

Summary
Salp15 is a tick saliva protein that inhibits CD4+ T cell differentiation through its interaction with CD4. The protein inhibits early signaling events during T cell activation and IL-2 production. Because murine experimental autoimmune encephalomyelitis development is mediated by central nervous system-infiltrating CD4+ T cells that are specific for myelin-associated proteins, we sought to determine whether the treatment of mice with Salp15 during EAE induction would prevent the generation of proinflammatory T cell responses and the development of the disease. Surprisingly, Salp15-treated mice developed more severe EAE than control animals. The treatment of EAE-induced mice with the tick saliva protein did not result in increased infiltration of T cells to the central nervous system, indicating that Salp15 had not affected the permeability of the blood-brain barrier. Salp15 treatment did not affect the development of antibody responses against the eliciting peptide or the presence of IFNγ in the sera. The treatment with Salp15 resulted, however, in the increased differentiation of Th17 cells in vivo, as evidenced by higher IL-17 production from PLP139-151-specific CD4+ T cells isolated from the central nervous system and the periphery. In vitro, Salp15 was able to induce the differentiation of Th17 cells in the presence of IL-6 and the absence of TGFβ These results suggest that a conductive milieu for the differentiation of Th17 cells can be achieved by restriction of the production of IL-2 during T cell differentiation, a role that may be performed by TGFβ and other immunosuppressive agents.
Keywords: Salp15, EAE, Th17 differentiation, IL-2, autoimmunity
Introduction
The tick salivary protein Salp15 inhibits the activation of CD4+ T cells during their encounter with cognate antigen by repressing the production of the autocrine growth factor IL-2 [1]. The effect of Salp15 is mediated by its interaction with the T cell co-receptor, CD4 [2]. The activity of Salp15 requires its carboxyl terminal residues and occurs through its interaction with the first two most-extracellular domains of CD4 [2,3]. The interaction results in a conformational change of CD4 that impedes the proper activation of the Src kinase, Lck [3], leading to the inhibition of early signaling cascades [2,4]. In vivo, Salp15 prevents the development of acute asthma in ovalbumin-induced mice [5] and the development of antibody and delayed-type sensitivity responses in KLH-immunized animals [1]. Thus, Salp15 has potential beneficial use in conditions in which CD4+ T cells are involved, including autoimmune diseases in which CD4+ T cells are major players.
Multiple sclerosis is a chronic inflammatory disease that affects the central nervous system. The inflammation associated with the disease is believed to be the consequence of the activation of inflammatory CD4+ T cells. Mice can also develop a demyelinating inflammatory disease of the central nervous system (CNS) known as Experimental Autoimmune Encephalomyelitis (EAE) that mimics MS [6]. The mice, when immunized with a specific peptide from the myelin proteolipid protein (PLP) or Myelin Basic Protein (MBP) [7,8,9] develop an MS-like syndrome with a course that is similar to that found in humans [10]. The inflammatory response associated with disease progression is caused by T helper cells that secrete interleukin 17 (IL-17A), known as Th17 cells, in combination with IFNγ-secreting Th1 cells [11,12,13,14]. Th17 cells participate directly in the development and pathogenesis of EAE [15]. Thus, the transfer of IL-17-producing T cells results in severe EAE [16] and mice deficient in IL-17 markedly suppress the development of EAE [15]. In addition, mice treated with a neutralizing monoclonal antibody specific for IL-17 demonstrated reduced CNS autoimmune inflammation [16].
In mice, the differentiation of Th17 cells requires the presence of transforming growth factor (TGF) β and the pleiotropic cytokine IL-6 [17,18,19]. In order for a Th17 response to prevail, the heterodimeric cytokine, IL-23, which shares the p40 subunit with IL-12, must be present to sustain Th17 cell survival and maintenance [16]. Th17 cell differentiation requires the orphan nuclear receptors, ROR-γt and RORα, two specific transcriptional factors that are selectively expressed in Th17 cells [20,21]. The importance of these transcriptional factors in the development of Th17 cells is underlined by experiments showing that RORγt/RORα double deficiency completely abrogates Th17 differentiation in vitro [21]. Furthermore, RORγt/RORα deficient mice do not develop EAE [21].
Because Salp15 is able to prevent the activation of antigen-specific CD4+ T cells in vivo [1,5] and prevent inflammation in a murine model of asthma [5], we postulated that the administration of the protein during the induction of EAE in mice would prevent the generation of autoreactive T cells and the development of CNS inflammation. Our results show that surprisingly, Salp15 induces the increased differentiation of Th17 effector cells both in vivo and in vitro. Our results suggest that immunosuppressive agents can mimic the role of TGFβ during the differentiation of Th17 cells through the repression of the production of IL-2 during the differentiation process.
Materials and Methods
Mice, Peptide and Induction of EAE
Female SJL/J mice 8 week old were purchased from Jackson laboratories (Bar Harbor, ME). The mice in groups of 3 to 5 were injected subcutaneously in both flanks with 100 μg of PLP peptide 139 – 151 (HSLGKWLGHPDKF) (Research Genetics, Huntsville, AL). The original Cysteine has been substituted by Serine for stability purposes. This change does not induce any alteration in the antigenicity of the peptide [22]. EAE was induced by immunizing the mice with 100 μg of the peptide dissolved in an emulsion of 200 μl total volume of 50% Complete Freund’s Adjuvant and 50% sterile normal saline containing 400 μg of heat killed Mycobacterium tuberculosis H37Rα (BD Diagnostics Systems, Sparks, MD). The mice also received an intraperitoneal injection of 200 ng of pertussis toxin on the day of immunization. All experiments involving animals were approved by the Institutional Animal Care and Use Committee at UMass Amherst.
Treatment with Salp15 and EAE clinical scores
Recombinant Salp15 was produced and purified using the Drosophila expression system as previously described [1]. Mice treated with Salp15 or Salp15ΔP11/vehicle control received 100 μl injections starting the day before immunization, on the day of immunization and day two-post immunization (days, −1, 0, 2). The mice were weighted and examined for EAE clinical symptoms daily based on the following scores; 0-no symptoms, 1-loss of tail tone, 2-hind limb weakness, 3-paralysis of both hind limbs, 4-moribund/dead. Mice with intermediate clinical symptoms were scored in 0.5 increments.
Purification and restimulation of splenic and spinal cord CD4+ T cells
CD4+ T cells were purified from the spleen by negative selection using biotinylated antibodies against CD8a, Ly6G, CD11b, B220, class II (I-Ak and I-Ek) and panNK cell surface molecules (BD Bioscience, San Jose, CA), followed by incubation with anti-biotin antibodies bound to microbeads and passage through a magnetic column (Miltenyi Biotec, Auburn, CA). The spinal cord of the immunized mice was removed, mechanically grinded in a solution of PBS containing 1% FCS. T lymphocytes were purified from the PBS solution by Ficoll-plaque gradient centrifugation. Splenic CD4+ T cells or lymphocytes purified from the spinal cord were mixed with mitomycin C-treated syngeneic splenocytes in a 1:1 ratio and incubated with 50 μg/ml of PLP139-151 in the absence or presence of 2 ng/ml of recombinant human IL-23 (eBioscience, San Diego, CA) for 96 h.
In vitro Th17 differentiation
2 × 106 purified splenic CD4+ T cells were activated with 5 μg/ml of plate-bound αCD3ε and 1 μg/ml of soluble αCD28 antibodies (Cell Signaling, Danvers, MA) in the presence of 20 ng/ml of recombinant mouse IL-6 (R&D systems, Minneapolis, MN) and either 5 ng/ml of recombinant human TGFβ (R&D systems), Salp15 (10 μg/ml) or Salp15ΔP11 (10 μg/ml, control). After 4 days, the cells were recovered, washed, counted and restimulated with 1 μg/ml of anti-CD3 for 20 h. Four-day activation and restimulation supernatants were evaluated by ELISA for IL-2 and IL-17 production, respectively.
Cytokine ELISA
Activation and restimulation supernatants, as well as pooled sera from EAE-induced mice were analyzed for IL-2, IL-17 and IFNγ by capture ELISA, as described [1] using antibody pairs and recombinant standards purchased from BD Biosciences and R&D Systems.
Antibody ELISA
The PLP139-151-specific antibody titers in the sera of EAE-induced mice were determined by ELISA using 2-fold serial dilutions starting at 1/100, as described [23].
Real-time RT-PCR
After 4 days of T cell differentiation, total RNA was prepared using the Trizol reagent (Invitrogen, Carslbad, CA), according to the manufacturer’s instructions. Complementary DNA (cDNA) was synthesized with Superscript™ II reverse transcriptase (Invitrogen). The expression of il-17 and ror-γt were analyzed by quantitative RT-PCR using SYBR® Premix Ex Taq™ (SYBR® Green Takara Bio Inc, Otsu, Shiga, Japan) in a MX3005P thermal cycler (Agilent Technologies, Santa Clara, CA). The relative fold induction of each gene was calculated using the 2−ΔΔCT method, using the β-actin gene as an internal control, as described [24]. The primers used were: il-17 (5′-CTC CAG AAG GCC CTC AGA CTA C-3′ and 5′-AGC TTT CCC TCC GCA TTG ACA CAG-3′); ror-γt (5′-TTT GGA ACT GGC TTT CCA TC-3′ and 5′-AAG ATC TGC AGC TTT TCC ACA-3′); b-actin (5′-GAC GAT GCT CCCCGG GCT GTA TTC-3′ and 5′-TCT CTT GCT CTG GGC CTC GTC ACC-3′).
Results and Discussion
Treatment with Salp15 during EAE induction results in increased pathology
Murine experimental autoimmune encephalomyelitis is mediated by the activation of CD4+ T cells that respond to self antigens present in the myelin sheath. Salp15 has been shown to prevent the development of ovalbumin-induced asthma in mice [5] and of KLH-specific antibody and DTH responses [1] through the specific inhibition of CD4+ T cell activation. We thus hypothesized that the administration of the tick salivary protein during induction of murine EAE would prevent the activation of myelin-specific CD4+ T cells and the development of disease. We induced EAE by immunizing SJL mice with the proteolipid peptide, PLP139-151. The mice were treated with Salp15 (50 μg/injection) two days before, on the day, and one day after EAE induction. We monitored PLP139-151-immunized SJL mice daily for clinical disease. Both control- and Salp15-treated mice developed evidences of disease at day 9 post-immunization, with an equivalent progression until day 13 (data not shown). However, mice that received Salp15 showed an increase in the mean clinical score (Figure 1A). The development of the paralyzing symptoms was dependent on immunization, since the treatment of mock-immunized (without peptide) mice with Salp15 did not result in disease (data not shown). The effect of Salp15 was also dependent on the presence of the C-terminal portion of the protein (P11) [2], since the use of a mutant version that lacks the last 15 amino acids (Salp15ΔP11) [4] failed to induce increased disease (data not shown).
Figure 1. Treatment with Salp15 results in increase pathology upon induction of EAE.
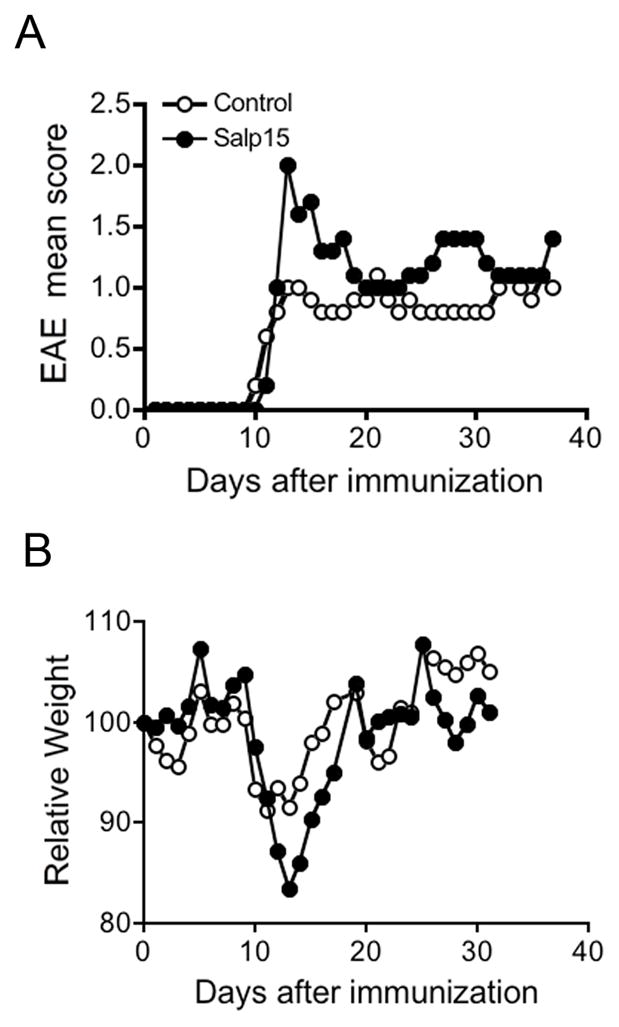
Groups of 5 mice were immunized with 100 μg of the peptide PLP139-151 in an emulsion containing complete Freund’s adjuvant supplemented with 400 μg of M. tuberculosis H37Rα. The mice were treated with 50 μg of Salp15 at days -2, 0, 1 and analyzed daily for pathological incidence (A), scores (B) and weight (C). Weights at the day of immunization were assigned a value of 100 in order to be able to compare between groups and the subsequent weights are relative to those at day 0. The experiment shown is representative of at least 7 experiments in which control animals were either treated with an inactive form of Salp15 (Salp15ΔP11) or the vehicle control.
The EAE-induced mice were also monitored daily for weight changes. In both groups of mice, the peak of disease corresponded with a depression of the average weight. However, the increased pathology in Salp15-treated mice correlated with a higher weight loss (Figure 1B). Thus, treatment with Salp15 induced increased pathology upon induction of EAE.
Treatment with Salp15 does not result in increased permeability of the blood-brain barrier or impaired B cell or Th1 responses
To determine whether the increased pathology as a result of the treatment with Salp15 had resulted from a potential enhancement in the permeability of the blood-brain barrier, we analyzed by flow cytometry the percentage of CD4+ T cells that had infiltrated the spinal cord of PLP139-151-immunized mice. At 12 and 40 days of immunization, there were no significant differences in the percentage of CD4+ T cells infiltrating the spinal cord between Salp15-treated and control mice (Figure 2A), suggesting that the treatment had not affected the permeability of the blood-brain barrier. The analysis of CD8+ T, B cells an macrophages also showed that the treatment with Salp15 had not affected cellular infiltration of the CNS upon EAE induction (data not shown).
Figure 2. Treatment with Salp15 during induction of EAE does not affect the permeability of the blood-brain barrier, B cell responses or Th1 responses.
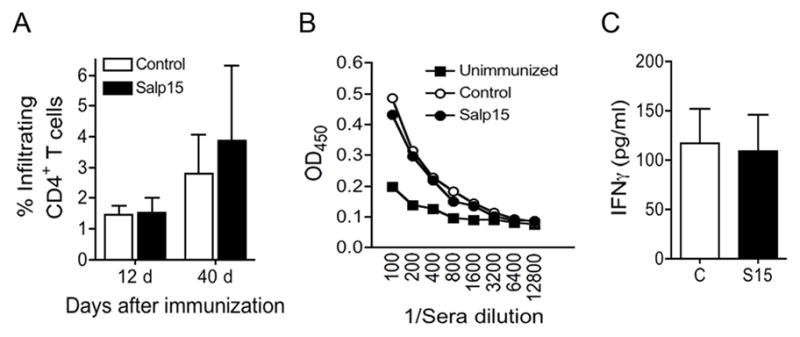
(A) Salp15-treated and control mice were induced EAE and analyzed at days 12 and 40 post-immunization for the infiltration of CD4+ T cells to the CNS by flow cytometry. The results represent the average of 5 mice per group and are representative of 2 experiments. (B) EAE mice were analyzed at 40 days post-immunization for serum levels of anti-PLP139-151 antibodies by ELISA. (B) Sera from Salp15-treated and control mice were pooled and analyzed for the levels of IFNγ by capture ELISA. The results shown represent the average of 4 individual experiments with 3–5 mice per group each.
The production of autoantibodies against the myelin sheath has been implicated in the pathogenesis of EAE [25]. We therefore analyzed the effect of the treatment with Salp15 on the generation of antibody responses against the eliciting peptide, PLP139-151. Sera from EAE-induced mice were analyzed for PLP139-151-specific antibody titers by ELISA. The levels of antibodies did not vary as a result of Salp15 treatment (Figure 2B), suggesting that at the dose used, Salp15 had not affected the development of B cell responses.
In order to evaluate the effect of the treatment with Salp15 on the differentiation of T cells during EAE induction, we also analyzed by ELISA the levels of IFNγ and IL-17 in pooled sera samples of EAE-induced and Salp15- or control-treated mice. The treatment with Salp15 did not induce a significant change in the serum levels of the Th1 cytokine, IFNγ (Figure 2C), suggesting that Th1 responses had not been affected by the treatment. The serum levels of IL-17 were in all cases below the detection limit of the assay (data not shown). We thus investigated the phenotype of PLP139-151-specific CD4+ T cells in restimulation experiments.
Salp15 augments antigen-specific Th17 differentiation in vivo
EAE is characterized by the increased differentiation of myelin-specific Th17 cells. We assessed recall responses of T cells from the spinal cord and spleens of the immunized mice to the inducing peptide (PLP139-151). The cells were restimulated in vitro with the peptide in the presence of mytomycin C-treated syngeneic splenocytes and assessed for IL-17 production. CD4+ T cells from Salp15-treated mice produced increased levels of the cytokine compared to control-treated mice (Figure 3A). Furthermore, splenic CD4+ T cells from Salp15-treated mice produced increased levels of IL-17 when restimulated in the presence of rmIL-23 (Figure 3B), a cytokine that is involved in the survival of effector Th17 cells [16]. Overall, these results suggested that the treatment with Salp15 had specifically induced an increased differentiation of antigen-specific Th17 cells and Th17-mediated spinal cord pathology. The strength of the signal given through the TCR affects the immunosuppressive activity of Salp15 [1]. Our results suggest that under the strong immunization conditions necessary to induce EAE, Salp15 affected CD4+ T cell differentiation rather than their activation.
Figure 3. Salp15 augments antigen-specific Th17 differentiation in vivo.
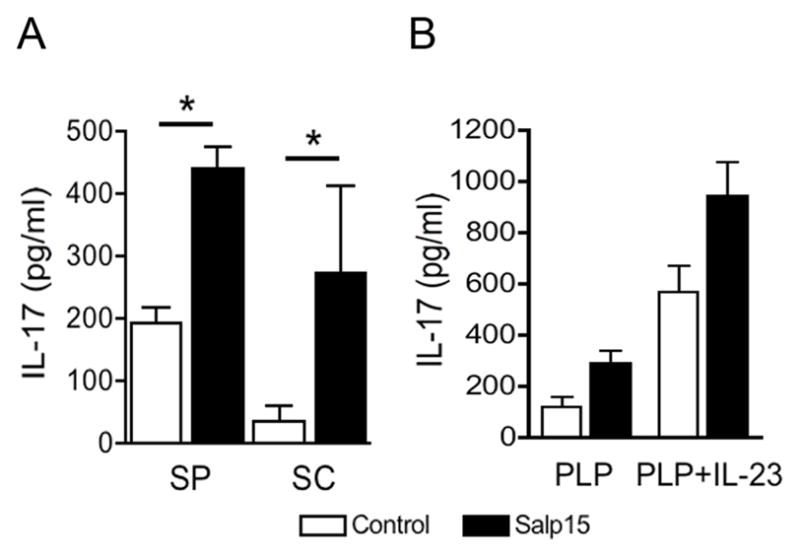
(A) CD4+ T cells purified from the spleen (SP) and ficoll-separated T cells from the spinal cord (SC) of PLP139-151-immunized mice were restimulated in the presence of syngeneic antigen presenting cells with 50 μg/ml of the peptide PLP139-151. After 40 h, IL-17 levels in the restimulation supernatants were assessed by capture ELISA. * P < 0.05 (student’s t test). (B) Purified splenic CD4+ T cells were also stimulated in the presence of 5 ng/ml of rmIL-23 and assessed for IL-17 production, as before. The results are representative of at least 3 independent experiments.
Salp15 increases the differentiation of Th17 cells in vitro
It has been demonstrated that IL-2 exerts an antagonistic effect on Th17 induction [26]. Since the tick saliva protein, Salp15, represses the expression of the il-2 gene through its interaction with the T cell co-receptor, CD4, we then determined whether the inhibition of IL-2 production during the activation of CD4+ T cells in the presence of IL-6 would enhance their differentiation to a Th17 phenotype. We differentiated purified splenic CD4+ T cells in the presence of IL-6 and in the absence or presence of TGFβ for 4 days. The cells were also incubated with a dose of Salp15 (10 μg/ml) that does not completely repress the production of IL-2 (at 50 μg/ml, the cells fail to activate, and die after 3 days of activation [1]). The activation of CD4+ T cells with 10 μg/ml of Salp15 resulted in the inhibition of IL-2 production at 4 days of activation (Figure 4A). Similarly, the presence of IL-6 and TGFβ during the activation period induced a reduction of IL-2 at the same time point (Figure 4A). The mechanism by which the tick protein represses the transcription of the il-2 gene is independent of TGFβ or TGFβ-like induced signals. Indeed, Salp15 did not induce tgf-β gene expression in T cells during the differentiation period (data not shown), demonstrating that is effect is independent of TGF-β. Similarly, the effect of Salp15 occurs in the absence of IL-1β induction, as determined in activation assays in the presence of purified splenic dendritic cells (data not shown). These results show, as described [1], that Salp15 does not influence cytokine production by antigen presenting cells. Rather, Salp15 binds to CD4 and prevents the activation of the Src kinase Lck [3] and thus, reduces the signals emanating from the TCR during the activation process.
Figure 4. Salp15 mimics the effect exerted by TGFβ during Th17 differentiation.
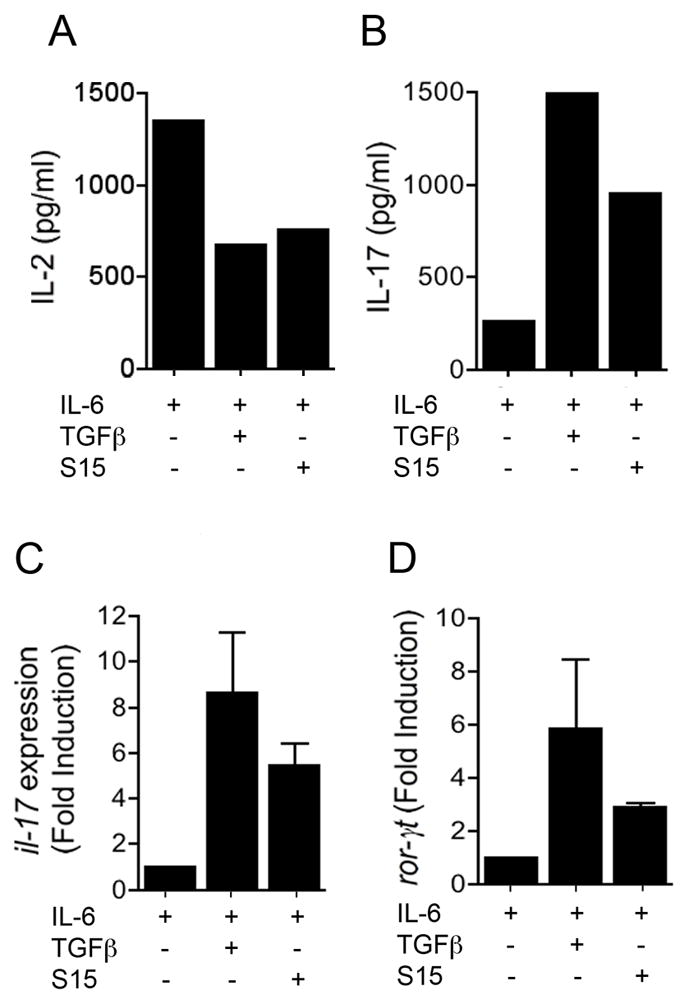
Purified splenic CD4+ T cells were differentiated in vitro with anti-CD3/CD28 in the presence of IL-6 (20 ng/ml) plus Salp15 (10 μg/ml), Salp15ΔP11 (10 μg/ml) or TGFβ (5 ng/ml). After 4 days, the cells were washed, counted and restimulated with anti-CD3 for 20 h. IL-2 was measured by capture ELISA at 4 days of activation (A), while IL-17 was measured in restimulation supernatants (B). At 4 days of activation, the cells were also used to extract RNA and assessed the expression of the il-17 (C) and ror-γt (D) genes by real-time RT-PCR, using β-actin as an internal control. The results presented are representative of 3–4 experiments.
After 4 days, the cells were washed and restimulated with αCD3 for 20 h. IL-17 was measured in the restimulation supernatants. The presence of Salp15 during the differentiation process resulted in increased levels of IL-17 (Figure 4B). The increased production of IL-17 corresponded to an increased transcription of the il-17 gene, determined by real-time RT-PCR (Figure 4C). The incubation of CD4+ T cells during their differentiation with Salp15 also resulted in increased gene expression of the transcription factor RORγt (Figure 4D), which is necessary to drive il-17 expression [20]. The action of Salp15 was not due to an effect on T regulatory cells, since the tick salivary protein did not affect the expression of the transcription factor, FoxP3 (not shown). These results demonstrate that at suboptimal levels, Salp15 does not prevent the activation of CD4+ T cells, while inducing the differentiation of effector Th17 cells.
Our results show that the tick salivary protein, Salp15 is able to induce the differentiation of Th17 cells in the presence of IL-6 through a TGFβ-independent mechanism. These results demonstrate that IL-6 is the main cytokine that induces Th17 differentiation while the role of TGFβ may be circumscribed to the prevention of excess levels of IL-2 that can antagonize this process. In contrast, in the absence of repression of IL-2 production, IL-6 induces Th2 differentiation [27], rather than Th17. Overall these results underscore that a careful balance between cytokines has to be maintained during activation/differentiation of CD4+ T cells. There are no reports associating tick feeding and the exacerbation of inflammatory diseases, including those of autoimmune origin. This could be due to the low levels of proteins deposited in the host. However, under the conditions used herein to test the potential use of Salp15 for the treatment of immune-mediated inflammation, our results show that the protein could affect the differentiation of CD4+ T cells. Importantly, our results also suggest that the capacity of Salp15 to induce Th17 differentiation may not be unique and could be achieved under conditions in which the production of IL-2 during T cell differentiation is repressed, such as with the use of immunosuppressive agents. Indeed, several studies have reported the increased incidence of autoimmune diseases in mice treated with immunosuppresive drugs such as cyclosporine A or rapamycin [28,29,30,31]. Since immunosuppressive drugs are routinely used in transplantation therapy, a careful evaluation of their overall impact in immune responses could be warranted.
Acknowledgments
This work was supported by the NINDS/NIH grant R21NS048433
We thank Dr. Cory Teuscher for his advice.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Anguita J, Ramamoorthi N, Hovius JW, Das S, Thomas V, Persinski R, Conze D, Askenase PW, Rincon M, Kantor FS, Fikrig E. Salp15, an Ixodes scapularis salivary protein, inhibits CD4+ T cell activation. Immunity. 2002;16:849–859. doi: 10.1016/s1074-7613(02)00325-4. [DOI] [PubMed] [Google Scholar]
- 2.Garg R, Juncadella IJ, Ramamoorthi N, Ashish, Ananthanarayanan SK, Thomas V, Rincon M, Krueger JK, Fikrig E, Yengo CM, Anguita J. Cutting edge: CD4 is the receptor for the tick saliva immunosuppressor, salp15. J Immunol. 2006;177:6579–6583. doi: 10.4049/jimmunol.177.10.6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ashish, Juncadella IJ, Garg R, Boone CD, Anguita J, Krueger JK. Conformational rearrangement within the soluble domains of the CD4 receptor is ligand-specific. J Biol Chem. 2008;283:2761–2672. doi: 10.1074/jbc.M708325200. [DOI] [PubMed] [Google Scholar]
- 4.Juncadella IJ, Garg R, Ananthanarayanan SK, Yengo CM, Anguita J. T cell signaling pathways inhibited by the tick saliva immunosuppressor, Salp15. FEMS Immunol Med Microbiol. 2007 doi: 10.1111/j.1574-695X.2007.00223.x. In press. [DOI] [PubMed] [Google Scholar]
- 5.Paveglio SA, Allard J, Mayette J, Whittaker LA, Juncadella I, Anguita J, Poynter ME. The tick salivary protein, Salp15, inhibits the development of experimental asthma. J Immunol. 2007;178:7064–7071. doi: 10.4049/jimmunol.178.11.7064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McRae BL, Kennedy MK, Tan LJ, Dal Canto MC, Picha KS, Miller SD. Induction of active and adoptive relapsing experimental autoimmune encephalomyelitis (EAE) using an encephalitogenic epitope of proteolipid protein. J Neuroimmunol. 1992;38:229–240. doi: 10.1016/0165-5728(92)90016-e. [DOI] [PubMed] [Google Scholar]
- 7.Lehmann PV, Forsthuber T, Miller A, Sercarz EE. Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature. 1992;358:155–157. doi: 10.1038/358155a0. [DOI] [PubMed] [Google Scholar]
- 8.McRae BL, Vanderlugt CL, Dal Canto MC, Miller SD. Functional evidence for epitope spreading in the relapsing pathology of experimental autoimmune encephalomyelitis. J Exp Med. 1995;182:75–85. doi: 10.1084/jem.182.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu M, Johnson JM, Tuohy VK. A predictable sequential determinant spreading cascade invariably accompanies progression of experimental autoimmune encephalomyelitis: a basis for peptide-specific therapy after onset of clinical disease. J Exp Med. 1996;183:1777–1788. doi: 10.1084/jem.183.4.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goverman J, Brabb T. Rodent models of experimental allergic encephalomyelitis applied to the study of multiple sclerosis. Lab Anim Sci. 1996;46:482–492. [PubMed] [Google Scholar]
- 11.Aggarwal S, Ghilardi N, XMH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 12.El-behi M, Rostami A, Ciric B. Current views on the roles of Th1 and Th17 cells in experimental autoimmune encephalomyelitis. J Neuroimmune Pharmacol. 2010;5:189–197. doi: 10.1007/s11481-009-9188-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iwakura Y, Ishigame H. IL-23/Il-17 axis in inflammation. J Clin Invest. 2006;116:1218–1222. doi: 10.1172/JCI28508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- 16.Langrish CL, Chen Y, Bumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 18.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 19.Veldhoem M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGF-beta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17 producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 20.Ivanov, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 21.Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang H, Chung Y, Ma L, Shah B, Panopoulos AD, Schluns K, Watowich SS, Tian Q, Jetten AM, Dong C. T helper 17 lineage differentiation is programmed by orphan nuclear receptor ROR alpha and ROR gamma. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tuohy VK, Lu Z, Sobel RA, Laursen RA, Lees MB. Identification of an encephalitogenic determinant of myelin proteolipid protein for SJL mice. J Immunol. 1989;142:1523–1527. [PubMed] [Google Scholar]
- 23.Olson CM, Jr, Bates TC, Izadi H, Radolf JD, Huber SA, Boyson JE, Anguita J. Local Production of IFN-{gamma} by Invariant NKT Cells Modulates Acute Lyme Carditis. J Immunol. 2009;182:3728–3734. doi: 10.4049/jimmunol.0804111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 25.Weber MS, Hemmer B, Cepok S. The role of antibodies in multiple sclerosis. Biochim Biophys Acta. 2010 doi: 10.1016/j.bbadis.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 26.Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, Shevach EM, O’Shea J. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 27.Rincon M, Anguita J, Nakamura T, Fikrig E, Flavell RA. Interleukin (IL)-6 directs the differentiation of IL-4-producing CD4+ T cells. J Exp Med. 1997;185:461–469. doi: 10.1084/jem.185.3.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakaguchi S, Sakaguchi N. Thymus and autoimmunity. Transplantation of the thymus from cyclosporine A-treated mice causes organ-specific autoimmune disease in athymic mice. J Exp Med. 1988;167:1479–1485. doi: 10.1084/jem.167.4.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blazar BR, Taylor PA, Snover DC, Sehgal SN, VDA Murine recipients of fully mismatched donor marrow are protected from lethal graph-versus-host disease by in vivo administration of rapamycin but develop an autoimmune-like syndrome. J Immunol. 1993;151:5726–5741. [PubMed] [Google Scholar]
- 30.Bucy RP, Xu XY, Li J, Huang G. Cyclosporin A-induced autoimmunity disease in mice. J Immunol. 1993;151:1039–1050. [PubMed] [Google Scholar]
- 31.Chen W, Thoburn C, Hess AD. Characterization of the pathogenic autoreactive T cells in cyclosporine-induced syngeneic graft-versus-host disease. J Immunol. 1998;15:7040–7046. [PubMed] [Google Scholar]