

Abstract
The solution structure of the Bacillus subtilis protein YndB has been solved using NMR in order to investigate proposed biological functions. The YndB structure exhibits the helix-grip fold, which consists of a β-sheet with two small and one long α-helix, forming a hydrophobic cavity that preferentially binds lipid-like molecules. Sequence and structure comparisons to proteins from eukaryotes, prokaryotes, and archaea suggest that YndB is very similar to the eukaryote protein Aha1, which binds to the middle domain of Hsp90 and induces ATPase activity. Based on these similarities, YndB has been classified as a member of the Activator of Hsp90 ATPase homolog-like protein (AHSA1) family with a function that appears to be related to stress response. An in silico screen of a compound library of ~18,500 lipids was used to identify classes of lipids that preferentially bind YndB. The in silico screen identified, in order of affinity, the chalcone/hydroxychalcone, flavanone, and flavone/flavonol classes of lipids, which was further verified by 2D 1H-15N HSQC NMR titration experiments with trans-chalcone, flavanone, flavone, and flavonol. All of these compounds are typically found in plants as precursors to various flavonoid antibiotics and signaling molecules. The sum of the data suggests an involvement of YndB with the stress response of B. subtilis to chalcone-like flavonoids released by plants due to a pathogen infection. The observed binding of chalcone-like molecules by YndB is likely related to the symbiotic relationship between B. subtilis and plants.
INTRODUCTION
The Bet v 1 protein from birch is a major allergen with high sequence similarity to the plant PR-10 pathogenesis-related proteins, which are involved in the response of plants towards microbial infection.1 Since the Bet v 1 protein structure was solved,2 numerous other proteins from among eukaryotes, archaea, and bacteria have been identified as having the same characteristic fold.3 The Bet v 1-like superfamily of proteins now contains approximately 10,135 sequences and consists of 13 unique families. The four largest families in the Bet v 1-like superfamily are the polyketide cyclases (3,475 sequences), the ring hydroxylases α-chain (2,022 sequences), the activator of Hsp90 ATPase homolog 1-like protein (AHSA1) family (1,762 sequences), and the StAR-related lipid transfer (START) family (1,026 sequences). The sequence similarity among the different Bet v 1-like families tends to be relatively low (0 to 38%), but all contain the same helix-grip fold that forms a hydrophobic cavity in between the long C-terminal α-helix and the antiparallel β-sheet.3 This hydrophobic cavity has been shown to preferentially bind to lipids, sterols, polyketide antibiotics, and other hydrophobic molecules.3
While the Bet v 1-like superfamily members share a similar fold, the biological functions vary across the different families. The ring hydroxylases degrade polycyclic aromatic hydrocarbons into non-aromatic cis-diols,4 the START family appears to be involved in steroidogenesis,5,6 while the polyketides cyclase family is involved with the biosynthesis of polyketide-based antibiotics and pigments.7 Members of the AHSA1 family are named after the human activator of Hsp90 ATPase protein (Aha1). While the proteins of this family have similar structures, the functions for most of the AHSA1 family members, except for its namesake, are ambiguous and are currently classified by UniProtKB8 as either a general stress protein or a conserved putative protein of unknown function. The eukaryotic protein Aha1 is proposed to interact with the middle domain of heat shock protein 90, which stimulates its ATPase activity.9,10 The domain organization of many homologous eukaryotic proteins in the AHSA1 family also suggests a function that is similar to Aha1. Conversely, homologous prokaryotic proteins have a much more diverse domain organization suggesting a wide range of possible functions.3
Of the 80 total structures solved for 59 members of the Bet v 1-like superfamily, 32 have ligands bound. The types of ligands that have been experimentally determined to bind Bet v 1-like proteins include membrane lipids, plant hormones, secondary metabolites, polycyclic aromatic hydrocarbons, and DNA/RNA.3 There are 12 total proteins in the AHSA1 family with known structures. The only protein in the AHSA1 family with a solved structure of its protein-ligand complex is the self-sacrificing resistance protein CalC from Micromonospora echinosporato,11 where CalC is shown bound to calicheamicin γ1,12 a potent antitumor antibiotic. Both Pfam13 and SCOP14 databases classify CalC as belonging to the AHSA1 family due to its 43 to 55% sequence similarity to other uncharacterized bacterial members of AHSA1. CalC, however, contains a break in the C-terminal helix that is uncharacteristic of most Bet v 1-like proteins and would likely indicate a new CalC-like family within the Bet v 1-like superfamily. This leaves only the human Aha1 with a proposed function within the AHSA1 family.
The Bacillus subtilis YndB protein is a protein of unknown biological function targeted for structural analysis by the Northeast Structural Genomics Consortium (NESG; http://www.nesg.org; NESG target: SR211). We previously reported the near complete NMR assignments for B. subtilis YndB,15 where the protein was originally identified as being a member of the START15,16 domain due to the similar helix-grip fold found in the structure of two homologous proteins and based on CATH comparisons.17 The NMR structures reported for Bacillus cereus protein BC4709 (PDB ID: 1xn6) and Bacillus halodurans protein BH1534 (PDB ID: 1xn5) led to their START domain classification.16 These two proteins are 64 and 57% homologous to YndB, respectively, inferring a similar annotation for YndB. However, the SCOP and Pfam databases have suggested that YndB, BC4709, and BH1534 belong to the AHSA1 family. Sequence similarity searches with YndB only identify proteins annotated as either AHSA1 or proteins of unknown function. The primary difference between the START domain and AHSA1 structures is that START domain proteins typically contain two additional N-terminal β-strands and an α-helix, which also makes the proteins larger. The structure of BC4709 and BH1534 do not have these additional structural components further supporting the AHSA1 classification.
Assigning a function to an uncharacterized protein like YndB can be a daunting task that involves obtaining a high-resolution structure18 combined with detailed studies that may include generating knockout libraries to analyze cell phenotypes, monitoring gene expression levels, or performing pull-down assays, all of which require in-depth bioinformatics analyses.19–23 Since the biological function of a protein is, by definition, derived from its interactions with other biomolecules or small molecules, identifying interacting partners is an alternative route to obtaining a functional annotation. One such technique, FAST-NMR,24,25 utilizes a small biologically-focused compound library combined with NMR high-throughput screening (HTS), rapid protein-ligand co-structures using AutoDock26 and chemical shift perturbations,27 and a comparison of protein active site structures (CPASS)28 to assist the functional annotation of proteins. However, the utility of FAST-NMR relies on structural homologs being found within the diverse functional chemical library. In the case of YndB, the known Bet v 1-like superfamily ligands combined with the expected hydrophobic cavity for YndB already suggests the protein is likely to bind lipid-like molecules. This eliminates the need for screening a diverse array of compounds found in the FAST-NMR compound library and instead requires an extensive screen against a focused lipid-like library. Due to the large number of biologically relevant lipid-like compounds29 and the corresponding limited commercial availability, an HTS assay is not practical or cost effective. Instead, an in silico screen30,31 provides an attractive alternative method to identify specific classes of compounds that may interact with YndB and to focus follow-up in vitro efforts.
To better understand the general biological role of AHSA1 proteins, the structure and putative biological function of the B. subtilis YndB protein was determined using NMR spectroscopy and an in silico ligand-binding screen. The three-dimensional solution structure of YndB (PDB ID: 2kte) is reported herein and is consistent with other AHSA1 proteins. Since most Bet v 1-like and AHSA1 proteins contain a hydrophobic ligand-binding pocket, an in silico screen of a ~18,500 lipid compound library29 was performed to identify a particular class of lipids that preferentially bind YndB and to provide insight into its biological function. The B. subtilis YndB protein was shown to experimentally bind trans-chalcone, a member of an important class of antibiotics and an important plant metabolite produced by chalcone synthase.32 Three other compounds similar in structure to chalcones (flavanone, flavone, and flavonol) and part of the same metabolic pathway were also shown to bind YndB, albeit weaker binders than trans-chalcone. These chalcone-like molecules are often found as precursors to flavonoids that play a key role in plant-microbe signaling and defense, where Bacillus strains have been shown to have a beneficial impact on plant health by protecting against fungal and bacterial pathogens.33 This suggests B. subtilis YndB may respond to a plant infection signal and induce a stress response.
MATERIALS AND METHODS
Solution Structure of B. subtilis YndB
Uniformly 13C, 15N-enriched YndB (152 amino acids with eight non-native residues LEHHHHHH at the C-terminus for purification) was purified following standard protocols used in the NESG consortium.34 The recombinant vector containing the gene for YndB was transformed into BL21 (DE3) pMGK cells. The soluble fraction of the lysed cells was collected and purified with a Ni-NTA affinity column (Qiagen) and gel filtration column (HiLoad 26/60 Superdex 75 pg, Amersham Biosciences) chromatography, described in full previously.15 The NMR sample was stored in 20 mM MES buffer, pH 6.5 (uncorrected) with 5% D2O, 0.02% NaN3, 10 mM DTT, 5 mM CaCl2, and 100 mM NaCl. The sample was stored in a sealed Shigemi tube (Shigemi Inc, Allison Park, PA) at 4°C when not actively collecting NMR data.
Initial structures of YndB were calculated using CYANA 2.1.35 The program assigned the NOEs utilizing a homology model in the first iteration, which was based on BC4709 and BH1534, the two protein structures with high sequence identity from Bacillus cereus (PDB ID: 1xn6) and Bacillus halodurans (PDB ID: 1xn5), respectively. CYANA assigned a total of 1,669 distance constraints. Additional data included 126 dihedral restraints from TALOS and 53 hydrogen bonds based on the secondary structure from TALOS.36 The 20 final structures of CYANA were manually inspected and two were removed due to an unusual conformation of the N-terminus. The final 18 structures were then further refined in explicit solvent using the RECOORD protocols in CNS.37,38 The final structures agree well with the NMR data, which is apparent from the low root-mean square deviations (rmsd) from the experimental distances and dihedral angles. To demonstrate that the final NOE assignments do not result in unreasonable atomic clashes, the normalized MOLPROBITY39 clash score of −2.08 is extremely low for NMR structures.40 Table 1 contains other important structural statistics related to the quality of the structure. According to MOLPROBITY 85% of the torsion angles of YndB are in the most favorable region of the Ramachandran Plot with 11.3% in the allowed regions. According to PSVS40 average rmsd for the ordered residues of 18 structures refined with explicit solvent was 0.8 Å for backbone atoms and 1.3 Å for all heavy atoms. The ensemble was deposited in the PDB (PDB ID: 2kte) (Figure 1A). Putative binding sites of YndB and homologous proteins BC4709 and BH1534 were investigated and compared using CASTp,41 which attempts to identify protein ligand binding sites and active sites by defining the molecular surface and determining surface accessible pockets. The three-dimensional structures of the proteins are represented here using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (http://www.cgl.ucsf.edu/chimera).42
Table 1.
Structural Statistics and Atomic rms Differences
| Structural Statistics for the 18 lowest energy conformations | |
|---|---|
| NOE distance restraintsa | |
| All | 1669 |
| interresidue sequential (|i-j| = 1) | 466 |
| interresidue short range (1 < |i-j| < 5) | 369 |
| interresidue long-range (|i-j| > 5) | 509 |
| intraresidue | 325 |
| H-bonds | 53 |
| Dihedral restraints | 126 |
| Ensemble RMSDb (Å) | |
| Secondary structure backbone | 0.8 |
| Secondary structure heavy | 1.3 |
| PSVS Z-scoresb | |
| Verify3D | −2.89 |
| ProsaII (−ve) | −1.12 |
| Procheck (phi-psi) | −2.83 |
| Procheck (all) | −3.90 |
| MolProbity Clash Score | −1.73 |
| RPF Scoresb | |
| Recall | 0.647 |
| Precision | 0.668 |
| F-measure | 0.657 |
| DP-score | 0.552 |
| Energy(kcal/mol)c | |
| total | −5504.8 ± 137.9 |
| bond | 79.8 ± 4.7 |
| angle | 272.2 ± 19.7 |
| dihedral | 714.0 ± 5.4 |
| impropers | 108.3 ± 8.4 |
| van der Waals | −604.0 ± 21.4 |
| Violationsc | |
| NOE > 0.5 Å | 0 |
| Dihedral > 5° | 2.6 |
| RMSD (experimental)c | |
| NOE (Å) | 0.0220 ± 0.001 |
| H-bonds (Å) | 0.023 ± 0.001 |
| Dihedral angles (°) | 1.18 ± 0.14 |
| RMSD (covalent geometry)c | |
| Bonds (Å) | 0.012± 0.0004 |
| Angles (°) | 1.3 ± 0.04 |
| Impropers (°) | 1.6 ± 0.08 |
| Ramachandran spaced | |
| Most Favored | 85.0% |
| Allowed | 11.3% |
| Disallowed | 3.7% |
Calculated with CYANA.
Calculated with PSVS.
Calculated with CNS.
Calculated with MOLPROBITY
Figure 1.

The NMR solution structure of Bacillus subtilis protein YndB (A) a backbone trace of the 18 lowest energy conformation models and (B) a ribbon diagram where the α-helices are colored red, the β-strands are colored yellow, and the loops are colored green.
Sequence and Structure Similarity to YndB
In order to identify homologous proteins and elucidate a possible function, multiple similarity comparisons were performed. The pair-wise sequence alignment of YndB to protein sequences in a non-redundant database was performed using BLASTP43–45 and the default BLOSUM62 scoring matrix. DaliLite v.346 was used to perform the structural similarity comparisons of YndB (model #10) with proteins from the RCSB Protein Data Bank. ClustalW47 was used to align the sequences of YndB and the two homologous proteins, BC4709 and BH1534, for a detailed analysis of conserved amino acid residues that make up functionally relevant components of each protein. The ClustalW sequence alignments used the default settings.
Virtual Screening of a Lipid Compound Library
An in silico screen of YndB against a lipid library was performed to identify classes of lipid molecules that are favored to bind the protein. The lipid library used in this study was obtained from the Nature Lipidomics Gateway (www.lipidmaps.org), which contains two-dimensional structures of 21,824 lipid molecules (as of January 2010) found in mammalian species.29,48,49 Clearly, the lipid library is not exhaustive and many lipid molecules found in non-mammalian organisms are not represented, but the goal of the virtual screening effort is to identify a structural homolog to the natural ligand or to identify a particular class of lipid that preferentially binds YndB. Eight major categories of lipids are represented in the Nature Lipidomics Gateway library: fatty acyls (3,476 structures), glycerolipids (3,012 structures), glycerophospholipids (1,958 structures), sphingolipids (3,376 structures), sterol lipids (2,125 structures), prenol lipids (1,156 structures), saccharolipids (13 structures), and polyketides (6,708 structures). The eight major categories are further divided into a total of 538 distinct subclasses of lipid compounds. The two-dimensional structure files provided by the Nature Lipidomics Gateway were converted into three-dimensional conformers using the program OMEGA 2.3.250 (OpenEye Scientific Software, Santa Fe, NM). OMEGA generates a database of multiple three-dimensional conformers for each ligand in the compound library using fragment assembly, ring conformation enumeration, and torsion driving. In this study, OMEGA was used to generate a maximum of 600 unique (>0.5 Å rmsd) conformers for each lipid molecule for a total searchable database consisting of ~10,000,000 conformers. OMEGA failed to generate conformers for 3306 of the lipid structure files. Most of these failures occurred during the processing of the sphingolipid category of lipids (3196 out of 3376 failed) due largely to the large size and number of branches/rotatable bonds of the molecules in this category.
The docking program FRED 2.2.551 (OpenEye Scientific Software, Santa Fe, NM) was used for the virtual screen of YndB against the lipid library. FRED is a rigid docking program which uses the multiple conformers of each ligand created in OMEGA and generates 100 docked poses within the defined binding site by rotating and translating the rigid molecule in order to optimize shape complementarity. The poses of each ligand conformer are then ranked using the built-in consensus scoring method, where only the top scoring pose is kept. Since the conformers are rigid during this docking process, FRED has been shown to be very fast as compared to other docking programs that allow for ligand flexibility.52 This speed is necessary in order to screen the large lipid-like library in a reasonable amount of time. While some accuracy may be lost due to rigid docking and a lack of a biologically relevant conformation for the ligand, FRED was primarily used to rapidly filter out compounds that could not fit into the YndB ligand-binding pocket. Prior to initiation of the docking, model 10 from the YndB PDB file (PDB ID: 2kte) was prepared using FRED Receptor 2.2.5 (OpenEye Scientific Software, Santa Fe, NM), where a high quality shape potential grid of 3403 Å3 was generated that encompassed the proposed binding cavity. Model 10 was selected as the target receptor for the virtual screen as it had the lowest violation energies during the structure calculations. The lipid library compounds were ranked using the default Chemgauss3 scoring function that includes descriptors of shape and molecular chemical properties. Chemgauss3 incorporates steric and hydrogen bond interactions, and protein and ligand desolvation parameters that are smooth using a Gaussian function. The relative enrichment for each lipid class within the top 1000, the top 500, the top 200, the top 100, and the top 50 ranked compounds were calculated according to the following equation:
| (1) |
where %RE is percent relative enrichment, %AbLib is the percent abundance of a lipid class in the Nature Lipidomics Gateway library, and %AbFRED is the percent abundance of a lipid class observed in either the top 1000, 500, 200, 100, or 50 ranked compounds by FRED.
NMR Titration Experiment
Based on the results of the virtual screen, three classes of lipid molecules were identified as possible binders: flavones/flavonols, flavanones, and chalcones/hydroxychalcones. Experimental validation of these possible binders was performed using chemical shift perturbations (CSPs) in 2D 1H-15N HSQC NMR spectra collected on a Bruker 500 MHz Avance spectrometer equipped with a triple-resonance, Z-axis gradient cryoprobe. The 2D 1H-15N HSQC NMR experiment was collected at 298K with 32 scans, 1024 data points and a sweep width of 15 ppm centered on the water peak at 4.693 ppm in the direct 1H-dimension and 128 data points with a sweep width of 36 ppm in the indirect 15N-dimension. The ligands selected to represent each of the three potential binding lipid classes were trans-chalcone, flavanone, flavone, and flavonol (Sigma-Aldrich, St. Louis, MO). Flavone and flavonol belong to the same class of lipids, but there was interest in how the binding would be affected with the addition of a polar functional group. The fatty acyl oleic acid (Sigma-Aldrich, St. Louis, MO) was also selected as a negative control. These compounds were selected based on availability, a simple scaffold that clearly represented the lipid class, and cost. Each compound was dissolved in “100%” deuterated DMSO-d6 (Sigma-Aldrich, St. Louis, MO) prior to titration. The titration analysis was performed with an 80 μM 15N-labeled YndB sample (20 mM MES buffer, pH 6.5 with 10% D2O, 0.02% NaN3, 10 mM DTT, 5 mM CaCl2, 100 mM NaCl, and 50 μM DSS) and increasing concentrations (ranging from 0 to 600 μM) of each ligand. The NMR data were processed with NMRpipe53 and the spectra viewed using NMRViewJ.54 Kaleidagraph 3.5 (Synergy Software) was used to fit the NMR data to eqn. (2),55,56
| (2) |
where CSPobs is the 2D 1H-15N HSQC chemical shift perturbations, [P] is the protein concentrations, [L] is the ligand concentration and KD is the dissociation constant.
B. subtilis YndB-ligand Co-structures
Co-structures of YndB bound to each compound used in the NMR titration experiment (trans-chalcone, flavanone, flavone, and flavonol) were generated to analyze the ligand-binding pocket. While a definitive identification of the binding pose of these ligands to YndB would require extensive NMR experiments and data analysis similar to the original effort to solve the apo-YndB structure, molecular docking can provide a rapid and reliable NMR-based model to examine the details of the binding interactions.
AutoDock 4.0126,57 with the AutoDockTools 1.5.2 (http://mgltools.scripps.edu) graphical interface was used to simulate 100 different binding poses for each YndB-ligand complex. AutoDock was used instead of FRED due to the accuracy gained from flexible ligand docking and because it is one of the most highly cited docking programs available.58 The grid map was generated with 0.375 Å spacing with xyz grid point dimensions of 50 × 58 × 48, which is of sufficient size to encompass the proposed binding pocket previously identified in CASTp. The docking calculations were performed using the Lamarckian genetic algorithm default settings with a population size of 300 and 5,000,000 energy evaluations.
RESULTS
Solution Structure of B. subtilis YndB
The observed secondary structure and fold for B. subtilis YndB are characteristic of the helix-grip fold found in the Bet v 1-like superfamily. The helix-grip fold consists of a β sheet with two small and one long α-helix. The β sheet is comprised of 5 strands instead of the normal 6. The missing short strand, which is normally β2, forms sheet like interactions in only 2 of the 18 structures in the ensemble and appears to protect the edge of β3; unprotected edges can be adventitious interaction sites for aggregation.59 However, the strands are annotated 1, 3, 4, 5, and 6 to facilitate comparisons with other family members: residues 12–18 (β1), 63–69 (β3), 73–78 (β4), 83–91 (β5), and 96–104 (β6). The three α-helices are comprised of residues 22–28 (α1), 33–36 (α2), and 120–143 (α3) (Figure 1B). There is significant variability in the loop regions of the protein corresponding to residues 37–63 and 105–120. These loops appear to be important for the structure of the hydrophobic ligand-binding cavity (Figure 1B).
Like other proteins with a helix-grip fold, YndB has an exposed hydrophobic core, likely used in the binding of lipid-like molecules. Analysis with CASTp shows that the volume of this putative binding cavity is 790 Å3. The core of YndB consists primarily of aromatic side chains. One element of the YndB binding pocket is the long α3-helix. This helix is anchored to the β sheet by residues W130, V134, and L138, which show NOE interactions to the sheet residues S15, T17, and L18. Helix α3 has previously been identified as being crucial to the function of the structurally related START domains.60,61 In both of these earlier studies, the removal of part of the α3-helix eliminated ligand binding. Based on the NMR solution structure for YndB, removing the C-terminal residues would result in α3 no longer being associated with the β-sheet and therefore, the protein would probably not be folded properly. Hence, we surmise that the previous results were likely due to protein instability and not the activity of specific residues to ligand binding.
The Bet v 1-like superfamily classification for YndB and the reliability of the NMR structure is further supported by the structural similarities to two homologous proteins, BC4709 and BH1534 (Figure 2A, B). The YndB protein exhibits a backbone rmsd of 1.1 Å and 1.2 Å to BC4709 and BH1534, respectively, when only secondary structural elements are included in the alignment. The main difference among the structures lies in the loop regions, where there appears to be a significant difference in the loop conformation of residues 37–63 and 105–120 for YndB. This difference affects the size of the hydrophobic cavity for YndB, where BC4709 and BH1534 have much smaller volumes (199 Å3 and 106 Å3, respectively) relative to YndB.
Figure 2.

(A) An overlay of the NMR solution structure of Bacillus subtilis protein YndB (yellow), with Bacillus cereus protein BC4709 (blue) (PDB ID: 1xn6) and Bacillus halodurans protein BH1534 (red) (PDB ID: 1xn5). The loops have the most divergence, where loop conformational changes have been linked to the mechanism of ligand binding. (B) The multiple sequence alignment from ClustalW of YndB, BC4709 and BH1534 with the 14 active site residues (< 5 Å from bound ligand) indicated in black rectangles.
As expected from the high sequence identity, the sequence compositions of the ligand binding sites are also similar (Figure 2C). Nine of the fourteen residues that line the binding cavity are identical and predominately hydrophobic (V29, G34, F76, W78, W83, V85, F87, W130, and L138) and two others show high similarity. While none of these residues exist within the loop regions, the large loop from residues 37–63 is very well conserved with 16 residues being identical among the three proteins, once again indicating the importance of these loops for ligand binding. The loop regions corresponding to residues 37–63 and 105–120 are predicted to be conformationally flexible based on the lack of NMR assignments.62 Of the 43 amino-acids that comprise the two loop regions, a total of 21 residues are unassigned. Correspondingly, these loop regions have a limited number of structural constraints resulting in the observed conformational variability in the ensemble of calculated structures (Figure 1A). While the lack of NMR assignments and NOEs suggests conformational flexibility, these observations are not sufficient to define the loops as dynamic and requires further experimental evidence for verification.63 Nevertheless, it is anticipated that a bound substrate would restrict the loop conformation near the YndB ligand binding site.
Sequence and Structure Similarity to YndB
The BLASTP search of YndB against a non-redundant protein sequence database identified 58 proteins with an E-value of 1.0 × 10−21 or lower that included BC4709 and BH1534. All of these proteins belong to the gram-positive organisms of the order Bacillales and have sequence identities > 39% and sequence similarities > 58%. There is a clear division in sequence similarity between the 58 Bacillales proteins and other gram-positive bacteria proteins homologous to YndB. The sequence similarity score drops significantly from E-values of 10−21 for Bacillales proteins to E-values of ≥10−9 for other gram-positive proteins. This sequence distinction may indicate a function specific to the Bacillales organisms.
The structural similarity search using DaliLite identified 590 proteins with a Z-score over 2.0. Once again, the two proteins with the greatest structural similarity are BC4709 and BH1534 with Z-scores of 14.0 and 14.2, respectively. The top 100 proteins with the highest structural similarities have Z-scores ranging from 9.0 to 14.2 with sequence identities < 25%, except for BC4709 and BH1534. All of the proteins identified in this range are either uncharacterized or members of the Bet v 1-like superfamily, which includes Bet v1-like proteins in plants.
Virtual Screening of a Lipid Compound Library
The in silico screen of YndB with the entire Nature Lipidomics Gateway lipid library took approximately 44 hours with the computation dispersed across 16 nodes of a Linux Beowulf cluster. Of the 18,518 structures in the library, FRED successfully docked 17,475 compounds to YndB. The relative enrichment (eqn. 2) of each lipid class from the FRED docking is plotted in Figure 3A. Only one lipid category, the polyketides, had a positive relative enrichment among the top 1000 docked lipid molecules. Polyketides represent 86.8% of the molecules in the top 1000, while they only make up 37.9% of the entire compound library for a relative enrichment of 129%. The polyketide representation increases significantly as the cutoff for the FRED scoring energy for molecules accepted in the top rankings is decreased. If only the top 50 docked compounds are considered, 98.0% of these compounds are polyketides, with only one hit being a member of the fatty acyl category. All of the polyketides identified belong to the flavonoid class of lipids. Within the flavonoids, three subclasses emerge as favorable hits from the virtual screen: chalcones/hydroxychalcones, flavanones, and flavones/flavonols.
Figure 3.

Summary of the FRED in silico screening results of the Nature Lipidomics Gateway lipid library and Bacillus subtilis protein YndB. ~18,500 lipid structures corresponding to ~10,000,000 conformers were docked into YndB. The compounds were ranked using the FRED Chemgauss3 scoring function. (A) A plot of the relative enrichment (eqn. 1) for each of the 8 major lipid categories within the top 1000 hits (red), the top 500 hits (green), the top 200 hits (purple), the top 100 hits (cyan), and the top 50 hits (orange). Only the polyketides were positively enriched in the virtual screen relative to their representation in the original lipid library. (B) The chemical structures of the four flavonoid compounds chosen to represent the three most enriched subclasses of lipids (chalcones/hydroxychalcones, flavanones, and flavones/flavonols) identified from the in silico screen.
The chalcone/hydroxychalcone subclass turns out to be the most significant hit since 44.9% of the flavonoids in the top 50 hits were chalcones while they only make up 9.4% of the library of flavonoids. The remaining flavonoids in the top 50 hits belong to the flavanone (28.6%) and flavone/flavonol (14.3%) subclasses. The molecules from these three subclasses all have very similar chemical structures, which consist of at least two benzene rings while containing only a few rotatable bonds (Figure 3B).
NMR Titration Experiment
While virtual screening appears to be a useful tool for identifying particular classes of lipids that have structural and chemical properties amenable to binding to YndB, these results require validation by experimental methods. NMR is routinely used to evaluate protein-ligand interactions, to measure dissociation constants (KD) and to identify ligand-binding sites through the observation of chemical shift perturbations (CSPs).27,64–66 CSPs were calculated by comparing the average 1H and 15N resonance changes between ligand-free and ligand-bound YndB 2D 1H-15N HSQC NMR spectra. The advantage of this approach is the speed and minimal amount of protein and ligand required. Unfortunately, access to the specific lipid compounds predicted to bind YndB is very limited due to low commercial availability and/or high cost. Based upon the in silico screening of YndB with a lipid library, chalcones/hydroxychalcones, flavanones, and flavones/flavonols were identified to be the most likely to bind YndB. Therefore, representative molecules were sought for each class containing the basic structural scaffold that would likely have characteristic binding properties. A member of the fatty acyl category of lipids was also sought for use as a negative control.
For the chalcone/hydroxychalcone subclass of lipids, trans-chalcone was selected to represents the basic structural scaffold for this class. The titration of YndB with trans-chalcone resulted in significant chemical shift perturbations (Figure 4A). Nine YndB residues with the most significant CSPs (greater than 2 standard deviation from the mean) were identified: Thr80, Trp105, Val111, Ile112, Val122, Met126, Trp130, Thr131, and Ile133. These residues line the opening of the proposed binding pocket (Figure 5A) identified by CASTp. Six more residues with significant perturbations (greater than 1 standard deviation from the mean) were also identified: Glu110, Val121, Arg123, Asp127, Gly128, and Asn135. These amino-acids reside in the long α3-helix and contribute to a portion of the ligand binding pocket. Their perturbation may indicate a structural change in α3-helix upon binding. The remaining loop residues that define the binding pocket are unassigned in apo-YndB.
Figure 4.

(A) Overlay of the 2D 1H-15N HSQC spectra of Bacillus subtilis protein YndB titrated with chalcone, where the chalcone concentration was increased from 0 μM (blue) to 160 μM (cyan). The significant chemical shift perturbations of the 9 assigned amino acid residues used to determine the dissociation constant (KD) are highlighted with a black oval and labeled accordingly. Not all of the perturbed peaks were assigned; these residues are likely from the loop regions. (B) NMR titration data for trans-chalcone (blue), flavanone (green), flavone (purple), and flavonol (orange). The normalized chemical shift perturbations for the nine most perturbed amino acid residues are plotted versus the protein-ligand concentration ratios. The titration curves were fit to a binding isotherm (eqn. 2) using Kaleidagraph 3.5 (Synergy Software). The best-fit curves are shown as a solid line. The theoretical curve displayed for trans-chalcone corresponds to a KD of 1 μM and represents the upper-limit for the KD. The measured KD values are 1 μM (trans-chalcone), 32 ± 3 μM (flavanone), 62 ± 9 μM (flavone), and 86 ± 16 μM (flavonol).
Figure 5.

(A) A representation of the Bacillus subtilis YndB protein surface using the NMR solution structure, where amino acid residues that exhibited NMR chemical shift perturbations (CSPs) caused by the titration of trans-chalcone, flavanone, flavone, and flavonol are colored red (≥ 2 standard deviations from mean) and blue (≥ 1 standard deviation from the mean). The residues with the largest CSPs can be found near the entrance to the ligand binding cavity, while the remaining residues are associated with helix α3. Shown within the ligand binding cavity are the docked conformations of the four ligands experimentally determined to bind YndB: chalcone (yellow), flavanone (green), flavone (purple), and flavonol (red). (B) The NMR solution structure of YndB docked with trans-chalcone (green). The sidechains for the 14 amino acid residues within 5 Å of the ligand are shown and labeled. Five aromatic sidechains surround the trans-chalcone molecule and form a hydrophobic pocket.
The normalized CSPs for each of the nine amino acid residues were plotted as a function of protein-ligand concentration ratios and fit to a binding isotherm (eqn. 2) to determine a dissociation constant. trans-Chalcone binds tightly to YndB with a KD 1 μM and a stoichiometry of 1:1. The binding stoichiometry is based on the observation that a two-site model does not fit the data as evident by the fact the chemical shift perturbations reaches a maximum at ~1:1 protein-trans-chalcone concentration ratio (Figure 4B). Calculating an exact KD for trans-chalcone was not possible given the YndB concentration (80 μM) used for the 2D 1H-15N HSQC titration experiments, significantly lowering the YndB concentration was not feasible. Superimposed on the trans-chalcone NMR titration data (Figure 4B) is a theoretical curve for a KD of 1 μM, implying an upper-limit for the trans-chalcone dissociation constant.
Representing the flavanone and flavone/flavonol subclasses, flavanone, flavone, and flavonol all showed chemical shift perturbations of the same residues found to be perturbed in the trans-chalcone titration, indicating that all the molecules bind in a similar manner. However, flavanone, flavone, and flavonol bound YndB significantly weaker than trans-chalcone with dissociation constants of 32 ± 3 μM, 62 ± 9 μM, and 86 ± 16 μM, respectively (Figure 4B). The range of the dissociation constants mirror the representation of each subclass in the virtual screen, where chalcones were the most abundantly ranked compounds, followed by flavanones and then the flavones/flavonols. Titration of oleic acid to YndB, which was used to represent the fatty acyl category of the lipids, showed no significant chemical shift perturbations and therefore no evidence of binding (data not shown).
B. subtilis YndB-ligand Co-structures
Using AutoDock, the three-dimensional structures of trans-chalcone, flavanone, flavone, and flavonol were each docked into the YndB binding pocket identified by CASTp and supported by NMR CSPs. The docking of each compound did not result in much variation between the poses for each of the ligands. Out of 100 docked poses for each ligand, at least 80 were within a 2.0 Å rmsd of each other. A comparison of the most energetically favorable poses for each ligand shows that the compounds essentially bind with the same orientation (Figure 5A). The docked structures are also consistent with the 1:1 binding stoichiometry predicted by the NMR titration experiments and CSPs. Binding two or more compounds in the YndB binding pocket is sterically prohibitive and the NMR CSPs do not identify a secondary ligand binding site.
The AutoDock predicted free energy of binding was essentially identical for each compound, averaging −7.2 kcal/mol and correlates with a dissociation constant of ~5 μM. With the exception of trans-chalcone, this is a stronger binding affinity than observed for the three compounds in the NMR titration experiments. But, predicting the actual free energy of binding using AutoDock has an estimated error of 2.2 kcal/mol.67 In the YndB-trans-chalcone modeled structure, there are 14 residues that reside within 5 Å of the docked trans-chalcone, where five of these residues are aromatic (Figure 5B). These aromatic residues presumably have a strong influence on ligand binding and selectivity, consistent with the hydrophobic and aromatic nature of trans-chalcone and the other flavonoids. Conversely, the binding of trans-chalcone to YndB does not appear to involve any hydrogen bonding interactions.
Most of the difficulty in generating an accurate protein-ligand co-structure for YndB stems from the suspected flexibility of the two loop regions that define the hydrophobic cavity. Any variation in the orientation of the loop sidechains directly results in changes in the binding site conformation that may be required to accommodate a ligand. This effect can be seen in some of the YndB structures found in the NMR ensemble. The YndB-chalcone model and the relative trans-chalcone orientation does correlate well with the binding site for the human phosphatidylcholine transfer protein (PC-TP) complexed with dilinoleoylphosphatidylcholine (PDB ID: 1ln1),68 a protein structure in the related START domain family (Figure 6). While the sequence identity between YndB and PC-TP is low (5%), both proteins have structural similarities (3.6 Å rmsd) with binding pockets located in the same region of the protein. However, the binding pocket of YndB is significantly smaller than the pocket found in PC-TP due to the tighter packing of the β-sheet with the loop regions and the long α3-helix. Nevertheless, the overlay of the YndB-chalcone model with the PC-TP complex indicates that chalcone and the other flavonoids bind within the large dilinoleoylphosphatidylcholine-binding pocket.
Figure 6.
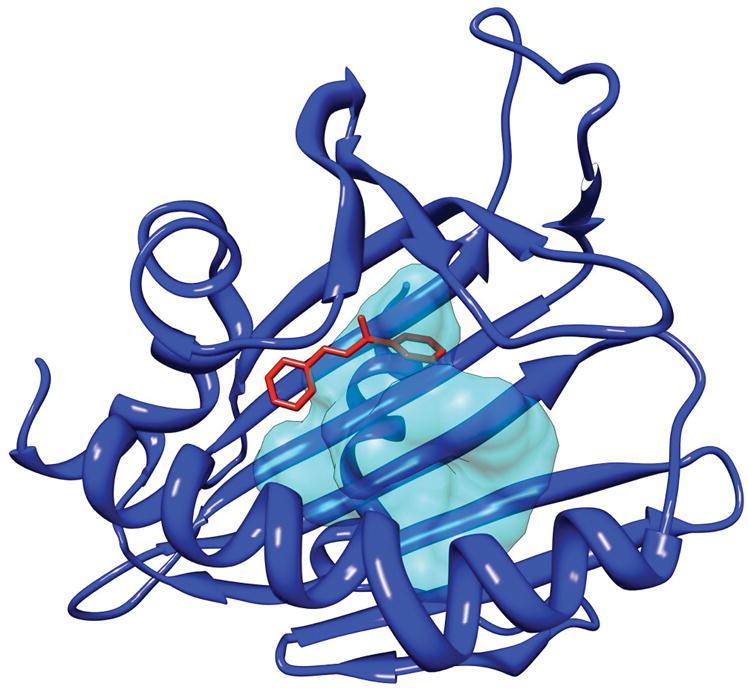
A structural alignment of the human phosphatidylcholine transfer protein (PC-TP) complexed with dilinoleoylphosphatidylcholine (PDB ID: 1ln1) with the Bacillus subtilis YndB-trans-chalcone NMR-based model. Only trans-chalcone is shown from the YndB-trans-chalcone structure. The structural alignment indicates the location of the docked trans-chalcone (red) relative to PC-TP (blue) and its ligand binding pocket represented by a transparent surface (cyan). The binding pockets of the two proteins are in the same region, indicating a reasonable docking of trans-chalcone to YndB.
DISCUSSION
The NMR structure for B. subtilis protein YndB indicates that the protein adopts a helix-grip fold and is clearly a member of the Bet v 1-like superfamily. The YndB structure contains an apparent hydrophobic cavity between the long C-terminal α-helix and the antiparallel β-sheet. Like other members of the Bet v 1-like superfamily the cavity suggests YndB binds to lipids, sterols, polyketide antibiotics, or other hydrophobic molecules as part of its biological function. The YndB protein was originally assigned as a START domain protein based on the high sequence similarity to B. cereus BC4709 and B. halodurans BH1534, which were assigned to START domains based on common structural features.15,16 Instead, SCOP and Pfam databases have suggested that YndB belongs to the closely related AHSA1 subfamily. Also, YndB, BC4709, and BH1534 do not have the additional N-terminal β-strands and the additional α-helix that are characteristic of a START domain structure.3 Similarly, a BLASTP sequence alignment search indicates YndB is more appropriately assigned as a member of AHSA1. The BLASTP search identified 58 proteins from organisms belonging to the gram-positive Bacillales order that are homologous to YndB with sequence identities > 39%. The functions for prokaryotic AHSA1 family members are typically classified as either a general stress protein or a conserved putative protein of unknown function. Similarly, the Dali search identified a large number of structural homologs to Bet v1-like proteins, AHSA1 family members and an abundance of hypothetical proteins or proteins of unknown function.
To further explore the potential functional annotation of YndB, an in silico screen against a ~18,500 lipid-like chemical library was conducted. The best binders identified from the in silico screen were from the three general lipid classes of flavones/flavonols, flavanones, and chalcones/hydroxychalcones. Representative compounds from all three classes were screened by NMR, where trans-chalcone, flavanone, flavone, and flavonol were all shown to bind in the YndB hydrophobic cavity with KD values of 1 μM, 32 μM, 62 μM, and 86 μM, respectively. The fact that all four molecules chosen from the in silico were shown to bind YndB is rather remarkable and indicative of the inherent value of our approach. The typical hit rate for a high-throughput screen is generally low (0.1–0.5%),69 where in silico screens may result in improved hit rates of up to 35–90%.70–72 A model for the YndB-chalcone complex was shown to be consistent with the binding of dilinoleoylphosphatidylcholine to human phosphatidylcholine transfer protein, a related START domain protein.
The binding of chalcone and flavanone to a B. subtilis protein is an intriguing observation since these molecules are primarily found in plants as precursors to flavonoid molecules used for antimicrobial defense, flower pigmentation, absorption of harmful UV radiation, and signaling between plants and beneficial microbes.73–77 A number of structural homologs to YndB identified by Dali were Bet v1-like proteins in plants. In plants, chalcones are often synthesized from a cinnamoyl-CoA molecule followed by malonyl-CoA additions. The conversion of the resulting molecule into chalcone is catalyzed by the protein chalcone synthase (CHS).32 Thus, chalcone is a key substrate for antibiotics or other flavonoid-based compounds. After the synthesis of chalcone, the chalcone isomerase (CHI) enzyme converts chalcone to flavanone, which is another compound identified to bind YndB. The other two binding compounds, flavone, and flavonol, are products of the various flavonoid synthesis pathways that initiate with the chalcone scaffold.32 Bacteria do not possess CHS or CHI proteins and thus don’t produce chalcone or flavanone. Some bacteria, including B. subtilis, do have proteins that appear to be homologous to the CHS proteins found in plants. These homologous proteins, known as type III polyketide synthases, appear to be pervasive in bacteria, indicating a possible mechanism for antimicrobial biosynthesis from chalcones, thus supporting the similarities of YndB to polyketide cyclases.7,78 Similarly, homologs of CHI have also been identified in bacteria.79 However, no type III polyketide synthase has been identified that is known to synthesize chalcones, and flavonoids have not been identified among the natural products of Bacillus. It seems unlikely that B. subtilis is producing chalcone-based antibiotics. These observations support the possibility of an exchange of genes between plants and bacteria, where these proteins are evolved for a unique bacterial function.80
The chalcone-binding property of YndB may be related to stress response as originally indicated based on the relationship to the eukaryotic Aha1 protein. Additionally, Bet v1-like proteins in plants, which were shown to be structural homologs of YndB, are also primarily related to a stress response caused by a pathogen infection. An evaluation of the genes found near YndB in B. subtilis supports the stress response explanation. While there are numerous uncharacterized membrane proteins identified in this cluster, one-gene codes for the membrane bound protein amino acid permease, which is involved in spore germination. The gene for protein BH1534 from B. halodurans also contains sporulation factors upstream and nucleotide metabolism downstream along with numerous putative membrane proteins. Similarly, the BC4709 gene from B. cereus has numerous membrane proteins in its cluster but also includes an ArsR transcriptional regulator and multidrug resistance proteins. The genes for all three of these proteins exist within regions containing stress response factors. Many other homologous proteins contain similar gene arrangements. These consistent gene arrangements hint at a likely stress-response mechanism, which is also supported by the similarities of these proteins to the eukaryotic Aha1 protein. Aha1 interacts with Hsp90 whereas prokaryotes have a homologous version of Hsp90 called HtpG, which has been shown to be induced under high heat stress conditions. It should be noted, however, there is currently no direct correlation between HtpG and YndB or spore formation.81
B. subtilis is a plant growth promoting rhizobacterium (PGPR), which is often found on the surface of plant roots and provides protection against pathogens through biofilm formation.82,83 Since chalcones are a key precursor to many antibiotics used by plants, it seems reasonable that B. subtilis has developed a mechanism of response towards chalcone-based compounds. Therefore, we hypothesize that the YndB protein, along with the homologous proteins BC4709 and BH1534, initiates a stress response-pathway when exposed to chalcone or chalcone-like compounds during the plant’s response to pathogen infection. Potentially, this stress response may either induce processes to help control plant pathogens84 and/or lead toward spore formation to protect B. subtilis from the impending release of antibiotics from the plant.85 Since flavonoids are routinely used as signaling molecules between plants and microbes during pathogen infections,86 it is reasonable to consider chalcone binding is part of the symbiotic relationship between B. subtilis and plants.
Acknowledgments
We thank Prof. Diana Murray for stimulating discussions regarding the NESG START domain project and Dr. John Cort at PNNL for providing the 750 MHz NMR data. This work was supported by a grant from the Protein Structure Initiative of the National Institutes of Health (U54 GM074958 to G.T.M.), supported, in part, by Award Number R21AI081154 from the National Institute of Allergy and Infectious Diseases Nebraska (R21AI081154), Tobacco Settlement Biomedical Research Development Fund and by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases. Research was performed in facilities renovated with support from the NIH under Grant RR015468-01. NMR spectra were acquired at the U.S. Department of Energy Office of Biological and Environmental Research) located at Pacific Northwest National Laboratory and operated for DOE by Battelle (contract KP130103).
References
- 1.van Loon LC, Rep M, Pieterse CM. Significance of inducible defense-related proteins in infected plants. Annu Rev Phytopathol. 2006;44:135–162. doi: 10.1146/annurev.phyto.44.070505.143425. [DOI] [PubMed] [Google Scholar]
- 2.Gajhede M, Osmark P, Poulsen FM, Ipsen H, Larsen JN, Joost van Neerven RJ, Schou C, Lowenstein H, Spangfort MD. X-ray and NMR structure of Bet v 1, the origin of birch pollen allergy. Nat Struct Biol. 1996;3(12):1040–1045. doi: 10.1038/nsb1296-1040. [DOI] [PubMed] [Google Scholar]
- 3.Radauer C, Lackner P, Breiteneder H. The Bet v 1 fold: an ancient, versatile scaffold for binding of large, hydrophobic ligands. BMC Evol Biol. 2008;8:286. doi: 10.1186/1471-2148-8-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Habe H, Omori T. Genetics of polycyclic aromatic hydrocarbon metabolism in diverse aerobic bacteria. Biosci Biotechnol Biochem. 2003;67(2):225–243. doi: 10.1271/bbb.67.225. [DOI] [PubMed] [Google Scholar]
- 5.Ponting CP, Aravind L. START: a lipid-binding domain in StAR, HD-ZIP and signalling proteins. Trends Biochem Sci. 1999;24(4):130–132. doi: 10.1016/s0968-0004(99)01362-6. [DOI] [PubMed] [Google Scholar]
- 6.Stocco DM. StAR protein and the regulation of steroid hormone biosynthesis. Annu Rev Physiol. 2001;63:193–213. doi: 10.1146/annurev.physiol.63.1.193. [DOI] [PubMed] [Google Scholar]
- 7.Ames BD, Korman TP, Zhang W, Smith P, Vu T, Tang Y, Tsai SC. Crystal structure and functional analysis of tetracenomycin ARO/CYC: implications for cyclization specificity of aromatic polyketides. Proc Natl Acad Sci U S A. 2008;105(14):5349–5354. doi: 10.1073/pnas.0709223105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Apweiler R, Bairoch A, Wu CH. Protein sequence databases. Curr Opin Chem Biol. 2004;8(1):76–80. doi: 10.1016/j.cbpa.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 9.Lotz GP, Lin H, Harst A, Obermann WM. Aha1 binds to the middle domain of Hsp90, contributes to client protein activation, and stimulates the ATPase activity of the molecular chaperone. J Biol Chem. 2003;278(19):17228–17235. doi: 10.1074/jbc.M212761200. [DOI] [PubMed] [Google Scholar]
- 10.Panaretou B, Siligardi G, Meyer P, Maloney A, Sullivan JK, Singh S, Millson SH, Clarke PA, Naaby-Hansen S, Stein R, Cramer R, Mollapour M, Workman P, Piper PW, Pearl LH, Prodromou C. Activation of the ATPase activity of hsp90 by the stress-regulated cochaperone aha1. Mol Cell. 2002;10(6):1307–1318. doi: 10.1016/s1097-2765(02)00785-2. [DOI] [PubMed] [Google Scholar]
- 11.Singh S, Hager MH, Zhang C, Griffith BR, Lee MS, Hallenga K, Markley JL, Thorson JS. Structural insight into the self-sacrifice mechanism of enediyne resistance. ACS Chem Biol. 2006;1(7):451–460. doi: 10.1021/cb6002898. [DOI] [PubMed] [Google Scholar]
- 12.Halcomb RL. Organic synthesis and cell biology: partners in controlling gene expression. Proc Natl Acad Sci U S A. 1994;91(20):9197–9199. doi: 10.1073/pnas.91.20.9197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer EL, Bateman A. The Pfam protein families database. Nucleic Acids Res. 2008;36(Database issue):D281–288. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andreeva A, Howorth D, Chandonia JM, Brenner SE, Hubbard TJ, Chothia C, Murzin AG. Data growth and its impact on the SCOP database: new developments. Nucleic Acids Res. 2008;36(Database issue):D419–425. doi: 10.1093/nar/gkm993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mercier KA, Mueller GA, Acton TB, Xiao R, Montelione GT, Powers R. (1)H, (13)C, and (15)N NMR assignments for the Bacillus subtilis yndB START domain. Biomol NMR Assign. 2009;3(2):191–194. doi: 10.1007/s12104-009-9172-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu G, Shen Y, Atreya HS, Parish D, Shao Y, Sukumaran DK, Xiao R, Yee A, Lemak A, Bhattacharya A, Acton TA, Arrowsmith CH, Montelione GT, Szyperski T. NMR data collection and analysis protocol for high-throughput protein structure determination. Proc Natl Acad Sci U S A. 2005;102(30):10487–10492. doi: 10.1073/pnas.0504338102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greene LH, Lewis TE, Addou S, Cuff A, Dallman T, Dibley M, Redfern O, Pearl F, Nambudiry R, Reid A, Sillitoe I, Yeats C, Thornton JM, Orengo CA. The CATH domain structure database: new protocols and classification levels give a more comprehensive resource for exploring evolution. Nucleic Acids Res. 2007;35(Database issue):D291–297. doi: 10.1093/nar/gkl959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Montelione GT, Arrowsmith C, Girvin ME, Kennedy MA, Markley JL, Powers R, Prestegard JH, Szyperski T. Unique opportunities for NMR methods in structural genomics. J Struct Funct Genomics. 2009;10(2):101–106. doi: 10.1007/s10969-009-9064-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.del Val C, Mehrle A, Falkenhahn M, Seiler M, Glatting KH, Poustka A, Suhai S, Wiemann S. High-throughput protein analysis integrating bioinformatics and experimental assays. Nucleic Acids Res. 2004;32(2):742–748. doi: 10.1093/nar/gkh257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joshi T, Chen Y, Becker JM, Alexandrov N, Xu D. Genome-scale gene function prediction using multiple sources of high-throughput data in yeast Saccharomyces cerevisiae. OMICS. 2004;8(4):322–333. doi: 10.1089/omi.2004.8.322. [DOI] [PubMed] [Google Scholar]
- 21.Lee YH, Moon IJ, Hur B, Park JH, Han KH, Uhm SY, Kim YJ, Kang KJ, Park JW, Seu YB, Kim YH, Park JG. Gene knockdown by large circular antisense for high-throughput functional genomics. Nat Biotechnol. 2005;23(5):591–599. doi: 10.1038/nbt1089. [DOI] [PubMed] [Google Scholar]
- 22.Oude Elferink R. One step further towards real high-throughput functional genomics. Trends Biotechnol. 2003;21(4):146–147. doi: 10.1016/S0167-7799(03)00029-5. discussion 147–148. [DOI] [PubMed] [Google Scholar]
- 23.Tucker CL. High-throughput cell-based assays in yeast. Drug Discov Today. 2002;7(18 Suppl):S125–130. doi: 10.1016/s1359-6446(02)02409-1. [DOI] [PubMed] [Google Scholar]
- 24.Mercier KA, Baran M, Ramanathan V, Revesz P, Xiao R, Montelione GT, Powers R. FAST-NMR: functional annotation screening technology using NMR spectroscopy. J Am Chem Soc. 2006;128(47):15292–15299. doi: 10.1021/ja0651759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Powers R, Mercier KA, Copeland JC. The application of FAST-NMR for the identification of novel drug discovery targets. Drug Discov Today. 2008;13(3–4):172–179. doi: 10.1016/j.drudis.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an emperical binding free energy function. J Comput Chem. 1998;19(14):1639–1662. [Google Scholar]
- 27.Stark J, Powers R. Rapid protein-ligand costructures using chemical shift perturbations. J Am Chem Soc. 2008;130(2):535–545. doi: 10.1021/ja0737974. [DOI] [PubMed] [Google Scholar]
- 28.Powers R, Copeland JC, Germer K, Mercier KA, Ramanathan V, Revesz P. Comparison of protein active site structures for functional annotation of proteins and drug design. Proteins. 2006;65(1):124–135. doi: 10.1002/prot.21092. [DOI] [PubMed] [Google Scholar]
- 29.Fahy E, Sud M, Cotter D, Subramaniam S. LIPID MAPS online tools for lipid research. Nucleic Acids Res. 2007;35(Web Server issue):W606–612. doi: 10.1093/nar/gkm324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghosh S, Nie A, An J, Huang Z. Structure-based virtual screening of chemical libraries for drug discovery. Curr Opin Chem Biol. 2006;10(3):194–202. doi: 10.1016/j.cbpa.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 31.Klebe G. Virtual ligand screening: strategies, perspectives and limitations. Drug Discov Today. 2006;11(13–14):580–594. doi: 10.1016/j.drudis.2006.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Austin MB, Noel JP. The chalcone synthase superfamily of type III polyketide synthases. Nat Prod Rep. 2003;20(1):79–110. doi: 10.1039/b100917f. [DOI] [PubMed] [Google Scholar]
- 33.Choudhary DK, Johri BN. Interactions of Bacillus spp. and plants--with special reference to induced systemic resistance (ISR) Microbiol Res. 2009;164(5):493–513. doi: 10.1016/j.micres.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 34.Acton TB, Gunsalus KC, Xiao R, Ma LC, Aramini J, Baran MC, Chiang YW, Climent T, Cooper B, Denissova NG, Douglas SM, Everett JK, Ho CK, Macapagal D, Rajan PK, Shastry R, Shih LY, Swapna GV, Wilson M, Wu M, Gerstein M, Inouye M, Hunt JF, Montelione GT. Robotic cloning and Protein Production Platform of the Northeast Structural Genomics Consortium. Methods Enzymol. 2005;394:210–243. doi: 10.1016/S0076-6879(05)94008-1. [DOI] [PubMed] [Google Scholar]
- 35.Herrmann T, Guntert P, Wuthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol. 2002;319(1):209–227. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- 36.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13(3):289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 37.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR System: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;D54(5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 38.Nederveen AJ, Doreleijers JF, Vranken W, Miller Z, Spronk CAEM, Nabuurs SB, Guentert P, Livny M, Markley JL, Nilges M, Ulrich EL, Kaptein R, Bonvin AMJJ. RECOORD: A recalculated coordinate database of 500+ proteins from the PDB using restraints from the BioMagResBank. Proteins. 2005;59(4):662–672. doi: 10.1002/prot.20408. [DOI] [PubMed] [Google Scholar]
- 39.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bhattacharya A, Tejero R, Montelione GT. Evaluating protein structures determined by structural genomics consortia. Proteins. 2007;66(4):778–795. doi: 10.1002/prot.21165. [DOI] [PubMed] [Google Scholar]
- 41.Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34(Web Server issue):W116–118. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 43.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 44.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gish W, States DJ. Identification of protein coding regions by database similarity search. Nat Genet. 1993;3(3):266–272. doi: 10.1038/ng0393-266. [DOI] [PubMed] [Google Scholar]
- 46.Holm L, Kaariainen S, Rosenstrom P, Schenkel A. Searching protein structure databases with DaliLite v.3. Bioinformatics. 2008;24(23):2780–2781. doi: 10.1093/bioinformatics/btn507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thompson JD, Gibson TJ, Higgins DG. Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinformatics. 2002;Chapter 2(Unit 2):3. doi: 10.1002/0471250953.bi0203s00. [DOI] [PubMed] [Google Scholar]
- 48.Fahy E, Subramaniam S, Brown HA, Glass CK, Merrill AH, Jr, Murphy RC, Raetz CR, Russell DW, Seyama Y, Shaw W, Shimizu T, Spener F, van Meer G, VanNieuwenhze MS, White SH, Witztum JL, Dennis EA. A comprehensive classification system for lipids. J Lipid Res. 2005;46(5):839–861. doi: 10.1194/jlr.E400004-JLR200. [DOI] [PubMed] [Google Scholar]
- 49.Fahy E, Subramaniam S, Murphy RC, Nishijima M, Raetz CR, Shimizu T, Spener F, van Meer G, Wakelam MJ, Dennis EA. Update of the LIPID MAPS comprehensive classification system for lipids. J Lipid Res. 2009;50 (Suppl):S9–14. doi: 10.1194/jlr.R800095-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bostrom J, Greenwood JR, Gottfries J. Assessing the performance of OMEGA with respect to retrieving bioactive conformations. J Mol Graph Model. 2003;21(5):449–462. doi: 10.1016/s1093-3263(02)00204-8. [DOI] [PubMed] [Google Scholar]
- 51.McGann MR, Almond HR, Nicholls A, Grant JA, Brown FK. Gaussian docking functions. Biopolymers. 2003;68(1):76–90. doi: 10.1002/bip.10207. [DOI] [PubMed] [Google Scholar]
- 52.Kellenberger E, Rodrigo J, Muller P, Rognan D. Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins. 2004;57(2):225–242. doi: 10.1002/prot.20149. [DOI] [PubMed] [Google Scholar]
- 53.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6(3):277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 54.Johnson BA. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol Biol. 2004;278:313–352. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- 55.Morton CJ, Pugh DJ, Brown EL, Kahmann JD, Renzoni DA, Campbell ID. Solution structure and peptide binding of the SH3 domain from human Fyn. Structure. 1996;4(6):705–714. doi: 10.1016/s0969-2126(96)00076-7. [DOI] [PubMed] [Google Scholar]
- 56.Fielding L. NMR methods for the determination of protein-ligand dissociation constants. Curr Top Med Chem. 2003;3(1):39–53. doi: 10.2174/1568026033392705. [DOI] [PubMed] [Google Scholar]
- 57.Huey R, Morris GM, Olson AJ, Goodsell DS. A semiempirical free energy force field with charge-based desolvation. J Comput Chem. 2007;28(6):1145–1152. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 58.Sousa SF, Fernandes PA, Ramos MJ. Protein-ligand docking: current status and future challenges. Proteins. 2006;65(1):15–26. doi: 10.1002/prot.21082. [DOI] [PubMed] [Google Scholar]
- 59.Richardson JS, Richardson DC. Natural beta-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc Natl Acad Sci U S A. 2002;99(5):2754–2759. doi: 10.1073/pnas.052706099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Arakane F, Sugawara T, Nishino H, Liu Z, Holt JA, Pain D, Stocco DM, Miller WL, Strauss JF., 3rd Steroidogenic acute regulatory protein (StAR) retains activity in the absence of its mitochondrial import sequence: implications for the mechanism of StAR action. Proc Natl Acad Sci U S A. 1996;93(24):13731–13736. doi: 10.1073/pnas.93.24.13731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Feng L, Chan WW, Roderick SL, Cohen DE. High-level expression and mutagenesis of recombinant human phosphatidylcholine transfer protein using a synthetic gene: evidence for a C-terminal membrane binding domain. Biochemistry. 2000;39(50):15399–15409. doi: 10.1021/bi001076a. [DOI] [PubMed] [Google Scholar]
- 62.Mercier KA, Mueller GA, Acton TB, Xiao R, Montelione GT, Powers R. 1H, 13C, and 15N NMR assignments for the Bacillus subtilis yndB START domain. J Biomol NMR. 2009 doi: 10.1007/s12104-009-9172-6. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Markwick PRL, Malliavin Trs, Nilges M. Structural Biology by NMR: Structure, Dynamics, and Interactions. PLoS Comput Biol. 2008;4(9):e1000168. doi: 10.1371/journal.pcbi.1000168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mercier Kelly A, Cort John R, Kennedy Michael A, Lockert Erin E, Ni S, Shortridge Matthew D, Powers R. Structure and function of Pseudomonas aeruginosa protein PA1324 (21–170) Protein Sci. 2009;18(3):606–618. doi: 10.1002/pro.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Montelione GT, Arrowsmith C, Girvin ME, Kennedy MA, Markley JL, Powers R, Prestegard JH, Szyperski T. Unique opportunities for NMR methods in structural genomics. J Struct Funct Genomics. 2009;10(2):101–106. doi: 10.1007/s10969-009-9064-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shortridge MD, Hage DS, Harbison GS, Powers R. Estimating protein-ligand binding affinity using high-throughput screening by NMR. J Comb Chem. 2008;10(6):948–958. doi: 10.1021/cc800122m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rosenfeld RJ, Goodsell DS, Musah RA, Morris GM, Goodin DB, Olson AJ. Automated docking of ligands to an artificial active site: augmenting crystallographic analysis with computer modeling. J Comput Aided Mol Des. 2003;17(8):525–536. doi: 10.1023/b:jcam.0000004604.87558.02. [DOI] [PubMed] [Google Scholar]
- 68.Roderick SL, Chan WW, Agate DS, Olsen LR, Vetting MW, Rajashankar KR, Cohen DE. Structure of human phosphatidylcholine transfer protein in complex with its ligand. Nat Struct Biol. 2002;9(7):507–511. doi: 10.1038/nsb812. [DOI] [PubMed] [Google Scholar]
- 69.Dove A, Marshall A. Drug screening-beyond the bottleneck. Nat Biotechnol. 1999;17(9):859–863. doi: 10.1038/12845. [DOI] [PubMed] [Google Scholar]
- 70.Doman TN, McGovern SL, Witherbee BJ, Kasten TP, Kurumbail R, Stallings WC, Connolly DT, Shoichet BK. Molecular docking and high-throughput screening for novel inhibitors of protein tyrosine phosphatase-1B. J Med Chem. 2002;45(11):2213–2221. doi: 10.1021/jm010548w. [DOI] [PubMed] [Google Scholar]
- 71.Konstantinou-Kirtay C, Mitchell JBO, Lumley JA. Scoring functions and enrichment: a case study on Hsp90. BMC Bioinf. 2007;8 doi: 10.1186/1471-2105-8-27. No pp. given. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ghosh S, Nie A, An J, Huang Z. Structure-based virtual screening of chemical libraries for drug discovery. Curr Opin Chem Biol. 2006;10(3):194–202. doi: 10.1016/j.cbpa.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 73.Forkmann G, Martens S. Metabolic engineering and applications of flavonoids. Curr Opin Biotechnol. 2001;12(2):155–160. doi: 10.1016/s0958-1669(00)00192-0. [DOI] [PubMed] [Google Scholar]
- 74.Steinkellner S, Lendzemo V, Langer I, Schweiger P, Khaosaad T, Toussaint JP, Vierheilig H. Flavonoids and strigolactones in root exudates as signals in symbiotic and pathogenic plant-fungus interactions. Molecules. 2007;12(7):1290–1306. doi: 10.3390/12071290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Winkel-Shirley B. Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol. 2001;126(2):485–493. doi: 10.1104/pp.126.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Long SR. Rhizobium-legume nodulation: life together in the underground. Cell. 1989;56(2):203–214. doi: 10.1016/0092-8674(89)90893-3. [DOI] [PubMed] [Google Scholar]
- 77.Perret X, Staehelin C, Broughton WJ. Molecular basis of symbiotic promiscuity. Microbiol Mol Biol Rev. 2000;64(1):180–201. doi: 10.1128/mmbr.64.1.180-201.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shen B, Hutchinson CR. Deciphering the mechanism for the assembly of aromatic polyketides by a bacterial polyketide synthase. Proc Natl Acad Sci USA. 1996;93(13):6600–6604. doi: 10.1073/pnas.93.13.6600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gensheimer M, Mushegian A. Chalcone isomerase family and fold: no longer unique to plants. Protein Sci. 2004;13(2):540–544. doi: 10.1110/ps.03395404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bode HB, Muller R. Possibility of bacterial recruitment of plant genes associated with the biosynthesis of secondary metabolites. Plant Physiol. 2003;132(3):1153–1161. doi: 10.1104/pp.102.019760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Versteeg S, Escher A, Wende A, Wiegert T, Schumann W. Regulation of the Bacillus subtilis heat shock gene htpG is under positive control. J Bacteriol. 2003;185(2):466–474. doi: 10.1128/JB.185.2.466-474.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rudrappa T, Bais HP. Arabidopsis thaliana Root Surface Chemistry Regulates in Planta Biofilm Formation of Bacillus subtilis. Plant Signal Behav. 2007;2(5):349–350. doi: 10.4161/psb.2.5.4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rudrappa T, Quinn WJ, Stanley-Wall NR, Bais HP. A degradation product of the salicylic acid pathway triggers oxidative stress resulting in down-regulation of Bacillus subtilis biofilm formation on Arabidopsis thaliana roots. Planta. 2007;226(2):283–297. doi: 10.1007/s00425-007-0480-8. [DOI] [PubMed] [Google Scholar]
- 84.Weller DM, Thomashow LS. Current challenges in introducing beneficial microorganisms into the rhizosphere. In: O’Gara F, Dowling DN, Boesten B, editors. Molecular Ecology of Rhizosphere Microorganisms: Biotechnology and the Release of GMOs. Weinheim: VCH; 1994. pp. 1–10. [Google Scholar]
- 85.Piggot PJ, Hilbert DW. Sporulation of Bacillus subtilis. Curr Opin Microbiol. 2004;7(6):579–586. doi: 10.1016/j.mib.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 86.Mabood F, Jung WJ, Smith DL. Soil Biology. Vol. 15. Berlin/Heidelberg: Springer; 2008. Signals in the Underground: Microbial Signaling and Plant Productivity. Molecular Mechanisms of Plant and Microbe Coexistence; pp. 291–318. [Google Scholar]