCID or C57BL/6 mice. Immediately after isolation these cells were treated in single cell suspension with escalating radiation doses (0–20 Gy). A minimum of 500 cells, stained with bis-benzimide, was scored for apoptosis 4 hours post irradiation. Data (mean±SEM) were collated from three independent experiments.
To confirm that the apoptotic response observed in the tumor endothelium in wild-type and SCID hosts was an autonomous feature of the endothelial cell, not affected by tissue microenvironmental factors, we purified tumor endothelial cell populations to greater than 95% purity as described (4) and exposed the cells to escalating doses of radiation ex vivo. Fig. 2B shows that at 4 hours after irradiation, there was little increase in apoptosis of tumor endothelial cells derived from either C57BL/6 or SCID hosts at doses up to 11 Gy, but at higher doses apoptosis increased significantly in tumor endothelial cells derived from either host, reaching similar rates of 37±3% and 33±5%, respectively (p=0.6) after 20 Gy. Combined, these studies indicate that the SCID mutation does not affect the sensitivity of SCID endothelium to radiation-induced apoptosis.
Host asmase−/− germ line mutation confers radioresistance in tumors transplanted in SCID mice
Whereas our studies showed that the irradiated SCID tumor endothelium expresses a wild type apoptotic phenotype in vivo and ex vivo, we tested the hypothesis that insertion of a germ line asmase loss-of-function mutation into SCID mice would confer radioresistance in wild-type tumors growing in SCIDasmase−/− mice. To achieve this phenotype, we backcrossed the asmase−/− allele into SCID mice as described in Materials and Methods. Fig. 3A depicts TUNEL/MECA-32 immunohistochemical double staining of B16 melanoma specimens at 6 hours after exposure to 20 Gy. Tumors in SCIDasmase+/+ displayed significant endothelial apoptosis, increasing from 4.9±1.9% in unirradiated controls to 26.3±6.8% after 20 Gy (p<0.001; Fig. 3B). In contrast, endothelium of tumors in SCIDasmase−/− hosts, was resistant to IR-induced apoptosis (3.5±1.0% vs. 2.8±0.5% apoptotic endothelial cells in unirradiated controls; p=0.4). Similar results were observed in MCA/129 fibrosarcomas (Fig. 3C), displaying a radiosensitive endothelial apoptotic phenotype in SCIDasmase+/+ hosts (apoptosis increase from 6.1±1.9% in unirradiated controls to 27.6±3.3 after 15 Gy; p<0.001), and an apoptosis-resistant phenotype in SCIDasmase−/− hosts (4.9±2.3% vs. 7.3±1.7% before and after 15 Gy, respectively; p=0.8).
Figure 3. Host asmase−/− but not the SCID mutation impacts radiation-induced apoptosis in implanted tumor endothelium.

A. Representative 5 µm histologic B16 melanoma sections of tumor obtained at 6 hours after exposure to 20 Gy and doubly-immunostained for TUNEL and MECA-32. Apoptotic endothelium manifest a red-brown TUNEL-positive nuclear signal surrounded by dark blue plasma membrane signal of MECA-32. B, C. Incidence of apoptotic endothelial nuclei in tissue sections of control and 20 Gy-irradiated B16 melanoma (B) or 15 Gy-irradiated MCA/129 fibrosarcoma (C). Data (means±SD) were compiled from 20 random hpfs (400X magnification) and collated from three independent experiments.
Fig. 4 shows MCA/129 fibrosarcoma tumors growing in SCIDasmase+/+ and SCIDasmase−/− littermates. Of 6 fibrosarcomas exposed to a single dose of 15 Gy in SCIDasmase+/+ hosts, 3 (50%) displayed a sustained complete response at 90 days (confirmed histologically to contain no residual tumor, consistent with a definition of local cure), while the rest showed a partial response with tumor growth delay of 16.3±3.3 days, reducing the mean growth rate from 55.1±6.7 to 5.0±1.0 mm3/day (p<0.005). In contrast, there were no significant tumor responses among fibrosarcomas growing in SCIDasmase−/− littermates showing a mean growth rate of 65.6±5.8 mm3/day compared to 79.5±8.5 mm3/day in unirradiated littermates (p=0.1). Similar results were observed with B16 melanomas displaying a partial response to 20 Gy in SCIDasmase+/+ hosts, with tumor growth delay of 10.6±1.4 days and a mean growth rate of 3.1±2.4 mm3/day, compared to no tumor growth delay in SCIDasmase−/− hosts (not shown).
Figure 4. Effect of the asmase−/− mutation on the radiation response of MCA/129 fibrosarcomas implanted in SCID mice.

Tumors were implanted in the right hind limb of SCIDasmase+/+ or SCIDasmase−/− hosts and irradiated (15 Gy) at the post-implantation day indicated by the arrow. Value at each data point is the mean±SEM. Number of mice in each group is in parentheses.
Discussion
The present studies confirm our working hypothesis that SCID endothelium exhibits a wild-type ASMase-mediated apoptotic phenotype in response to high-dose IR. The apoptotic sensitivities of SCID and wild-type endothelia in vitro and in vivo were essentially identical in time- and dose-dependence, and were similarly abrogated by genetic ASMase deficiency. The vulnerability of endothelium to IR-induced apoptosis appears related to 20-fold higher ASMase expression in endothelium than in any other mammalian cell (19), and to a preferential expression of a specialized secretory form of ASMase, residing under resting conditions in acidic vesicles that abut the inner surface of the plasma membrane (15, 20). While the mechanism by which radiation elicits the ASMase response is uncertain, our recent studies showed that IR induces, within seconds, translocation of ASMase to the plasma membrane exoplasmic leaflet to initiate apoptotic signaling (15). Being activated at the plasma membrane, this endothelial response is inert to radiation-induced DNA damage and to defective DSB repair associated with the SCID mutation, but is sensitive to ASMase modulation and abrogated by the asmase−/− mutation. Consistent with this biology, the present studies show that tumors transplanted in SCID hosts engage a normal endothelial component in response to SDRT, similar to that operating in tumors transplanted in DNA-PK wild-type hosts, while MCA/129 fibrosarcomas implanted in apoptosis-resistant SCIDasmase−/− hosts are completely resistant to the curative effects SDRT, as confirmed histologically in SCIDasmase+/+ mice. This model provides a mechanistic explanation for the phenomena originally reported by Budach et al. (2).
While the SCID mutation does not radiosensitize endothelium to ASMase-mediated apoptosis, it was reported to radiosensitize, as anticipated, endothelial cell lethality via DSB-mediated post-mitotic (clonogenic) death (21). Endothelial lethality by this mode arguably leads to reduction of microvascular density in tumor stroma, detected at 5 days after 15 Gy single dose in SCID, but not in NCr(nu/nu), hosts (6). It was suggested that this mode of endothelial damage is associated with an increase in post-radiation tumor growth delay (but not tumor cure as measured by TCD50) in SCID as compared to NCr(nu/nu) hosts (2, 5, 6). However, our studies failed to show enhanced tumor growth delay in SCID hosts for either MCA/129 fibrosarcomas or B16 melanomas, raising a question whether the radiosensitized clonogenic cell death of SCID endothelium and the prolonged tumor growth delay reported in some studies are indeed causally associated. Regardless of this issue, the present studies are consistent with the notion that clonogenic death of irradiated endothelium does not impact tumor cure by SDRT, as previously suggested (2). Rather, the data provide compelling support for the hypothesis that ASMase-mediated apoptotic lethality of the microvascular endothelium is a crucial regulator of tumor SCC lethality after high-dose IR exposure in both wild-type and SCID hosts. As such, this response model represents a paradigm shift from basic concepts of classical radiobiology, which argue that stromal damage plays little to no role in radiation-induced tumor cure, presumably occurring via autonomous radiation-induced SCC lethality.
Our genetic and pharmacologic studies suggest an alternative mechanism, demonstrating that exposure to clinically-relevant SDRT simultaneously activates two tumor tissue elements as primary targets, inducing potentially lethal lesions in parenchymal tumor stem cell DNA and triggering ASMase/ceramide-mediated endothelial apoptosis in the tumor microvascular network. Our present and previously published data provide evidence that functional linkage of these two-target response mechanisms is mandatory for tumor cure with SDRT, and that endothelial cell ceramide elevation serves as a rheostat linking the two pathophysiologic processes (3, 4, 22). The endothelial apoptotic component evolves immediately after irradiation and precipitates a myriad of tissue responses. While the pathophysiology of these responses is only partially defined, recent studies report an acute transient hypoxia followed by spontaneous re-oxygenation, demonstrated by contrast-enhanced dynamic magnetic resonance imaging (DCE-MRI) with gadopentate dimeglumine (Gd-DTPA) (23, 24) or electron paramagnetic resonance (EPR) oximetry (25, 26), activation of signaling and metabolic pathways within parenchymal tumor cells (27, 28), and mobilization of bone marrow-derived cells that populate the tumor and trigger reconstruction and remodeling of the tumor microvascular network via vasculogenesis and angiogenesis (4, 29). Our present data showing conversion of MCA/129 fibrosarcoma into a radioresistant tumor phenotype when treated with SDRT in SCIDasmase−/− hosts suggests that microvascular dysfunction triggered by SDRT in wild-type mice epigenetically impairs repair of potentially lethal radiation lesions in the DNA of tumor SCCs. Consistent with this notion, our recent unpublished studies on the resolution kinetics of DNA repair foci post SDRT demonstrated that the microvascular dysfunctional component of SDRT represses DSB repair in MCA/129 fibrosarcoma and B16 melanoma, leading to tumor SCC lethality and tumor cure (Thin, Kolesnick and Fuks, unpublished). Thus, a cell membrane ceramide rheostat that confers endothelial apoptosis, rather than DNA damage-mediated endothelial clonogenic lethality, triggers microvascular dysfunction upstream of a complex pathophysiologic pathway that results in SDRT tumor cure. This model provides an explanation for the wild-type SDRT responses reported in tumors implanted in SCID mice.
Acknowledgments
Financial Support: This research was supported by NIH R01 # CA105125 to A. H-F., NIH R01 # CA085704 to R.K., and a gift from the Virginia and D.K. Ludwig Fund for Cancer Research to Z.F.
Footnotes
The authors have declared that no conflicts of interest exist.
References
- 1.Brown M, Bristow R, Glazer P, et al. Comment on "Tumor response to radiotherapy regulated by endothelial cell apoptosis" (II) Science. 2003;302:1894. doi: 10.1126/science.1089517. author reply. [DOI] [PubMed] [Google Scholar]
- 2.Budach W, Taghian A, Freeman J, Gioioso D, Suit HD. Impact of stromal sensitivity on radiation response of tumors. J Natl Cancer Inst. 1993;85:988–993. doi: 10.1093/jnci/85.12.988. [DOI] [PubMed] [Google Scholar]
- 3.Fuks Z, Kolesnick R. Engaging the vascular component of the tumor response. Cancer Cell. 2005;8:89–91. doi: 10.1016/j.ccr.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 4.Garcia-Barros M, Paris F, Cordon-Cardo C, et al. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science. 2003;300:1155–1159. doi: 10.1126/science.1082504. [DOI] [PubMed] [Google Scholar]
- 5.Gerweck LE, Vijayappa S, Kurimasa A, Ogawa K, Chen DJ. Tumor cell radiosensitivity is a major determinant of tumor response to radiation. Cancer Res. 2006;66:8352–8355. doi: 10.1158/0008-5472.CAN-06-0533. [DOI] [PubMed] [Google Scholar]
- 6.Ogawa K, Boucher Y, Kashiwagi S, Fukumura D, Chen D, Gerweck LE. Influence of tumor cell and stroma sensitivity on tumor response to radiation. Cancer Res. 2007;67:4016–4021. doi: 10.1158/0008-5472.CAN-06-4498. [DOI] [PubMed] [Google Scholar]
- 7.Suit HD, Willers H. Comment on "Tumor response to radiotherapy regulated by endothelial cell apoptosis" (I) Science. 2003;302:1894. doi: 10.1126/science.1089918. author reply. [DOI] [PubMed] [Google Scholar]
- 8.Fulop GM, Phillips RA. The scid mutation in mice causes a general defect in DNA repair. Nature. 1990;347:479–482. doi: 10.1038/347479a0. [DOI] [PubMed] [Google Scholar]
- 9.Biedermann KA, Sun JR, Giaccia AJ, Tosto LM, Brown JM. scid mutation in mice confers hypersensitivity to ionizing radiation and a deficiency in DNA double-strand break repair. Proc Natl Acad Sci U S A. 1991;88:1394–1397. doi: 10.1073/pnas.88.4.1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang C, Biedermann KA, Mezzina M, Brown JM. Characterization of the DNA double strand break repair defect in scid mice. Cancer Res. 1993;53:1244–1248. [PubMed] [Google Scholar]
- 11.Bedford JS, Dewey WC Radiation Research Society. 1952–2002. Historical and current highlights in radiation biology: has anything important been learned by irradiating cells? Radiat Res. 2002;158:251–291. doi: 10.1667/0033-7587(2002)158[0251:hachir]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 12.Gerweck LE, Zaidi ST, Zietman A. Multivariate determinants of radiocurability. I: Prediction of single fraction tumor control doses. Int J Radiat Oncol Biol Phys. 1994;29:57–66. doi: 10.1016/0360-3016(94)90226-7. [DOI] [PubMed] [Google Scholar]
- 13.Koch U, Krause M, Baumann M. Cancer stem cells at the crossroads of current cancer therapy failures-Radiation oncology perspective. Semin Cancer Biol. 2010 doi: 10.1016/j.semcancer.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 14.Peters LJ, Withers HR, Thames HD, Jr, Fletcher GH. Tumor radioresistance in clinical radiotherapy. Int J Radiat Oncol Biol Phys. 1982;8:101–108. doi: 10.1016/0360-3016(82)90392-3. [DOI] [PubMed] [Google Scholar]
- 15.Gulbins E, Kolesnick R. Raft ceramide in molecular medicine. Oncogene. 2003;22:7070–7077. doi: 10.1038/sj.onc.1207146. [DOI] [PubMed] [Google Scholar]
- 16.Haimovitz-Friedman A, Kan CC, Ehleiter D, et al. Ionizing radiation acts on cellular membranes to generate ceramide and initiate apoptosis. J Exp Med. 1994;180:525–535. doi: 10.1084/jem.180.2.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rotolo JA, Mesicek J, Maj J, et al. Regulation of ceramide synthase-mediated crypt epithelium apoptosis by DNA damage repair enzymes. Cancer Res. 2010;70:957–967. doi: 10.1158/0008-5472.CAN-09-1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim JH, Alfieri AA, Kim SH, Young CW. Potentiation of radiation effects on two murine tumors by lonidamine. Cancer Res. 1986;46:1120–1123. [PubMed] [Google Scholar]
- 19.Marathe S, Schissel SL, Yellin MJ, et al. Human vascular endothelial cells are a rich and regulatable source of secretory sphingomyelinase. Implications for early atherogenesis and ceramide-mediated cell signaling. J Biol Chem. 1998;273:4081–4088. doi: 10.1074/jbc.273.7.4081. [DOI] [PubMed] [Google Scholar]
- 20.Grassme H, Schwarz H, Gulbins E. Molecular mechanisms of ceramide-mediated CD95 clustering. Biochem Biophys Res Commun. 2001;284:1016–1030. doi: 10.1006/bbrc.2001.5045. [DOI] [PubMed] [Google Scholar]
- 21.Shinohara ET, Geng L, Tan J, et al. DNA-dependent protein kinase is a molecular target for the development of noncytotoxic radiation-sensitizing drugs. Cancer Res. 2005;65:4987–4992. doi: 10.1158/0008-5472.CAN-04-4250. [DOI] [PubMed] [Google Scholar]
- 22.Truman J-P, Garcia-Barros M, Kaag M, et al. Endothelial Membrane Remodeling is Obligate for Anti-angiogenic Radiosensitization during Tumor Radiosurgery. PLoS ONE. 2010 doi: 10.1371/annotation/6e222ad5-b175-4a00-9d04-4d120568a897. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Keyzer F, Vandecaveye V, Thoeny H, et al. Dynamic contrast-enhanced and diffusion-weighted MRI for early detection of tumoral changes in single-dose and fractionated radiotherapy: evaluation in a rat rhabdomyosarcoma model. Eur Radiol. 2009;19:2663–2671. doi: 10.1007/s00330-009-1451-1. [DOI] [PubMed] [Google Scholar]
- 24.Goda F, Bacic G, O'Hara JA, Gallez B, Swartz HM, Dunn JF. The relationship between partial pressure of oxygen and perfusion in two murine tumors after X-ray irradiation: a combined gadopentetate dimeglumine dynamic magnetic resonance imaging and in vivo electron paramagnetic resonance oximetry study. Cancer Res. 1996;56:3344–3349. [PubMed] [Google Scholar]
- 25.Bratasz A, Pandian RP, Deng Y, et al. In vivo imaging of changes in tumor oxygenation during growth and after treatment. Magn Reson Med. 2007;57:950–959. doi: 10.1002/mrm.21212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goda F, O'Hara JA, Rhodes ES, et al. Changes of oxygen tension in experimental tumors after a single dose of X-ray irradiation. Cancer Res. 1995;55:2249–2252. [PubMed] [Google Scholar]
- 27.Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer. 2008;8:425–437. doi: 10.1038/nrc2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moeller BJ, Cao Y, Li CY, Dewhirst MW. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell. 2004;5:429–441. doi: 10.1016/s1535-6108(04)00115-1. [DOI] [PubMed] [Google Scholar]
- 29.Kioi M, Vogel H, Schultz G, Hoffman RM, Harsh GR, Brown JM. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest. 2010;120:694–705. doi: 10.1172/JCI40283. [DOI] [PMC free article] [PubMed] [Google Scholar]