

Abstract
BACKGROUND
Live attenuated vaccine strain of measles virus (MV) has promising antitumor activity and is undergoing clinical testing in three different phase I cancer trials. The virus uses one of two receptors, CD46 which is ubiquitously expressed on all nucleated cells or CD150 which is expressed on immune cells, to infect cells. To minimize potential toxicity due to indiscriminate infection of normal cells, we have generated a fully retargeted MV that infects cells exclusively through the prostate-specific membrane antigen (PSMA) receptor, which is overexpressed on prostate cancer cells and tumor neovasculature.
METHODS
A single-chain antibody (scFv) specific for the extracellular domain of PSMA (J591) was inserted as a C-terminal extension on the MV attachment protein. Specificity of infection by the PSMA targeted virus was evaluated in parallel with the parental MV and a control virus which binds to CD38, a myeloma antigen. Antitumor activity of the PSMA retargeted virus was tested in both LNCaP and PC3-PSMA tumor xenograft models, with and without low dose external beam radiation.
RESULTS
Replication of the PSMA targeted virus was comparable to the parental MV. The PSMA scFv efficiently redirected virus infection and cytopathic killing exclusively to PSMA positive prostate cancer cells and not PSMA negative cells. There was an additive effect on cell killing from radiation treatment and virotherapy. The PSMA virus induced tumor regression of LNCaP and PC3-PSMA tumor xenografts. Extensive areas of MV infection and apoptosis were seen in virus treated tumors.
CONCLUSIONS
The PSMA retargeted virus warrants further investigation as a virotherapy agent.
Keywords: oncolytic measles virus, PSMA targeting, radiation, prostate cancer
INTRODUCTION
Prostate cancer accounts for 33% of all cancer diagnoses in American men (218,890 cases in 2007) as well as 9% of cancer deaths (27,050 in 2007) in men [1]. With the widespread use of prostate-specific antigen (PSA) testing, patients now present at a younger age, with a lower serum PSA, and a higher proportion of organ confined tumors. Primary treatment can be radical prostatectomy, external beam radiation therapy, brachytherapy, combination of external beam radiation therapy and brachytherapy, hormonal therapy (androgen deprivation) or watchful waiting [2]. Early stage androgen dependent prostate cancer responds well to conventional therapies. Even patients with metastatic prostate cancer initially respond to chemical or surgical castration although the response lasts only for a median duration of 18–24 months. Relatively few treatment options exist for patients with hormone-refractory prostate cancer. Median survival is poor at approximately 20 months from the time of initiation of standard docetaxel-based chemotherapy [3]. Clearly, novel targeted therapeutics are needed for the treatment of androgen-refractory disease.
In this study, we investigated the potential of a replication-competent attenuated vaccine strain (Edmonston) of measles virus for tumor selective virotherapy of prostate cancer. Oncolytic measles virus has promising antitumor activity against a variety of tumor types, including prostate cancer [4–8]. The virus exerts its antitumor activity by infecting human cancer cells via one of the two measles receptors, CD46 or SLAM, and inducing extensive intercellular fusion between the infected cell and neighboring cells to form non-viable multinucleated structures (syncytia), generating a large bystander killing [9,10]. Virotherapy using attenuated measles virus is potentially safe; millions of doses of measles vaccine have been given worldwide in very effective measles vaccination programs and the vaccine does not cause measles illness in immunocompetent individuals [9]. Two recombinant measles viruses expressing marker proteins (MV-CEA and MV-NIS) are being tested in three different phase I clinical trials [11,12]. These “first in human use” dose escalation trials test the safety of intratumoral, intraperitoneal, and intravenous delivery of measles viruses in patients with ovarian cancer, glioblastoma, and multiple myeloma, respectively. In particular, the myeloma trial incorporates administration of cyclophosphamide as an immunosuppressive agent to dampen the host immune responses against measles virus to enhance or prolong viral replication [13]. However, co-administration of an immunosuppressive agent with virotherapy might result in toxicity due to indiscriminate infection of normal tissues by the CD46/SLAM tropic virus. The measles receptor CD46 is expressed ubiquitously on all nucleated human cells [14], albeit at significantly lower levels than on tumor cells [15], while SLAM is expressed on immune cells [16]. Lack of specificity in receptor usage might impose a limitation on the highest tolerated dose level that can be used in measles virotherapy. To generate a tumor specific virus, we have developed a strategy to engineer the coat protein of measles virus to bind exclusively to a specific antigen. These fully retargeted viruses are ablating for CD46 and SLAM interactions and has an additional targeting moiety inserted as a C terminal extension on the measles attachment hemagglutinin (H) protein [17,18].
In the current study, we have generated a fully retargeted virus that recognizes the prostate-specific membrane antigen (PSMA) which is expressed in high numbers on surfaces of prostate cancer cells to restrict virus infection exclusively through this receptor. PSMA is a type II glycoprotein and is expressed strongly in high-grade prostate cancers, metastatic lesions, and androgen-independent disease [19]. In addition, PSMA is expressed abundantly on the tumor neovasculature of human tumors, making it an attractive target for monoclonal antibody (mAb) therapy even for non-prostate cancers [20,21]. Several PSMA targeted therapeutics (mAb, tumor specific cytotoxic T cells) have been developed and some are in clinical testing [22,23]. In particular, the J591 mAb binds to the extracellular domain of PSMA and has been shown to bind to prostate cancer cells and endothelial cells in the tumor vasculature. The J591 single-chain antibody (scFv) was cloned by Gong and colleagues and was used very successfully to target cytotoxic T lymphocytes (CTL) for prostate cancer therapy [20,24]. In this study, we demonstrate that the recombinant measles virus that is retargeted to infect via human PSMA specifically infects PSMA positive cells in vitro and in vivo, and has potent antitumor activity against two human prostate tumor xenograft models. In addition, application of low dose external beam radiation enhanced the antitumor activity of the PSMA retargeted measles virus. As such, radiation therapy can potentially be used in combination with measles virotherapy for control of androgen refractory prostate cancer.
MATERIALS ANDMETHODS
Cell Lines and Culture
Cell lines were purchased from the American Type Cell Culture (ATCC, Manassas, VA): prostate cancer cell lines LNCaP (ATCC CRL-1740) and PC3 (ATCC CRL-1435), Jurkat T cell leukemia (ATCC TIB-152), Raji B cell lymphoma (ATCC CCL-86), and Vero African green monkey kidney cells (ATCC CCL-81). KAS 6/1 multiple myeloma cells were a kind gift from Dr. Diane Jelinek (Mayo Clinic). LNCaP, Jurkat, KAS 6/1, and Raji cells were cultured in RPMI-1640; PC3 and Vero (5% FBS) cells were cultured in DMEM medium supplemented with 10% FBS and 100 U/ml of penicillin and 100 μg/ml streptomycin. KAS 6/1 cells also required recombinant human IL-6 at 1 ng/ml. PC3/PIP was generated by transduction of PC3 cells using VSV-G pseudotyped lentiviral vector expressing human PSMA as described previously [25]. A modified single cell deposition assay was used to select for high PSMA expression colonies [26]. PC3/PIP cells were plated into 96-well plates at a density of 0.3 cell/well. Individual wells with only one single cell were expanded. The selected colonies were examined for PSMA expression using Western blot and an anti-PSMA antibody (Santa Cruz, SC-10269), and a high PSMA expression colony was used in all subsequent experiments. Vero α-His cells are Vero cells stably transfected to express a membrane anchored single-chain antibody that recognizes a six-histidine peptide [18].
Generation of PSMA-Retargeted Measles Virus
The fully retargeted measles virus was generated as previously described [18,27]. Alanine substitutions at residues 481 and 533 of the measles H protein ablated viral interactions with CD46 and SLAM, respectively. The cDNA encoding the single-chain antibody J591 against PSMA [28], was inserted as a SfiI-NotI PCR fragment between the mutant H gene and its C-terminal H6 peptide (Fig. 1). The chimeric H cDNA was then subcloned via PacI-SpeI restriction sites into the full-length infectious cDNA clone of measles virus encoding an enhanced green fluorescent protein (eGFP) reporter gene. Finally, the PSMA-retargeted measles virus was rescued and propagated on a pseudoreceptor system via the 6-histidine peptide tag and Vero cells expressing a single-chain antibody against His6 [18]. To test specificity of the PSMA-retargeted virus (MVG-αPSMA), another fully retargeted measles virus displaying a scFv against the plasma cell marker CD38 (MVG-αCD38) was used as a control [18]. All viruses were propagated on Vero-αHis cells (multiplicity of infection, MOI 0.02) and titers of viral stocks were determined by TCID50 titrations as described previously [29].
Fig. 1.
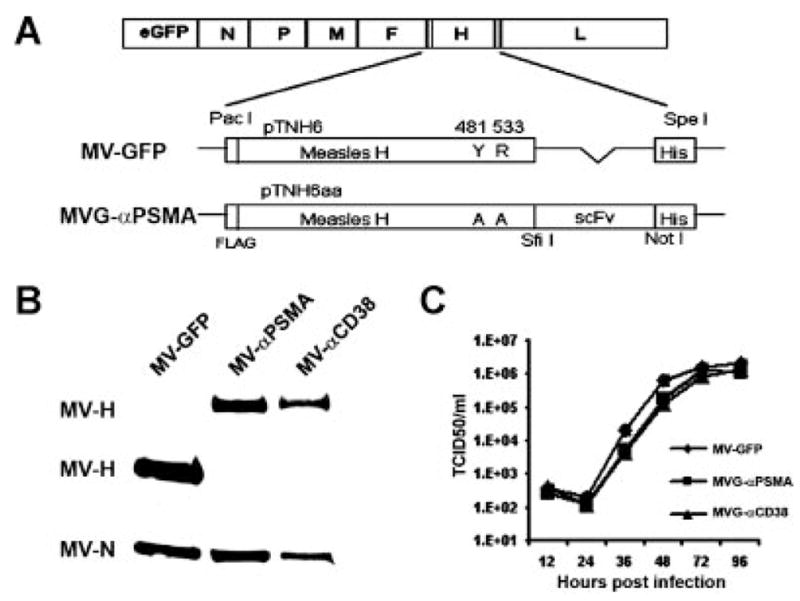
Construction and characterization of PSMA-retargeted measles virus. A: Schematic representation of the parental MV-GFP and PSMA fully retargeted measles viral genomes. The 481Y → A and 533R → Amutations in Hablate CD46/SLAM interaction. The anti-PSMA (or CD38) single-chain antibody (scFv) is inserted at the COOH-terminal of mutated H followed by a six-histidine peptide (H6). B: Immunoblotting of MV-GFP, MVG-αPSMA and MVG-αCD38 virions using anti-H and anti-N antibodies. 5 ×104 TCID50 of each virus was loaded. The chimeric H glycoproteins of MVG-αPSMA and MVG-αCD38 (lanes 2 and 3) have higher molecular weights compared to that of MV-GFP (lane 1). C: One-step growth kinetics of MV-GFP, MVG-αPSMA and MVG-αCD38 on Vero-αHis cells.
Characterization of the PSMA Retargeted Virus
Immunoblot analysis for measles H proteins
Immunoblotting was performed on the viral particles to confirm correct incorporation of the anti-PSMA scFv into the H protein. An aliquot (104 TCID50) of viral samples were mixed with an equal volume of 2× SDS loading buffer (Bio-Rad, Hercules, CA), denatured for 5 min at 95 °C, and separated in a 7.5% SDS–polyacrylamide gel. The proteins were transferred to nitrocellulose membrane (Amersham, Piscataway, NJ), and the N proteins were detected with a monoclonal mouse anti-measles N antibody (1:5,000 dilution, Novus Biologicals, Littleton, CO) while the H proteins were detected using a polyclonal rabbit anti-measles H protein antibody at 1:10,000 dilution [29]. Secondary antibody was applied to the respective blots, for the anti-N blot we used a goat anti-mouse-HRP (KPL, Gaithersburg, MD) at 1:5,000 dilution and for the anti-H blot, goat anti-rabbit-HRP (Calbiochem, San Diego, CA) antibody was used at 1:5,000 dilution. The blots were developed using the SuperSignal West Pico Chemiluminescent Substrate kit (Pierce Chemical, St. Louis, MO) according to manufacturer’s instructions.
Virus growth kinetics
The growth characteristics of the recombinant viruses were compared with the parent virus MV-GFP [18]. All the retargeted double blind (CD46 and SLAM ablated) viruses display a six-histidine peptide (His6) to enable their propagation and spread via a pseudoreceptor on Vero cells stably expressing a membrane anchored anti-His6 scFv [18]. Vero-αHis cells were infected with the viruses at a MOI of 3.0 in Opti-MEM (Life Technologies, Rockville, MD) medium for 2 hr at 37 °C after which the virus inoculum was removed and cells were incubated at 32 °C in standard growth medium. The cells were harvested at 12, 24, 36, 48, and 72 hr after infection by scraping them into 1 ml Opti-MEM, and cell-associated viruses were released by two freeze–thaw cycles. Viral titers were determined by TCID50 titration on Vero-αHis cells [29].
In Vitro Specificity of Viral Infection
The presence or absence of PSMA expression on prostate cancer cell lines LNCaP, PC3, PC3/PIP, and a panel of hematopoietic cancer cell lines KAS 6/1 (multiple myeloma), Jurkat (T cell lymphoma) and Raji (B cell lymphoma) was determined by immunoblotting cell lysates with mouse anti-human PSMA antibody (Santa Cruz Biotechnology, sc-10269, Santa Cruz, CA). The CD38 and CD46 expression of the above cell lines were examined by flow cytometry using PE-conjugated mouse anti-human CD38 antibody (BD Pharmingen #555460, San Jose, CA) and FITC-conjugated anti-human CD46 antibody (BD Pharmingen #555949), respectively.
For infection assays, cells (1–2 ×105 adherent cells or 106 suspension cells) were incubated with the viruses at an MOI of 0.5 for 3 hr at 37 °C, and the inoculum was replaced by the growth medium. For FACS analysis, the medium contained 40 μg/ml of a fusion inhibitory peptide (Z-D-Phe-L-Phe-Gly-OH, Bachem, Torrance, CA) that blocks H/F induced cell-to-cell fusion. At 48 hr after infection, cells were photographed under fluorescence microscopy, or harvested for flow cytometry to determine the percentage of GFP positive virus infected cells.
In Vitro Cell Killing by the PSMA Retargeted Virus
Crystal violet staining for cytopathic effect (CPE) caused by the PSMA retargeted virus
LNCaP cells were mock infected or infected with MVG-αPSMA at MOI of 0.25 for 2 hr at 37 °C, after which the virus inoculum was removed and the cells were incubated overnight at 37 °C. The next day, the cells received no radiation or irradiation at 2, 4, or 8 Gy (137Cs source). At 96 hr post-irradiation, cells were fixed with 4% paraformaldehyde, stained with 2% crystal violet, and photographed using a microscope.
Trypan blue exclusion assay
LNCaP cells were infected with MVG-αPSMA at MOI of 0.1, 0.25, or 0.5 for 2 hr at 37 °C as described above. The next day, the cells received no radiation or were irradiated at 2 or 5 Gy (137Cs), and incubated further at 37 °C. At 48, 72, and 96 hr after infection, the cells were trypsinized, washed and 0.25% trypan blue was added. The numbers of viable cells were counted using a light microscope.
Luciferase activity assay
LNCaP cells stably expressing firefly luciferase were infected with viruses at MOI of 0.1, 0.25, or 0.5 as described above. The next day, cells received no radiation, or 2 or 5 Gy of 137Cs radiation. Cells were incubated at 37 °C for different periods of time after which cell viability was determined by measuring firefly luciferase activity. Cells were trypsinized and resuspended in the same volume of medium as untreated cells (cell concentration was 104/50 μl). Fifty microliters of each sample and 15 μl of D-luciferin (GBT, St. Louis, MO, concentration 15 mg/ml) were added into 96-well black plates, and the relative light units (RLU) was determined using a Microplate scintillation & luminescence counter (Model: c9906, Perkin-Elmer, Downers Grove, IL). Cell viability was presented as RLU of luciferase activity/104 cells.
Immunohistochemical Staining for MV-N Protein and Apoptosis in Tumors
Tumors were sectioned into halves and frozen immediately in optimal cutting temperature (OCT) medium. Cryosections (5 μm in thickness) were fixed in −20 °C acetone for 5–10 min, permeabilized using 0.01% Tween-20-PBS for 15 min, exposed to blocking medium (5% horse serum in PBS) for 30 min and incubated with biotinylated primary antibody against measles N protein (Mab 8906, Chemicon) for 1 hr. The slides were developed using Vector ABC HRP DAB substrate kits (Vector Labs, Burlingame, CA) per manufacturer’s instructions and counterstained with Gill’s Hematoxylin. The presence of apoptotic areas in the tumor sections was detected using a terminal deoxynucleotidyl transferase-mediated nick end labeling (TUNEL) apoptosis detection kit per manufacturer’s instructions (Roche, Indianapolis, IN). DAB was used as a chromogen.
In Vivo Experiments
All animal procedures were approved by the Institutional Animal Care and Use Committee of Mayo Foundation. To determine the specificity of virus infection in vivo, male athymic mice (5–6 weeks of age; Taconic Laboratory, Germantown, NY) were implanted with 5 ×106 LNCaP or PC3/PIP cells subcutaneously (s.c.) in the right flank. When the tumors reached 300–400 mm3, mice were randomized into four treatment groups of three mice each. Mice in each group were injected intratumorally with 100 μl of saline or 2 ×106 TCID50 of each MV (MVG-αCD38, MVG-αPSMA, and MV-GFP) in 100 μl of Opti-MEM medium. Ninety-six hours post-MV administration, the mice were euthanized and the tumors were embedded in OCT and sectioned. The sections were checked for GFP expression under fluorescence microscopy and immunostained for MV-N protein expression.
Two models of PSMA positive human prostate cancer cell lines were used in the therapy experiments: LNCaP and PC3/PIP. For the LNCaP therapy experiments, 8 ×106 LNCaP cells were mixed with Matrigel (50%), and implanted s.c. in the right flank of male athymic mice. When the tumors reached 200–300 mm3 (about 5–7 weeks), mice were randomized into six treatment groups of six mice each. Mice in each treatment groups were given one intratumoral dose of 2 ×106TCID50 of each MV diluted in 100 μl of Opti-MEM. Treatment groups were heat-inactivated MV-GFP, active MV-GFP, MVG-αCD38, MVG-αPSMA, external beam radiation (RT) and MVG-αPSMA + RT combination. Low dose external beam radiation (2 Gy) was administered at the tumor site 24 hr post-MV administration as described previously [30]. Mice were observed twice a week following the treatments. Tumor size and body weight of the mice were measured. Tumor volumes were calculated as 0.5 × length × width2. Mice were euthanized when the tumor size exceeded 1,000 mm3 or if tumors ulcerate.
For therapy experiments in the PC3/PIP model, 4 ×106 PC3/PIP cells were implanted s.c. in the right flank of male athymic mice. When the tumors reached 100–200 mm3 (about 2–3 weeks), mice were randomized into six treatment groups of 6–12 mice each. Mice received two intratumoral injections (1 week interval) of 2 ×106 TCID50 of each MV diluted in 100 μl of Opti-MEM medium; heat-inactivated MV-GFP, MV-GFP, MVG-αCD38, MVG-αPSMA, external beam radiation (RT) and MVG-αPSMA + RT combination. Low dose external beam radiation (2 Gy) was administered at the tumor site 48 hr post-MV administration as described previously [30]. Mice were observed twice a week following the treatments and tumor size and body weight of the mice were measured. Mice were euthanized when the tumor size exceeded 1,000 mm3 or if tumors ulcerate.
Statistical Analysis
The differences in tumor burden (tumor volume cm3) in each group were analyzed by ANOVA. Kaplan–Meier survival curves were plotted and the log-rank test was used to examine the significance of differences in the survival between groups. We used GraphPad Prism (GraphPad Software, San Diego, CA) for the statistical analysis. A P value of < 0.05 is considered to be significant.
RESULTS
Characterization of the PSMA Retargeted Measles Virus
To target measles virus specifically to prostate cancer, a human PSMA specific scFv [28] was displayed as a C-terminal extension on the mutated measles H protein (HAA) which has alanine substitutions at residues 481 and 533 to ablate H interaction with the native measles receptors, CD46 and SLAM. Receptor binding of the retargeted virus is now dependent on the displayed scFv. The scFv was inserted as a SfiI-NotI PCR fragment between the CD46/SLAM ablated HAA gene and a six-histidine peptide (His6) and then subcloned as a PacI-SpeI fragment into the full-length infectious cDNA clone of measles virus encoding a GFP reporter gene (Fig. 1A). Presence of the His6 tag allows rescue and propagation of fully retargeted viruses on Vero producer cells stably expressing a membrane anchored scFv specific for His6 [18]. Immunoblotting of virions using an anti-measles H antibody demonstrated correct incorporation of the anti-PSMA scFv on HAA (Fig. 1B). Chimeric H proteins with a displayed scFv (against PSMA or CD38) have a higher molecular weight compared to the parental H protein in MV-GFP (Fig. 1B). Display of the scFv on the H protein did not negatively impact incorporation of the chimeric H-scFv proteins into virions. The blots were also probed for the most abundant measles structural protein, nucleocapsid N. As shown from the anti-N and anti-H immunoblots, the relative intensity of H:N is higher in the parental virus compared to either chimeric viruses, indicating higher incorporation of the unmodified H proteins per virion. However, the infectivity of the scFv displaying viral stocks is well-maintained and reaches titers comparable to the parental MV-GFP, suggesting that unmodified measles viruses contain excess of H-protein relative to their requirements, and that H abundance is not limiting for virus entry (Fig. 1B). Growth kinetics of the PSMA retargeted virus was compared against the parental MV-GFP virus and the previously published CD38 retargeted MVG-αCD38 virus [29]. As shown in Figure 1C, the one-step growth curves showed comparable replication kinetics and most importantly, the one-step growth kinetics and final viral titers were not significantly compromised by the displayed anti-PSMA scFv.
Specificity of MVG-αPSMA Infection In Vitro
Prostate cancer cells LNCaP, PC3, PC3/PIP (PC3 transfected to stably express the human PSMA protein) and CD38 positive Jurkat, KAS 6/1 and Raji hematopoietic cancer cell lines were chosen to test the specificity and oncolytic activity of MVG-αPSMA. Specificity control was provided by the CD38 retargeted virus MVG-αCD38. Expression of the PSMA protein in each of the cell lines was determined by immunoblotting. As shown in Figure 2, PSMA was strongly expressed by LNCaP and PC3/PIP cells but was negative in PC3 cells and the hematopoietic cell lines. Presence of CD38 and CD46 receptors were determined by flow cytometry using FITC-conjugated anti-human CD38 or anti-human CD46 antibodies. Raji, KAS 6/1 and Jurkat cells express high levels of CD38 receptors. In contrast, the prostate cancer cells were negative for CD38 (Fig. 2). As expected, all of the human cancer cell lines express abundant levels of the measles receptor, CD46 (Fig. 2).
Fig. 2.

Receptor expression profiles of prostate cancer and hematopoietic cancer cell lines. A:Immunoblotting of various prostate and hematopoietic cancer cell lines with anti-PSMA antibody. Beta-actin was used as a loading control. B:Expression of CD38 and CD46 receptors on the respective cell lines were determined by flow cytometry. Isotype control = Empty histograms.
Specificity of PSMA usage by MVG-αPSMA was analyzed by infectivity assays on the panel of PSMA negative or PSMA positive cell lines (Fig. 3). The panel of six cell lines was infected with MV-GFP, MVG-αPSMA or MVG-αCD38 viruses at MOI of 0.5 for 2 hr and examined using a fluorescence microscopy at 48 hr post-infection. All cell lines were efficiently infected by the parental MV-GFP virus giving rise to the characteristic measles induced cytopathic effect (CPE) of syncytial formation (Fig. 3). In contrast, the CD38 or PSMA fully retargeted CD46/SLAM ablated viruses infected and caused CPE only in cells that express the relevant targeted receptor (Fig. 3). Thus, the MVG-αPSMA virus induced syncytia formation only in LNCaP and PC3/PIP cells and not others. Virus infectivity was further evaluated in Vero-αHis virus producer cells, LNCaP, PC3 and PC3/PIP cell lines by analyzing the numbers of GFP positive single infected cells using flow cytometry (Fig. 3B). MV-GFP, MVG-αPSMA and MVG-αCD38 efficiently infected Vero-αHis viral producer cells resulting in more than 90% GFP positive cells at 48 hr post-infection (MOI 0.5). Infectivity of MV-GFP was lower on LNCaP cells (~60%) than on PC3 or PC3/PIP (>90%). Similarly, the MVG-αPSMA virus infected ~30% of the LNCaP cells and >80% of PC3/PIP cells and showed no detectable levels of infection in PC3 cells which do not express human PSMA.
Fig. 3.

Specificity of infection by the MVG-αPSMA virus. A:PSMA positive (LNCaP, PC3/PIP) and PSMA negative (PC3) prostate cancer cell lines and hematopoietic cancer cell lines were infected with the respective viruses at MOI of 0.5. Photographs of virus infected cells were taken at 48 hr post-infection using a fluorescence microscope. B:Flow cytometry analysis of numbers of GFP expressing virus infected cells at 48 hr post-infection.
Oncolytic Activity of MVG-αPSMA Virus in Combination With RT
Radiation therapy (RT), administered either as external beams generated by a linear accelerator or implanted as radioactive seeds in the tumor (interstitial brachytherapy), is an important treatment option for localized prostate cancer [31]. Hence, we explored the possibility that the oncolytic activity of the MVG-αPSMA virus might be enhanced in combination with RT. Androgen sensitive LNCaP and androgen resistant PC3/PIP cells were mock infected or exposed to a low dose of MVG-αPSMA virus (MOI 0.1, 0.25, or 0.5). The next day, the cells were irradiated with a137Cs source. The cells were then incubated further after which the amount of cytotoxicity was determined. As shown in Figure 4, crystal violet staining showed that there was more cytotoxic killing of measles infected and irradiated cells than cells that received RT alone or virus alone (MOI 0.25, no RT). To obtain a quantitative readout of the extent of cytotoxicity, cell viability was determined by trypan blue exclusion assay (Fig. 4B,C) and firefly luciferase activity of LNCaP and PC3/PIP cells stably expressing firefly luciferase (Fig. 4D,E). There was enhanced cytotoxic killing in both LNCaP and PC3/PIP cells exposed to combination therapy of MVG-αPSMA and RT (Fig. 4B,C). There was dose-dependent effect on viral dose and radiation, with increased cell killing at the high viral or radiation doses.
Fig. 4.
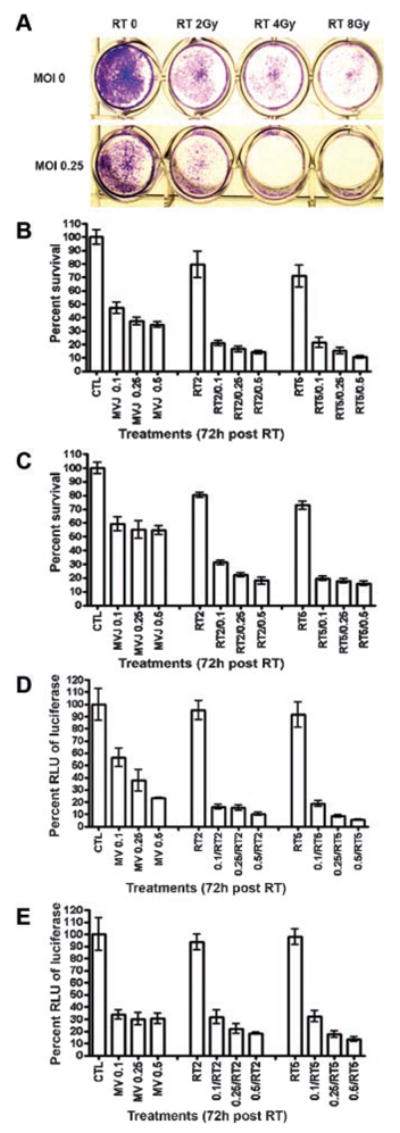
Quantitation of in vitro cytotoxic killing of prostate cancer cells by MVG-αPSMA in combination with radiotherapy (RT). A:LNCaP cells were mock infected (MOI 0) or infected with MVG-αPSMA (MOI 0.25) and 24 hr later, were irradiated (Gy) with a137Cs source. Crystal violet staining of viable cells was performed at 96 hr post-RT. B–E: LNCaP and PC3/PIP cells were mock infected (CTL) or infected with MVG-αPSMA (MV/MOI 0.1, 0.25, or 0.5) and were irradiated (RT) at 2 or 5 Gy 24 hr later. The numbers of viable cells were estimated by trypan blue exclusion assay (B,C) or by firefly luciferase activity (D,E) at 72hr post-RT and cell survival is calculated as a percentage of mock infected non-irradiated cells (CTL = 100%). The error bars represent SD, average from three replicate experiments.
Anti-Tumor Activity of MVG-αPSMA Virus In Vivo
The antitumor potential of different therapy options, virus alone, RT alone or virus +RT combination were tested in PC3/PIP and LNCaP xenograft models in male athymic mice. Tumors received one intratumoral injection of test article (2 ×106 TCID50 active virus) or 2 Gy RT delivered locally. In the LNCaP xenograft model, there was no antitumor activity due to administration of heat inactivated MV-GFP, 2 Gy RT alone or an equivalent dose of active MVG-αCD38 negative control virus. In contrast, both MVG-αPSMA and parental MV-GFP have significant and comparable antitumor activities against the tumor xenografts, resulting in regression or growth inhibition of tumors (Fig. 5A,B). Combination treatment using MVG-αPSMA virus and RT (2 Gy) 24 hr post-virus administration further enhanced the antitumor activity of MVG-αPSMA. In the survival curve analysis, MVG-αPSMA significantly enhanced (P < 0.005) survival of mice as compared with MVG-αCD38 and the heat-inactivated MV-GFP (Fig. 5C). Body weights of the mice were also monitored over time (Fig. 5D). Mice that received MVG-αPSMA, MV-GFP, and combination MVG-αPSMA/RT treatments showed an increase in body weight (Fig. 5D). In contrast, mice that received heat inactivated MV-GFP, MVG-αCD38 or radiation alone had significant tumor burden and did not demonstrate weight gain as the responsive groups.
Fig. 5.

In vivo anti-tumor activity of MVG-αPSMA in LNCaP model. Six groups of mice (n = 6 mice per group) were implanted subcutaneously with LNCaP xenografts. When tumor volumes reached150–350 mm3, each mouse received one intratumoral dose of active virus at 2 ×106 TCID50 and/or 2 Gy RT or heat inactivated MV-GFP (HIMV-GFP). A: Average tumor volumes of mice in each treatment group. B: Individual profiles of tumor growth over time for each mouse in the respective treatment groups. C: Kaplan–Meier survival curve of each virus treatment group. D: Average body weight of mice in each treatment group. Error bars represent SD. Asterisk (*) denotes statistically significant with P < 0.001.
The antitumor potential of MVG-αPSMA was also evaluated in the androgen resistant prostate cancer model, PC3/PIP cells which express human PSMA. PC3/PIP tumors did not respond to therapy using heat inactivated MV-GFP, RT (2 Gy) alone or MVG-αCD38 virus (Fig. 6A). In contrast, tumor volumes were significantly lower (P < 0.001) if the mice were given intratumoral injections of 4 ×106 TCID50 MV-GFP or MVG-αPSMA or if they received combination therapy using MVG-αPSMA and RT. The survival of mice were compared and shown to be significantly enhanced (P < 0.001) if they received MV-GFP, MVG-αPSMA or MVG-αPSMA + RT therapies (Fig. 6B) but this experiment is not powered to detect differences between each of these three treatment groups.
Fig. 6.

In vivo anti-tumor activity of MVG-αPSMA in PC3/PIP model. Six treatment groups of 6–12 mice per group were implanted subcutaneously with 5 ×106 of PC3/PIP cells. When tumor volumes reached100–300 mm3, each mouse received two intratumoral doses of virus at 2 ×106 TCID50/dose given with a 2-week interval. A: Average tumor volumes of mice per treatment group over time are shown. Error bars represent SD. B: Kaplan–Meier survival curve of mice in each treatment group. The statistical significance between survival curves of mice in the respective treatment groups were compared with the control heat inactivated MV-GFP group. Asterisk (*) denotes statistically significant with P < 0.001.
Immunohistochemical Analysis of Virus Infection and Spread in Tumors
Tumors from another cohort of mice treated similarly with the viruses were harvested at 4 days post-injection and examined using a fluorescence microscopy under low power (40×). There were areas with bright GFP fluorescence in MVG-αPSMA and MV-GFP treated groups whereas no GFP positive areas were detected in tumors treated with heat-inactivated and MVG-αCD38 treated groups (data not shown). Cryosections of tumors were also immunostained with a antibody against the measles virus N protein. As expected, the retargeted viruses retained their specificity in vivo (Fig. 7). There were extensive areas of measles infection (positive brown color staining) in both LNCaP and PC3/PIP xenografts treated with the PSMA specific MVG-αPSMA virus and the parental MV-GFP virus (Fig. 7). In contrast, no or minimal MV-N protein staining was seen in LNCaP or PC3/PIP tumors injected with heat-inactivated MV or the negative control virus MVG-αCD38 (Fig. 7).
Fig. 7.

In vivo specificity and infection efficiency of MVG-αPSMA virus. Tumor xenografts were injected intratumorally with one dose of 2 ×106TCID50 virus and harvested 4 days later. Tumors were cryosectioned and immunostaining for MV-nucleocapsid N protein (brown color) was performed. MVG-αPSMA virus efficiently infected PSMA positive LNCaP and PC3/PIP tumors. In contrast, PSMA negative tumor PC3 showed little or no infection by the MVG-αPSMA virus.
To elucidate the mechanism of cell death in vivo, LNCaP tumors were sectioned and stained with anti-measles N antibody and TUNEL for apoptosis. There were areas of positive N staining in the MV-GFP and MVG-αPSMA treated groups but not in the heat inactivated MV-GFP, RT or MVG-αCD38 groups (Fig. 8). These tumor sections also stained positive for nicked DNA by TUNEL staining, indicating apoptosis in the tumor cells (Fig. 8). Closer examination of the sections indicates that some of the MV-N areas are positive for TUNEL in the corresponding slide (blue arrows). In other cryosections such as the one shown for MVG-αPSMA treated tumor, the areas of tumor destruction stained strongly positive for TUNEL but not for MV-N, suggesting that the MV replication has already ceased in those apoptotic areas (green arrows).
Fig. 8.

Measles virus infected prostate tumor cells die by apoptosis in vivo. LNCaP tumors that received one dose virus and/or 2 Gy RT were harvested 4 days and immunostained for MV-N protein or presence of nicked DNA by TUNEL staining. Green arrows depict corresponding area that is negative for MV-N staining but positive for TUNEL staining. Blue arrows depict corresponding area that is positive for MV-N staining indicating active viral gene expression, but positive for TUNEL staining.
DISCUSSION
We have generated a PSMA retargeted oncolytic measles virus and demonstrated that this virus specifically infected and caused cytopathic killing of PSMA positive prostate cancer cells and not PSMA negative cells. When administered intratumorally to mice bearing PSMA positive LNCaP or PC3/PIP tumor xenografts, the virus inhibited the growth or caused regression of established tumors. Virus infected tumors expressed high levels of viral antigen and stained positive in the TUNEL assay for apoptosis. The PSMA targeted virus was not compromised by the displayed scFv and its antitumor activity was comparable to the parental MV-GFP virus.
Virotherapy has shown promise as an anti-cancer modality since its initial use in the 1950s to 1970s [32]. Viruses from diverse Families were used as oncolytics; typically these viruses preferentially kill tumor cells in part due to the deregulated innate cellular antiviral immune responses in tumor cells [33]. Virotherapy using live attenuated measles virus from the Edmonston vaccine lineage is appealing from several aspects [34]. There is extensive safety data in humans from the worldwide vaccination programs that use the Edmonston vaccine. Millions of doses of measles vaccine have been administered and the virus has not reverted to the wild type strain or caused measles [9]. Using a virus to which the majority of the population has pre-existing antibodies protects the general population and prevents virus transmission between the patient and care givers. However, it also poses a formidable barrier to systemic use of this virus in patients who have been vaccinated or been infected by the wild type virus. Intratumoral administration can potentially circumvent virus neutralization and result in a positive antitumor outcome even in patients with antiviral antibodies [35]. However, preexisting antiviral antibodies can quickly neutralize the virus post-intravascular administration. Thus, we and others are currently investigating strategies to more effectively deliver viruses in the face of preexisting antiviral antibodies or innate immunity. Strategies include hiding the virus in cell carriers that could deliver the virus to tumor sites [36–38], shielding of the virus using polymers [39,40] or use of immunosuppressive drugs such as cyclophosphamide to dampen the innate immunity [41].
Measles is a fusogenic virus, the infected cell fuses with its neighbors to form nonviable multinucleated syncytia. Hence, in combination with replicative spread of the virus, there is significant bystander killing compared to cytotoxic killing by a non-replicating vector. Another attractive feature of measles virus is the availability of a rescue system to generate recombinant viruses that express additional genes for monitoring viral gene expression [42] or enhancing viral potency through prodrug activating enzymes or concentration of radioisotopes [12,43]. A potentially important genetic modification of oncolytic measles virus is alteration of virus tropism to enhance tumor specificity and/or delivery to tumor sites. The measles viruses currently being tested in phase I clinical trials use either of its two native receptors, CD46 or SLAM. CD46, a regulator of complement activation, is expressed at low levels on all human nucleated cells but is overexpressed on tumor cells [14] while SLAM is found in immune cells [16]. While CD46 is overexpressed on many tumor cells and is an attractive target for cancer therapy [15,44], interaction with SLAM is associated with measles virus induced immunosuppression [45,46]. To overcome the issue of non-specificity and potential toxicity from indiscriminate infection of normal cells by the virus, we recently developed a versatile system for generation of fully retargeted measles viruses whose tropism is dependent on the extraviral scFv displayed as a C-terminal extension on the measles H attachment protein [18]. The displayed scFv plays an important role in defining the performance of the retargeted virus as an oncolytic agent. Using a panel of viruses displaying anti-HER-2/neu scFvs with dissociation constants ranging from 10−6 to 10−11 M, we demonstrated that affinity of the scFv for its target receptor determines the fusogenicity and cytopathic potency of the HER-2/neu retargeted measles viruses [47].
The scFv display technology on the measles H platform is robust and versatile; numerous scFvs specific for various target receptors have been successfully displayed on the H protein to efficiently redirect cell entry of measles viruses and lentiviral vectors [34,48,49]. Considerations for choice of receptor include relative abundance of the target on tumor cells versus normal cells (very few “tumor-associated” receptors are truly tumor specific), availability of the binding ligand cDNA as well as affinity of the ligand for the receptor. PSMA is a surface membrane antigen that is expressed at low levels in benign prostatic epithelium but is over-expressed on prostate cancer cells [50]. The sensitivity and specificity of PSMA immunoreactivity in distinguishing prostate cancer from any other type of malignancy is 65.9% and 94.5%, respectively [50]. PSMA expression is up-regulated with disease progression where it is weakly positive in well-differentiated tumors and becoming highest in metastatic, hormone-refractory disease [51]. Indeed, adenocarcinomas with Gleason primary patterns 4 and 5 showed staining in 100% of the cells [52]. PSMA is not entirely tumor specific; it is expressed weakly in small intestine epithelial cells, proximal renal tubule cells, salivary glands although its expression in these tissues is 100–1,000-fold less than in normal prostate tissues, and even less compared to its expression in cancer tissues [22]. During cancer progression from androgen-dependence to androgen-independence, the overall expression of PSMA is up-regulated [53], and MVG-αPSMA virotherapy could be an excellent alternative in cases of androgen-depletion therapy failure.
Another appealing feature of PSMA targeting is that the antigen is abundantly expressed on tumor vasculature but not normal vasculature [25,54,55]. Using a panel of monoclonal antibodies that bind to the extracellular PSMA domain (including J591mAb used in this study) or intracellular domain, Chang et al. [25] determined that all the mAbs reacted strongly with neovasculature of a wide spectrum of malignant neoplasms including renal cell carcinoma, colonic adenocarcinoma, glioblastoma multiforme, malignant melanoma, non-small cell lung carcinoma, breast carcinoma and prostatic carcinoma. Antigens uniquely expressed on tumor vasculature are highly sought after for vascular targeting; these antigens are not restricted to a certain tumor type, and thus have broad applicability, and are readily accessible from the lumenal side of the tumor blood vessel to localize and potentially enhance delivery of the therapeutic. As such, MVG-αPSMA virotherapy can be used for oncolytic virotherapy of other cancer types in addition to prostate cancer. Indeed, taking advantage of high specificity and broad range appeal of PSMA targeting, several promising PSMA targeted therapeutics are being developed in academia and by the industry, including radio-labeled antibodies against PSMA for targeted tumor tissue imaging and PSMA targeted cytotoxic T lymphocytes for immune mediated tumor cell killing [19,23].
In summary, our results demonstrated equivalent potency of MVG-αPSMA to the parental virus in viral proliferation, infection efficiency and cyotoxicity but with greatly improved specificity to prostate cancer both in vitro and in vivo. Interestingly, we also noted that MVG-αPSMA virus showed better cytotoxicity and infection (MV-N immunostaining) in vivo compared to in vitro. This may be explained in part by the increased expression of PSMA in vivo in tumor tissue than in tissue culture. Immunoblotting showed higher PSMA expression in tumors harvested from mice compared to tumor cells in tissue culture when equivalent amounts of protein were loaded (data not show). We also explored the effect of low dose external beam irradiation in combination with measles virotherapy for prostate cancer. There is an additive effect, resulting in enhanced cytotoxic cell killing by combining MVG-αPSMA virotherapy with irradiation at 2 or 5 Gy in LNCaP and PC3/PIP cells in vitro. Low dose RT at 2 Gy did not inhibit the growth of LNCaP or PC3/PIP tumors in mice. However, there was significant extension of survival of mice treated intratumorally with MVG-αPSMA virus alone or MVG-αPSMA +2 Gy RT compared to control mice treated with heat-inactivated virus although we did not detect a significant difference in the Kaplan–Meier survival of mice in these two treatment groups. Future experiments involving larger cohorts of mice to give sufficient power to detect this (potentially small) difference as well as optimizing the timing or dose of the radiation therapy in conjunction with virotherapy will be required. However, based on these promising in vitro and in vivo studies, we plan to evaluate the PSMA targeted virus as a single agent in a dose escalation intratumoral phase I clinical trial for patients with androgen refractory prostate cancer.
Acknowledgments
This work is supported by the Alliance for Cancer Gene Therapy and the National Institutes of Health/National Cancer Institute (CA118488 and CA129193).
Grant sponsor: Alliance for Cancer Gene Therapy; Grant sponsor: National Institutes of Health/National Cancer Institute; Grant numbers: CA118488, CA129193.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J Clin. 2008;58(2):71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Boorjian SA, Blute ML. Surgical management of high risk prostate cancer: The Mayo Clinic experience. Urol Oncol. 2008;26(5):530–532. doi: 10.1016/j.urolonc.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 3.Petrylak DP. Future directions in the treatment of androgen-independent prostate cancer. Urology. 2005;65(6 Suppl):8–12. doi: 10.1016/j.urology.2005.04.020. [DOI] [PubMed] [Google Scholar]
- 4.Peng KW, Ahmann GJ, Pham L, Greipp PR, Cattaneo R, Russell SJ. Systemic therapy of myeloma xenografts by an attenuated measles virus. Blood. 2001;98(7):2002–2007. doi: 10.1182/blood.v98.7.2002. [DOI] [PubMed] [Google Scholar]
- 5.Peng KW, TenEyck CJ, Galanis E, Kalli KR, Hartmann LC, Russell SJ. Intraperitoneal therapy of ovarian cancer using an engineered measles virus. Cancer Res. 2002;62(16):4656–4662. [PubMed] [Google Scholar]
- 6.Phuong LK, Allen C, Peng KW, Giannini C, Greiner S, TenEyck CJ, Mishra PK, Macura SI, Russell SJ, Galanis EC. Use of a vaccine strain of measles virus genetically engineered to produce carcinoembryonic antigen as a novel therapeutic agent against glioblastoma multiforme. Cancer Res. 2003;63(10):2462–2469. [PubMed] [Google Scholar]
- 7.Blechacz B, Splinter PL, Greiner S, Myers R, Peng KW, Federspiel MJ, Russell SJ, LaRusso NF. Engineered measles virus as a novel oncolytic viral therapy system for hepatocellular carcinoma. Hepatology. 2006;44(6):1465–1477. doi: 10.1002/hep.21437. [DOI] [PubMed] [Google Scholar]
- 8.Msaouel P, Iankov ID, Allen C, Morris JC, von Messling V, Cattaneo R, Koutsilieris M, Russell SJ, Galanis E. Engineered measles virus as a novel oncolytic therapy against prostate cancer. Prostate. 2009;69(1):82–91. doi: 10.1002/pros.20857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Griffin D. Measles virus. In: Griffin D, Lamb R, Martin M, Roizman B, Straus S, editors. Field’s virology. Vol. 1. Philadelphia: Lippincott Williams & Wilkins; 2001. p. 1401. [Google Scholar]
- 10.Galanis E, Bateman A, Johnson K, Diaz RM, James CD, Vile R, Russell SJ. Use of viral fusogenic membrane glycoproteins as novel therapeutic transgenes in gliomas. Hum Gene Ther. 2001;12(7):811–821. doi: 10.1089/104303401750148766. [DOI] [PubMed] [Google Scholar]
- 11.Peng KW, Facteau S, Wegman T, O’Kane D, Russell SJ. Non-invasive in vivo monitoring of trackable viruses expressing soluble marker peptides. Nat Med. 2002;8(5):527–531. doi: 10.1038/nm0502-527. [DOI] [PubMed] [Google Scholar]
- 12.Dingli D, Peng KW, Harvey ME, Greipp PR, O’Connor MK, Cattaneo R, Morris JC, Russell SJ. Image-guided radiovirotherapy for multiple myeloma using a recombinant measles virus expressing the thyroidal sodium iodide symporter. Blood. 2004;103(5):1641–1646. doi: 10.1182/blood-2003-07-2233. [DOI] [PubMed] [Google Scholar]
- 13.Myers RM, Greiner SM, Harvey ME, Griesmann G, Kuffel MJ, Buhrow SA, Reid JM, Federspiel M, Ames MM, Dingli D, Schweikart K, Welch A, Dispenzieri A, Peng KW, Russell SJ. Preclinical pharmacology and toxicology of intravenous MV-NIS, an oncolytic measles virus administered with or without cyclophosphamide. Clin Pharmacol Ther. 2007;82(6):700–710. doi: 10.1038/sj.clpt.6100409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liszewski MK, Post TW, Atkinson JP. Membrane cofactor protein (MCP or CD46): Newest member of the regulators of complement activation gene cluster. Annu Rev Immunol. 1991;9:431–455. doi: 10.1146/annurev.iy.09.040191.002243. [DOI] [PubMed] [Google Scholar]
- 15.Ong HT, Timm MM, Greipp PR, Witzig TE, Dispenzieri A, Russell SJ, Peng KW. Oncolytic measles virus targets high CD46 expression on multiple myeloma cells. Exp Hematol. 2006;34(6):713–720. doi: 10.1016/j.exphem.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 16.Tatsuo H, Yanagi Y. The morbillivirus receptor SLAM (CD150) Microbiol Immunol. 2002;46(3):135–142. doi: 10.1111/j.1348-0421.2002.tb02678.x. [DOI] [PubMed] [Google Scholar]
- 17.Nakamura T, Peng KW, Vongpunsawad S, Harvey M, Mizuguchi H, Hayakawa T, Cattaneo R, Russell SJ. Antibody-targeted cell fusion. Nat Biotechnol. 2004;22(3):331–336. doi: 10.1038/nbt942. [DOI] [PubMed] [Google Scholar]
- 18.Nakamura T, Peng KW, Harvey M, Greiner S, Lorimer IA, James CD, Russell SJ. Rescue and propagation of fully retargeted oncolytic measles viruses. Nat Biotechnol. 2005;23(2):209–214. doi: 10.1038/nbt1060. [DOI] [PubMed] [Google Scholar]
- 19.Wang X, Yin L, Rao P, Stein R, Harsch KM, Lee Z, Heston WD. Targeted treatment of prostate cancer. J Cell Biochem. 2007;102(3):571–579. doi: 10.1002/jcb.21491. [DOI] [PubMed] [Google Scholar]
- 20.Gong MC, Chang SS, Sadelain M, Bander NH, Heston WD. Prostate-specific membrane antigen (PSMA)-specific monoclonal antibodies in the treatment of prostate and other cancers. Cancer Metastasis Rev. 1999;18(4):483–490. doi: 10.1023/a:1006308826967. [DOI] [PubMed] [Google Scholar]
- 21.Milowsky MI, Nanus DM, Kostakoglu L, Sheehan CE, Vallabhajosula S, Goldsmith SJ, Ross JS, Bander NH. Vascular targeted therapy with anti-prostate-specific membrane antigen monoclonal antibody J591 in advanced solid tumors. J Clin Oncol. 2007;25(5):540–547. doi: 10.1200/JCO.2006.07.8097. [DOI] [PubMed] [Google Scholar]
- 22.Nanus DM, Milowsky MI, Kostakoglu L, Smith-Jones PM, Vallabahajosula S, Goldsmith SJ, Bander NH. Clinical use of monoclonal antibody HuJ591 therapy: Targeting prostate specific membrane antigen. J Urol. 2003;170(6 Pt 2):S84–S88. doi: 10.1097/01.ju.0000095151.97404.7c. discussion S88–S89. [DOI] [PubMed] [Google Scholar]
- 23.Olson WC, Heston WD, Rajasekaran AK. Clinical trials of cancer therapies targeting prostate-specific membrane antigen. Rev Recent Clin Trials. 2007;2(3):182–190. doi: 10.2174/157488707781662724. [DOI] [PubMed] [Google Scholar]
- 24.Gade TP, Hassen W, Santos E, Gunset G, Saudemont A, Gong MC, Brentjens R, Zhong XS, Stephan M, Stefanski J, Lyddane C, Osborne JR, Buchanan IM, Hall SJ, Heston WD, Riviere I, Larson SM, Koutcher JA, Sadelain M. Targeted elimination of prostate cancer by genetically directed human T lymphocytes. Cancer Res. 2005;65(19):9080–9088. doi: 10.1158/0008-5472.CAN-05-0436. [DOI] [PubMed] [Google Scholar]
- 25.Chang SS, Reuter VE, Heston WD, Bander NH, Grauer LS, Gaudin PB. Five different anti-prostate-specific membrane antigen (PSMA) antibodies confirm PSMA expression in tumor-associated neovasculature. Cancer Res. 1999;59(13):3192–3198. [PubMed] [Google Scholar]
- 26.Metharom P, Liu C, Wang S, Stalboerger P, Chen G, Doyle B, Ikeda Y, Caplice NM. Myeloid lineage of high proliferative potential human smooth muscle outgrowth cells circulating in blood and vasculogenic smooth muscle-like cells in vivo. Atherosclerosis. 2008;198(1):29–38. doi: 10.1016/j.atherosclerosis.2007.09.020. [DOI] [PubMed] [Google Scholar]
- 27.Hasegawa K, Nakamura T, Harvey M, Ikeda Y, Oberg A, Figini M, Canevari S, Hartmann LC, Peng KW. The use of a tropism-modified measles virus in folate receptor-targeted virotherapy of ovarian cancer. Clin Cancer Res. 2006;12(20 Pt 1):6170–6178. doi: 10.1158/1078-0432.CCR-06-0992. [DOI] [PubMed] [Google Scholar]
- 28.Gong MC, Latouche JB, Krause A, Heston WD, Bander NH, Sadelain M. Cancer patient T cells genetically targeted to prostate-specific membrane antigen specifically lyse prostate cancer cells and release cytokines in response to prostate-specific membrane antigen. Neoplasia. 1999;1(2):123–127. doi: 10.1038/sj.neo.7900018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hadac EM, Peng KW, Nakamura T, Russell SJ. Reengineering paramyxovirus tropism. Virology. 2004;329(2):217–225. doi: 10.1016/j.virol.2004.08.036. [DOI] [PubMed] [Google Scholar]
- 30.Liu C, Sarkaria JN, Petell CA, Paraskevakou G, Zollman PJ, Schroeder M, Carlson B, Decker PA, Wu W, James CD, Russell SJ, Galanis E. Combination of measles virus virotherapy and radiation therapy has synergistic activity in the treatment of glioblastoma multiforme. Clin Cancer Res. 2007;13(23):7155–7165. doi: 10.1158/1078-0432.CCR-07-1306. [DOI] [PubMed] [Google Scholar]
- 31.Pisansky TM, Gold DG, Furutani KM, Macdonald OK, McLaren RH, Mynderse LA, Wilson TM, Hebl JR, Choo R. High-dose-rate brachytherapy in the curative treatment of patients with localized prostate cancer. Mayo Clin Proc. 2008;83(12):1364–1372. doi: 10.1016/S0025-6196(11)60785-4. [DOI] [PubMed] [Google Scholar]
- 32.Kelly E, Russell SJ. History of oncolytic viruses: Genesis to genetic engineering. Mol Ther. 2007;15(4):651–659. doi: 10.1038/sj.mt.6300108. [DOI] [PubMed] [Google Scholar]
- 33.Russell SJ, Peng KW. Viruses as anticancer drugs. Trends Pharmacol Sci. 2007;28(7):326–333. doi: 10.1016/j.tips.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakamura T, Russell SJ. Oncolytic measles viruses for cancer therapy. Expert Opin Biol Ther. 2004;4(10):1685–1692. doi: 10.1517/14712598.4.10.1685. [DOI] [PubMed] [Google Scholar]
- 35.Hu JC, Coffin RS, Davis CJ, Graham NJ, Groves N, Guest PJ, Harrington KJ, James ND, Love CA, McNeish I, Medley LC, Michael A, Nutting CM, Pandha HS, Shorrock CA, Simpson J, Steiner J, Steven NM, Wright D, Coombes RC. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin Cancer Res. 2006;12(22):6737–6747. doi: 10.1158/1078-0432.CCR-06-0759. [DOI] [PubMed] [Google Scholar]
- 36.Munguia A, Ota T, Miest T, Russell SJ. Cell carriers to deliver oncolytic viruses to sites of myeloma tumor growth. Gene Ther. 2008;15(10):797–806. doi: 10.1038/gt.2008.45. [DOI] [PubMed] [Google Scholar]
- 37.Thorne SH, Contag CH. Integrating the biological characteristics of oncolytic viruses and immune cells can optimize therapeutic benefits of cell-based delivery. Gene Ther. 2008;15(10):753–758. doi: 10.1038/gt.2008.42. [DOI] [PubMed] [Google Scholar]
- 38.Power AT, Bell JC. Taming the Trojan horse: Optimizing dynamic carrier cell/oncolytic virus systems for cancer biotherapy. Gene Ther. 2008;15(10):772–779. doi: 10.1038/gt.2008.40. [DOI] [PubMed] [Google Scholar]
- 39.O’Riordan CR, Lachapelle A, Delgado C, Parkes V, Wadsworth SC, Smith AE, Francis GE. PEGylation of adenovirus with retention of infectivity and protection from neutralizing antibody in vitro and in vivo. Hum Gene Ther. 1999;10(8):1349–1358. doi: 10.1089/10430349950018021. [DOI] [PubMed] [Google Scholar]
- 40.Carlisle RC, Benjamin R, Briggs SS, Sumner-Jones S, McIntosh J, Gill D, Hyde S, Nathwani A, Subr V, Ulbrich K, Seymour LW, Fisher KD. Coating of adeno-associated virus with reactive polymers can ablate virus tropism, enable retargeting and provide resistance to neutralising antisera. J Gene Med. 2008;10(4):400–411. doi: 10.1002/jgm.1161. [DOI] [PubMed] [Google Scholar]
- 41.Fulci G, Breymann L, Gianni D, Kurozomi K, Rhee SS, Yu J, Kaur B, Louis DN, Weissleder R, Caligiuri MA, Chiocca EA. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc Natl Acad Sci USA. 2006;103(34):12873–12878. doi: 10.1073/pnas.0605496103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peng KW, Hadac EM, Anderson BD, Myers R, Harvey M, Greiner SM, Soeffker D, Federspiel MJ, Russell SJ. Pharmacokinetics of oncolytic measles virotherapy: Eventual equilibrium between virus and tumor in an ovarian cancer xenograft model. Cancer Gene Ther. 2006;13(8):732–738. doi: 10.1038/sj.cgt.7700948. [DOI] [PubMed] [Google Scholar]
- 43.Ungerechts G, Springfeld C, Frenzke ME, Lampe J, Johnston PB, Parker WB, Sorscher EJ, Cattaneo R. Lymphoma chemovirotherapy: CD20-targeted and convertase-armed measles virus can synergize with fludarabine. Cancer Res. 2007;67(22):10939–10947. doi: 10.1158/0008-5472.CAN-07-1252. [DOI] [PubMed] [Google Scholar]
- 44.Fishelson Z, Donin N, Zell S, Schultz S, Kirschfink M. Obstacles to cancer immunotherapy: Expression of membrane complement regulatory proteins (mCRPs) in tumors. Mol Immunol. 2003;40(2–4):109–123. doi: 10.1016/s0161-5890(03)00112-3. [DOI] [PubMed] [Google Scholar]
- 45.Schneider-Schaulies J, Schneider-Schaulies S. Receptor interactions, tropism, and mechanisms involved in morbillivirus-induced immunomodulation. Adv Virus Res. 2008;71:173–205. doi: 10.1016/S0065-3527(08)00004-3. [DOI] [PubMed] [Google Scholar]
- 46.Ohno S, Ono N, Seki F, Takeda M, Kura S, Tsuzuki T, Yanagi Y. Measles virus infection of SLAM (CD150) knockin mice reproduces tropism and immunosuppression in human infection. J Virol. 2007;81(4):1650–1659. doi: 10.1128/JVI.02134-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hasegawa K, Hu C, Nakamura T, Marks JD, Russell SJ, Peng KW. Affinity thresholds for membrane fusion triggering by viral glycoproteins. J Virol. 2007;81(23):13149–13157. doi: 10.1128/JVI.01415-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frecha C, Costa C, Negre D, Gauthier E, Russell SJ, Cosset FL, Verhoeyen E. Stable transduction of quiescent T cells without induction of cycle progression by a novel lentiviral vector pseudotyped with measles virus glycoproteins. Blood. 2008;112(13):4843–4852. doi: 10.1182/blood-2008-05-155945. [DOI] [PubMed] [Google Scholar]
- 49.Funke S, Maisner A, Muhlebach MD, Koehl U, Grez M, Cattaneo R, Cichutek K, Buchholz CJ. Targeted cell entry of lentiviral vectors. Mol Ther. 2008;16(8):1427–1436. doi: 10.1038/mt.2008.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mhawech-Fauceglia P, Zhang S, Terracciano L, Sauter G, Chadhuri A, Herrmann FR, Penetrante R. Prostate-specific membrane antigen (PSMA) protein expression in normal and neoplastic tissues and its sensitivity and specificity in prostate adenocarcinoma: An immunohistochemical study using mutiple tumour tissue microarray technique. Histopathology. 2007;50(4):472–483. doi: 10.1111/j.1365-2559.2007.02635.x. [DOI] [PubMed] [Google Scholar]
- 51.Su SL, Huang IP, Fair WR, Powell CT, Heston WD. Alternatively spliced variants of prostate-specific membrane antigen RNA: Ratio of expression as a potential measurement of progression. Cancer Res. 1995;55(7):1441–1443. [PubMed] [Google Scholar]
- 52.Bostwick DG, Pacelli A, Blute M, Roche P, Murphy GP. Prostate specific membrane antigen expression in prostatic intraepithelial neoplasia and adenocarcinoma: A study of 184 cases. Cancer. 1998;82(11):2256–2261. doi: 10.1002/(sici)1097-0142(19980601)82:11<2256::aid-cncr22>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 53.Laidler P, Dulinska J, Lekka M, Lekki J. Expression of prostate specific membrane antigen in androgen-independent prostate cancer cell line PC-3. Arch Biochem Biophys. 2005;435(1):1–14. doi: 10.1016/j.abb.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 54.Silver DA, Pellicer I, Fair WR, Heston WD, Cordon-Cardo C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin Cancer Res. 1997;3(1):81–85. [PubMed] [Google Scholar]
- 55.Liu H, Moy P, Kim S, Xia Y, Rajasekaran A, Navarro V, Knudsen B, Bander NH. Monoclonal antibodies to the extracellular domain of prostate-specific membrane antigen also react with tumor vascular endothelium. Cancer Res. 1997;57(17):3629–3634. [PubMed] [Google Scholar]