

Abstract
The hepatocyte growth factor (HGF)/Met signalling pathway is up-regulated in many cancers, with downstream mediators playing a role in DNA double strand break repair. Previous studies have shown increased radiosensitization of tumours through modulation of Met signalling by genetic methods. We investigated the effects of the anti-HGF monoclonal antibody, AMG102, on the response to ionizing radiation in a model of glioblastoma multiforme in vitro and in vivo. Radiosensitivity was evaluated in vitro in the U-87 MG human glioma cell line. Met activation was measured by Western blot, and the effect on survival following radiation was evaluated by clonogenic assay. Mechanism of cell death was evaluated by apoptosis and mitotic catastrophe assays. DNA damage was quantitated by γH2AX foci and neutral comet assay. Growth kinetics of subcutaneous tumours was used to assess the effects of AMG102 on in vivo tumour radiosensitivity. AMG102 inhibited Met activation after irradiation. An enhancement of radiation cell killing was shown with no toxicity using drug alone. Retention of γH2AX foci at 6 and 24 hrs following the drug/radiation combination indicated an inhibition of DNA repair following radiation, and comet assay confirmed DNA damage persisting over the same duration. At 48 and 72 hrs following radiation, a significant increase of cells undergoing mitotic catastrophe was seen in the drug/radiation treated cells. Growth of subcutaneous tumours was slowed in combination treated mice, with an effect that was greater than additive for each modality individually. Modulation of Met signalling with AMG102 may prove a novel radiation sensitizing strategy. Our data indicate that DNA repair processes downstream of Met are impaired leading to increased cell death through mitotic catastrophe.
Keywords: glioblastoma, AMG102, radiation, HGF, Met
Introduction
Of the ≍1.5 million patients diagnosed with cancer in the United States annually, the vast majority receive radiation therapy at some point in the course of treatment. More specifically, radiation has proved central to treating both primary and metastatic brain tumours, in particular offering the most effective non-surgical therapy for malignant gliomas, including Glioblastoma multiforme (GBM) [1]. Normal tissue toxicity is the dose limiting factor for this treatment modality, and as such, end-points of local control or cure can prove elusive. Taking advantage of radiobiology unique to tumours, pharmaceutical-based radiation sensitizers have the potential to increase radiation’s impact on neoplastic tissue specifically, while ideally leaving normal tissue unaffected.
The cell surface receptor tyrosine kinase, Met, is well characterized as the target for the diimeric polypeptide, hepatocyte growth factor (HGF) [2–6]. Met activation plays an important physiologic role in a diverse range of processes including embryogenesis and tissue regeneration [7, 8]; however, aberrant Met signalling has long been associated with the pathogenesis and pathophysiology of a wide variety of human malignancies [9–11]. Activated Met signalling results in a complex genetic program collectively referred to as ‘invasive growth’ [12] mediated by multiple downstream pathways including increased cell survival, proliferation, motility/migration, invasion and angiogenesis [9].
Met protein expression is typically up-regulated in human gliomas, with the degree of up-regulation correlating with tumour grade; further, Met signalling often becomes up-regulated in the context of an autocrine loop [13–15]. In addition to influencing tumour biology, the Met pathway has been implicated as a determinant of glioma cell response to ionizing radiation (IR). Two previously reported studies have shown that delivery of ribozymes or antisense gene constructs targeting HGF and/or Met gene expression enhances the radiosensitivity of glioma cell lines in vitro and in vivo. In both cases greater than additive responses to drug plus IR therapy were realized, resulting in decreased cell proliferation and tumour volumes, as well as increased tumour-free survival. However, these studies were limited insofar as they required direct, local injection of agents and adjuvant reagents that currently have no clinical precedent [16, 17]. An alternative to these genetic approaches is to inhibit the Met pathway via the fully human monoclonal anti-HGF antibody, AMG102. Currently in phase II clinical trials, this agent has been shown to have a sub-nanomolar affinity for the HGF ligand, as well as the ability to modulate the Met signalling cascade over a broad range of concentrations [18, 19]. Previous studies have shown, either as a mono- or combination therapy, the ability of AMG102 to inhibit growth of tumours, including gliomas, both in vitro and in vivo [18, 20–22]. No published studies to date have combined a clinically relevant pharmaceutical agent with IR. In the results presented here, we have shown that targeting the Met signalling axis with a clinically relevant dose of AMG102 enhances the in vitro and in vivo radiosensitivity of GBM cells. Further, such modulation was shown to enhance the effects of IR, with persistent, unrepaired DNA damage likely leading to increased cell death. These results further support the potential of modulating Met signalling as a target for tumour radiosensitization.
Materials and methods
Cell lines and treatment
The U-87 MG human GBM cell line (ATCC, Manassas, VA, USA) was grown in Dulbecco's Modified Eagle Medium (DMEM) (Invitrogen, Carlsbad, CA, USA) with 10% foetal bovine serum (FBS), and maintained at 37°C, 5% CO2. Cells were used within 6 months, and were authenticated by the supplier using short tandem repeat (STR) profiling, isoenzyme analysis, karyotype analysis, morphologic analysis and contamination testing. AMG102, human IgG2 and HGF, generously provided by Amgen, Inc. (Thousand Oaks, CA, USA), were diluted in phosphate-buffered solution (PBS) and stored at −80°C. For all in vitro experiments, growth media was supplemented with HGF at 1 ng/ml, and AMG102 or IgG2 were used at 1 μg/ml; cells were plated 24 hrs prior to drug treatment, and were exposed to drug for 24 hrs prior to irradiation. Cultures and animals were irradiated using an X-RAD 320 X-ray source (Pantak, Solon, OH, USA) at a dose rate of 2.54 Gy/min.
Western blot
Cell pellets were lysed on ice in RIPA buffer (Pierce, Rockford, IL, USA) supplemented with complete mini ethylenediaminetetraacetic acid free protease inhibitor cocktail (Roche, Indianapolis, IN, USA) and phosphatase inhibitor cocktail (Sigma, St. Louis, MO, USA). Protein concentration was determined by Bradford assay (Bio-Rad, Hercules, CA, USA). Protein (40 μg) were diluted 1:1 in Tris-Glycine SDS sample buffer with 5% beta mercaptoethanol (BME) added, boiled at 100°C for 8 min., electrophoresed on a 4–20% Tris-Glycine gel and wet-transferred overnight to a 0.2-μm-pore nitrocellulose membrane (Invitrogen). Membrane was blocked in 5% membrane blocking agent (GE Healthcare, Piscataway, NJ, USA), incubated with primary antibody overnight at 4°C, incubated with horseradish peroxidase (HRP)-coupled secondary antibody 2 hrs at room temperature, developed with Visualizer Western Blot Detection Kit (Millipore, Billerica, MA, USA) and visualized on a LAS-4000 imager (Fujifilm, Edison, NJ, USA). Membrane was stripped with Re-Blot Plus Mild (Millipore) and re-blocked and probed for additional proteins of interest. The following antibodies and dilutions were utilized: rabbit anti-human p-Met (Tyr1234/35) (1:500); mouse anti-human Met (1:300) (Cell Signaling, Danvers, MA, USA); mouse anti-actin (1:2500) (Millipore); goat anti-rabbit-HRP (1:5000) and goat antimouse-HRP (1:2000) (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Clonogenic assay
Cells were seeded into 6-well tissue culture plates and allowed to attach for 6 hrs. AMG102 or IgG2 control was added to the culture media and the plates were irradiated 16 hrs later. Ten to 14 days after seeding, colonies were stained with crystal violet, the number of colonies containing at least 50 cells was determined and the surviving fractions were calculated. Survival curves were generated after normalizing for the cytotoxicity generated by AMG102 alone.
Cell cycle analysis
Cell cycle phase distribution and G2-checkpoint integrity were evaluated by flow cytometry. Positive controls were treated 24 hrs with 0.2 μg/ml nocodazole (Sigma). Cells were trypsinized, fixed, and stained per manufacturer’s instructions with Cell Cycle Reagent and analysed on an EasyCyte Plus flow cytometre (Guava Technologies, Hayward, CA, USA). G2-checkpoint integrity was evaluated as previously reported [23] utilizing mouse anti-phospho-H3 histone (Millipore) at 1:133, and staining with a goat antimouse-fluorescein isothiocyanate (FITC) secondary antibody (Jackson ImmunoResearch, West Grove, PA, USA) at 1:30 to distinguish mitotic cells.
Apoptotic cell death
Apoptotic fraction was evaluated by flow cytometry. Positive controls were treated 24 hrs with 1 μM staurosporine (Sigma). Cells were trypsinized and stained per manufacturer’s instructions with Nexin Reagent and analysed on an EasyCyte Plus flow cytometre (Guava Technologies). The% double-positive annexin-V/7-AAD cells, representing early/late apoptosis respectively, were used to calculate the apoptotic indices presented here.
Mitotic catastrophe
Cells were grown in 4-well chamber slides, fixed with methanol for 15 min. at −20°C, washed three times with PBS, blocked with 1% bovine serum albumin (BSA) three times for 10 min. and stained overnight at 4°C with mouse anti-α-tubulin antibody (Sigma) at 1:1000. Cells were washed three times with 1% BSA, and were stained with goat antimouse-Texas Red antibody (Jackson ImmunoResearch) at 1:200 for 2 hrs at room temperature. Cells were washed three times with 1% BSA, and nuclei were counterstained with 0.8 mg/ml 4′,6-diamidino-2-phenylindole (DAPI) (Sigma) for 30 min., followed by two washes with PBS and one with ddH2O. Cover slips were mounted with VectaShield anti-fade solution (Vector Labs, Burlingame, CA, USA) and were visualized on a Leica DMRXA fluorescent microscope with a 20× objective (Wetzlar, Germany). Digital images were captured by a MicroPublisher 3.3 camera (QImaging, Surrey, Canada), overlaid in Adobe Photoshop CS (San Jose, CA, USA) and counted in ImageJ (NIH, Bethesda, MD, USA). The presence of nuclei fragmented with ≥2 lobes was the criterion for defining cells undergoing mitotic catastrophe. For each treatment condition 150–200 cells were scored, and the average of three separate counts of the same cells was taken.
Immunofluorescent staining for γH2AX
Cells were grown in 4-well chamber slides. Cells were fixed 15 min. at room temperature with 2% paraformaldehyde, washed three times with PBS, permeablized 5 min. on ice with 1% Triton X-100, washed three times with PBS and blocked three times for 10 min. with 1% BSA. Mouse anti-γH2AX antibody (Millipore) was added at 1:500 and incubated overnight at 4°C. Cells were washed three times with 1% BSA and a goat antimouse-FITC antibody (Jackson ImmunoResearch) was added at 1:100 and incubated 1 hr at room temperature. Cells were washed three times with 1% BSA, were counterstained with DAPI (Sigma) at 0.8 mg/ml for 30 min., and finally were washed twice with PBS and once with ddH2O. Cover slips were mounted with VectaShield anti-fade solution (Vector Labs) and slides were examined on a Leica microscope as previously described. For each treatment condition, γH2AX foci were quantitated in 50 cells by a blinded evaluator, taking the average of three separate counts of the same cells.
Neutral comet assay
DNA fragmentation was assessed using the CometAssay Kit (Trevigen, Gaithersburg, MD, USA) according to manufacturer’s instructions. Briefly, cells were trypsinized on ice until detached, pelleted, resuspended at 1 × 105 cells/ml in cold PBS, combined with melted agarose at a 1:10 ratio and immediately pipetted onto slides. Agarose was allowed to set for 30 min. at 4°C and then immersed in lysis solution for 1 hr. Slides were run in a horizontal electrophoresis apparatus for 10 min. at 19 V covered by exactly 5 mm of cold 1× TBE. Slides were rinsed twice in ddH2O and fixed in 70% ethanol for 5 min. Slides were air dried for 4 hrs and stained with 25 μg/ml propidium iodide (Guava Technologies). Slides were visualized on a Leica microscope as previously described. CASP 1.2.2 software (http://www.casplab.com; accessed 04/06/2009) was used to record Olive Tail Moment ([Tail mean – Head mean]× Tail%DNA/100) for each cell. For each treatment condition 25–50 cells were analysed by a blinded evaluator. Experiments were repeated three times with significance determined for each replicate individually; one representative figure is shown.
In vivo tumour growth
All animal studies were conducted in accordance with the principles and procedures outlined in the NIH Guide for the Care and Use of Animals. Four- to 6-week-old, female, Athymic NCr nu/nu, nude mice (NCI Animal Production Program, Frederick, MD, USA) were used for all in vivo studies. Animals were caged in groups of five or fewer and fed animal chow and water ad libitum. A single cell suspension (10 × 106) of U-87 MG cells was implanted on the lateral aspect of the rear leg. When tumours reached ≍100 mm3 ([L × W2]/2), animals were randomized into four groups: untreated controls, AMG102 (15 μg i.p.), IR (3 Gy) or AMG102 + IR (15 μg i.p., 3 Gy). Treatment with AMG102 was initiated at randomization, and irradiation of tumours took place 16 hrs later, with animals restrained in custom lead jigs. Tumours were measured three times per week until they reached ≥1000 mm3. Time taken for control tumours to undergo three doublings was determined. Tumour volumes were interpolated from fit spline curves for all groups the day control tumours had undergone exactly three doublings. Group means ± standard error are reported. Experiment was repeated two times with significance determined for each replicate individually; a representative figure is shown.
Statistical analysis
Data presented are the mean ± the standard error from three independent experiments unless indicated otherwise. Significance was assessed by t-test (two-tailed with 95% confidence intervals) using Prism 5.0 software (GraphPad Software, San Diego, CA, USA); Unpaired with Welch’s correction was utilized for in vitro studies (a parametric test assuming Gaussian distribution), and Mann-Whitney for in vivo studies (a non-parametric test not requiring the assumption of Gaussian distribution, chosen due to the smaller sample size of the in vivo cohorts).
Results
Previous studies have shown that radiation induces Met phosphorylation, indicative of receptor activation, in pancreatic carcinoma and neuroblastoma cell lines [24, 25]. As shown here, exposure of U-87 MG cells to the clinically relevant dose of 2 Gy results in a clear increase in the levels of phospho-Met at 6 and 24 hrs after irradiation (Fig. 1). To determine the effects of AMG102 on basal and radiation-induced phospho-Met expression, cells were treated (1 μg/ml) for 24 hrs before irradiation. Whereas exposure to AMG102 only had no effect on the levels of total Met, a clear decrease in the levels of phospho-Met was apparent in unirradiated cells. In addition, AMG102 reduced the level of radiation-induced phospho-Met at 6 and 24 hrs after IR.
Fig 1.

The influence of AMG102 on Met activation in U-87 MG cells. Cells were exposed to AMG102 (1 μg/ml) for 24 hrs, irradiated (2 Gy) and collected at 1, 6 and 24 hr time-points.
To determine the effects of AMG102 on tumour cell radiosensitivity, a clonogenic survival assay was performed (Fig. 2). U-87 MG cells were pre-treated with AMG102 or IgG2 control (1 μg/ml) for 16 hrs prior to IR and colony forming efficiency was determined 10–14 days later. AMG102 pre-treatment increased radiosensitivity with a dose enhancement factor of 1.2 at a surviving fraction of 10%. Further, a plating efficiency of 96% in unirradiated cells was observed. These data indicate that at this dose level, AMG102 enhances radiation-induced cell killing in GBM cells when combined with a clinically relevant IR exposure, with negligible toxicity derived from anti-HGF treatment alone.
Fig 2.

The influence of AMG102 on U-87 MG tumour cell radiosensitivity. U-87 MG cells were exposed to AMG102 or IgG2 control (1 μg/ml) for 16 hrs and exposed to between 0 and 8 Gy IR. Colony-forming efficiency was determined 10–14 days later and survival curves generated after normalizing for cytotoxicity due to AMG102 alone. PE: plating efficiency; DEF0.1: dose enhancement factor at 10% surviving fraction; •: IgG2 control; ▪: AMG102. n = 3; points: mean; bars: ±S.E.
Alteration of cell cycle kinetics, whereby cells are placed in a more radiosensitive phase, is a well-known mechanism by which radiosensitizers can exert their effects [26]. Cell cycle distribution following 24 hrs pre-treatment with AMG102 (1 μg/ml) was examined in U-87 MG cells, with no difference in phase distribution observed (data not shown). Moreover, an abrogation of the IR-induced G2 checkpoint following exposure to certain drugs has been shown to contribute to radiation-induced cytotoxicity [27, 28]. Integrity of this checkpoint was investigated by distinguishing G2- versus M-phase cells in the 4N cell population following IR. U-87 MG cells were pre-treated with AMG102 (1 μg/ml), exposed to 2 Gy, and stained for the phospho-H3 histone as an M-phase marker [23]. The addition of drug treatment did not alter the mitotic index of cells, as compared with their control or IR-only counterparts (data not shown). In particular, the low mitotic index seen in combination treated cells mirrored that of IR-only cells, indicating an intact G2 checkpoint. Taken together, these data indicate that AMG102-induced radiosensitization does not result from cell cycle redistribution, nor is it a result of G2-checkpoint abrogation.
To define the mode of cell death contributing to the dose enhancement detected by the clonogenic survival assay, U-87 MG cells pre-treated with AMG102 (1 μg/ml) for 24 hrs and exposed to 2 Gy were analysed for both apoptosis and mitotic catastrophe. AMG102 exposure alone had no effect on the level of U-87 MG apoptosis. When combined with radiation, a slight increase in apoptotic index at 24 and 72 hrs after IR was detected in AMG102 treated cells, which was not statistically significant (Fig. 3A). For solid tumour cells the most frequent mode of radiation-induced death is mitotic catastrophe [29]. Cells undergoing mitotic catastrophe are detectable at 48 and 72 hrs after irradiation with 2 Gy. AMG102 exposure alone did not induce U-87 MG mitotic catastrophe. However, irradiation of AMG102 treated cells resulted in a significant increase in cells undergoing mitotic catastrophe at 48 and 72 hrs after IR as compared with cells that received radiation alone (Fig. 3B). The AMG102-mediated increase in radiation-induced mitotic catastrophe suggests that inhibition of Met activation modifies the induction and/or the repair of radiation-induced DNA damage.
Fig 3.


The influence of AMG102 on mechanism of cell death in U-87 MG cells. (A) Apoptosis. U-87 MG cells were exposed to AMG102 (1 μg/ml), irradiated (2 Gy) and collected at 24 and 72 hr time-points for analysis by flow cytometry. Apoptotic Index,% annexin-V + 7-AAD+ cells. n = 3; columns: mean; bars: ±S.E. (B) Mitotic catastrophe. U-87 MG cells were exposed to AMG102 (1 μg/ml) for 24 hrs, irradiated (2 Gy) and fixed at 24, 48 and 72 hr time-points. Immunofluorescence staining was performed. Nuclei from each replicate scored three separate times in the same 150–200 cells, with the average taken. Mitotic Catastrophe Index,% cells present with ≥2 nuclear fragments. n = 3; columns: mean; bars: ±S.E.; * P = 0.0310, ** P = 0.0026 (unpaired t-test, IR versus AMG102 + IR).
The formation of DNA double strand breaks (DSB) following exposure to IR is central to the cytotoxicity associated with radiation therapy [30]. The measurement of the phospho-histone 2AX (γH2AX) nuclear foci has been established as a sensitive indicator of radiation-induced DSBs, as well as a reliable predictor of cell killing when defined at 24 hrs following radiation exposure [31–33]. To quantitate nuclear γH2AX foci, immunofluorescence staining of U-87 MG cells pre-treated for 24 hrs with AMG102 (1 μg/ml) followed by 2 Gy was performed (Fig. 4A). AMG102 treatment alone had no effect on the number of γH2AX foci. At 1 hr after IR there was no difference in the number of foci in drug treated and control groups, suggesting that AMG102 has no effect on the initial level of radiation-induced DSBs. However, at 6 and 24 hrs after IR there was a significantly greater number of γH2AX foci in the drug treated groups. These data suggest that treatment with AMG102 inhibits the repair of IR-induced DNA DSBs.
Fig 4.


The influence of AMG102 on radiation-induced DNA damage in U-87 MG cells. (A) γH2AX foci. U-87 MG cells were exposed to AMG102 or vehicle control IgG2 (1 μg/ml) for 24 hrs, irradiated (2 Gy), and fixed at 1, 6 and 24 hr time-points. Immunofluorescence staining was performed. Nuclear foci from each replicate counted three separate times in the same 50 cells, with the average taken. n = 3; columns: mean; bars: ±S.E.; *P = 0.0002, **P < 0.0001 (unpaired t-test, IR versus AMG102 + IR). (B) Neutral comet assay. U-87 MG cells were exposed to AMG102 (1 μg/ml) for 24 hrs, irradiated (10 Gy) and collected at 0, 1, 6 and 24 hr time-points. 25–50 cells analysed per condition. Olive Tail Moment = (Tail mean – Head mean) × Tail%DNA/100; •: untreated control; ▪: AMG102; ○: IR; □: AMG102 + IR. Representative figure, repeated three times; points: mean; bars: ±S.E.; *,** P < 0.0001 (unpaired t-test, IR versus AMG102 + IR).
To further investigate the effects of AMG102 on radiation-induced DSBs and their repair we performed the neutral comet assay (Fig. 4B) [34]. U-87 MG cells were exposed to AMG102 (1 μg/ml) for 24 hrs, irradiated with 10 Gy to induce sufficient DNA DSBs and collected for analysis at 0–24 hrs later. As indicated by the Olive Tail Moment, the most widely used method of quantitating DNA fragmentation observed with this assay, AMG102 treatment had no effect on the initial level of radiation-induced DSBs (0 hr). However, significantly more damage remained at both 6 and 24 hrs after IR in the AMG102-treated groups when compared with those exposed to IR alone. Consistent with the γH2AX results, these data suggest that exposure to AMG102 inhibits the repair of IR-induced DNA DSBs.
To determine whether the radiosensitizing properties of AMG102 would translate to an in vivo model, U-87 MG cells were implanted subcutaneously into mice and tumour growth was monitored. Tumours grown to ≍100 mm3 were randomized to one of four treatment arms: untreated control, AMG102 only (15 μg i.p.), IR only (3 Gy), AMG102 and IR (15 μg i.p., 3 Gy). Average growth rates are shown below (Fig. 5 and Table 1). No difference was detected in tumour volumes between control or single modality treated mice (i.e. IR or AMG102 alone) when compared on the day that control tumours had undergone three doublings. AMG102/IR combination treated mice had significantly smaller tumours (≍30%) on this day when compared with both control and single modality groups. These results suggest that AMG102 enhances the radiosensitivity of U-87 MG cells grown in vivo as subcutaneous xenografts.
Fig 5.
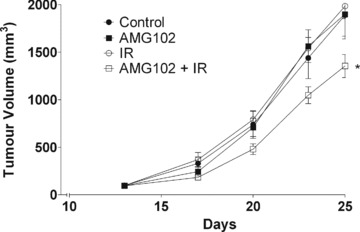
The influence of AMG102 on U-87 MG tumour growth. U-87 MG subcutaneous xenograft tumours grown to ≍100 mm3 before randomization to four groups: untreated controls, AMG102, IR, AMG102 + IR. AMG102 (15 μg) was injected i.p. and tumours irradiated (3 Gy) 16 hrs later with animals shielded in custom lead jigs. Tumour volume: [L × W2]/2; •: untreated control; ▪: AMG102; ○: IR; □: AMG102 + IR. Representative figure, repeated two times; points: mean; bars: ±S.E.; * P < 0.0241 (unpaired t-test, AMG102 + IR versus all other groups).
Table 1.
The influence of AMG102 on U-87 MG tumour growth
| Control | AMG102 | IR | AMG102 ± IR | |
|---|---|---|---|---|
| Mean | 807.1 | 793.1 | 865.5 | 536.6 |
| S.E. | 151.8 | 101.3 | 86.55 | 58.23 |
U-87 MG subcutaneous xenograft tumours grown to ≍100 mm3 before randomization to four groups: untreated controls, AMG102, IR, AMG102 + IR. AMG102 (15 μg) was injected i.p. and tumours irradiated (3 Gy) 16 hrs later with animals shielded in custom lead jigs. Tumour volumes were interpolated from fit spline curves for all groups the day control tumours had undergone exactly three doublings (20.3 days after randomization).
Discussion
GBM is the most common primary brain tumour in adults [35]. With current standard of care, radiation plus concomitant temozolomide therapy, median survival still remains at 14.6 months [36]. Identification of novel molecular targets that would allow for radiosensitization of GBM cells remains a major focus of research as even modest improvements in progression-free survival represent significant gains over the current clinical reality. The studies presented here provide additional support for a role of HGF/Met signalling as a target for enhancing GBM radiosensitivity.
Inhibition of Met signalling has previously been shown to radiosensitize glioma cells in vitro and in vivo [16, 17]. However, these results required manipulation of HGF/Met at the level of gene expression utilizing either ribozyme- or antisense oligonucleotide-based approaches; neither of these techniques are currently in routine clinical practice. AMG102 is currently in phase II clinical trials in GBM as a monotherapy [37]. The experiments described here show that AMG102, a fully human anti-HGF monoclonal antibody, enhances the radiosensitivity of U-87 MG glioma cells grown in vitro and in vivo; thus this study represents the first report of radiosensitization through modulation of the HGF/Met signalling axis with a clinically relevant pharmaceutical agent.
Activated Met signalling is mediated by numerous downstream effectors including Ras/Raf/mitogen-activated protein kinase (MAPK), PI3K/Akt, Myc, NF-κB and Signal Transducers and Activators of Transcription (STAT) pathways [12, 38]. Many of these effectors have been shown to have roles in the cellular radiation response [39–41]. It has also been noted that clinically relevant doses of IR have been shown to activate Met signalling [24, 25, 42] and moreover, activated Met signalling has been directly shown to enhance DNA repair following exposure to IR [43]. The persistence of DNA DSBs is well known to be an important mediator of cytotoxicity following exposure to IR [30]. The data presented here show induction of DNA DSBs by IR was found to be equal between irradiated groups with and without drug treatment; however, repair of DSBs was inhibited in AMG102/IR combination treated cells. Taken together, these results indicate that inhibition of Met-mediated DSB repair after IR contributes to AMG102-mediated radiosensitization. This study is among the first published reports directly linking the inhibition of Met activation with a change in the kinetics of DSB repair [44, 45].
Effector molecules downstream of activated Met are commonly considered to be anti-apoptotic [43]. In previous studies combining Met modulation with IR, studies investigating the use of Met axis modulators as monotherapies [11, 46, 47], and studies examining AMG102 itself [18, 20], therapeutic efficacy has been attributed to inhibition of proliferation and increased cell death by apoptosis. In contrast, the results presented here indicate that AMG102 has no significant effect on radiation-induced apoptosis but does enhance radiation-induced mitotic catastrophe. Although designed in a similar fashion to the previous studies cited above, the work presented here has a number of key differences that may account for this discrepancy with respect to mechanism of cell death. First, in these studies, the level of Met activation was controlled by adding a predetermined amount of exogenous HGF, whereas previous studies have relied on endogenous levels of receptor activation by autocrine produced HGF. Downstream mediators may play a more significant role with respect to DNA DSB repair when compared with their anti-apoptotic effects in cells with increased Met activation by exogenous HGF stimulation. Secondly, when compared to previous work examining AMG102 in particular, the studies presented here utilize a lower concentration of drug, with differences in drug/IR dosing schedules. Lastly, none of the studies presented above assayed for cells undergoing mitotic catastrophe; it may be that this was a significant mechanism of Met-mediated cell death that has not been examined previously examined.
Subcutaneous xenografted U-87 MG tumours treated with sub-maximal AMG102 in conjunction with IR exhibited an inhibition of growth that was more than additive when compared with control and single modality treated groups. When considered in the context of previous studies this result supports the finding that the Met signalling axis is a valid target for radiosensitization in vivo.
AMG102 possesses a number of distinct advantages as a Met modulating drug. Most importantly, monoclonal antibodies are a well-established therapeutic modality, and further, this agent in particular is currently in a variety of phase II monotherapy and combination clinical trials. Moreover, at the dose level used in these studies, toxicity due to antibody alone was negligible as demonstrated by the robust plating efficiency seen on clonogenic assay. As opposed to merely a radiation modifier, this indicates that the effect of AMG102 in combination with IR is that of a true radiosensitizer, with minimal toxicity expected to normal tissue from drug alone. The use of a fully human monoclonal antibody targeted against HGF is beneficial for three reasons – first, targeting HGF prevents the phenomenon of antibody-mediated, ligand-independent dimerization seen with bivalent antibodies against the Met receptor itself [10]; second, being fully human, this molecule carries little risk of being targeted by a host immune response; lastly, such antibodies possess plasma half-lives in the order of 15 days, as opposed to hours for small molecules, thus allowing for a comparative simplification of dosing regimens with less frequent administrations [48]. It is also of note that delivery of similar therapeutics to intracranial tumours has been shown to be possible, thus indicating that the blood–brain and blood–tumour barriers do not block passage of such molecules [46, 49].
Conclusion
The data presented here indicate the capacity of AMG102, the fully human anti-HGF monoclonal antibody, to radiosensitize U-87 MG GBM cells in vitro and in vivo. We propose the mechanism of radiosensitization involves the inhibition of DNA repair processes downstream of activated Met, leading to increased cell death by mitotic catastrophe.
Acknowledgments
This research was supported in part by the Intramural Research Program of the National Institutes of Health, National Cancer Institute and by the Howard Hughes Medical Institute through the Howard Hughes Medical Institute-National Institutes of Health, Research Scholar Program.
Conflict of interest
TLB is an employee of Amgen, Inc., the manufacturer and patent holder of the drug used in the studies presented above.
References
- 1.Pazdur R, Wagman LD, Camphausen KA, et al. Oncology, editor. 11 ed. Lawrence, KS, USA: Medica; 2008. Cancer management: a multidiciplinary approach, medical, surgical, & radiation oncology. [Google Scholar]
- 2.Cooper C, Park M, Blair D, et al. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature. 1984;311:29–33. doi: 10.1038/311029a0. [DOI] [PubMed] [Google Scholar]
- 3.Stoker M, Gherardi E, Perryman M, et al. Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature. 1987;327:239–42. doi: 10.1038/327239a0. [DOI] [PubMed] [Google Scholar]
- 4.Nakamura T, Nishikawa T, Hagiya M, et al. Molecular cloning and expression of human hepatocyte growth factor. Nature. 1989;342:440–3. doi: 10.1038/342440a0. [DOI] [PubMed] [Google Scholar]
- 5.Weidner KM, Arakaki N, Hartmann G, et al. Evidence for the identity of human scatter factor and human hepatocyte growth factor. Proc Natl Acad Sci USA. 1991;88:7001–5. doi: 10.1073/pnas.88.16.7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bottaro D, Rubin J, Faletto D, et al. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251:802–4. doi: 10.1126/science.1846706. [DOI] [PubMed] [Google Scholar]
- 7.Higuchi O, Nakamura T. Identification and change in the receptor for hepatocyte growth factor in rat liver after partial hepatectomy or induced hepatitis. Biochem Biophys Res Commun. 1991;176:599–607. doi: 10.1016/s0006-291x(05)80226-8. [DOI] [PubMed] [Google Scholar]
- 8.Watanabe S, Hirose M, Wang XE, et al. Hepatocyte growth factor accelerates the wound repair of cultured gastric mucosal cells. Biochem Biophys Res Commun. 1994;199:1453–60. doi: 10.1006/bbrc.1994.1394. [DOI] [PubMed] [Google Scholar]
- 9.Birchmeier C, Birchmeier W, Gherardi E, et al. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–25. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 10.Christensen JG, Burrows J, Salgia R. c-Met as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Let. 2005;225:1–26. doi: 10.1016/j.canlet.2004.09.044. [DOI] [PubMed] [Google Scholar]
- 11.Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7:504–16. doi: 10.1038/nrd2530. [DOI] [PubMed] [Google Scholar]
- 12.Boccaccio C, Comoglio PM. Invasive growth: a MET-driven genetic programme for cancer and stem cells. Nat Rev Cancer. 2006;6:637–45. doi: 10.1038/nrc1912. [DOI] [PubMed] [Google Scholar]
- 13.Koochekpour S, Jeffers M, Rulong S, et al. Met and hepatocyte growth factor/scatter factor expression in human gliomas. Cancer Res. 1997;57:5391–8. [PubMed] [Google Scholar]
- 14.Rosen EM, Laterra J, Ansamma J, et al. Scatter factor expression and regulation in human glial tumors. Int J Cancer. 1996;67:248–55. doi: 10.1002/(SICI)1097-0215(19960717)67:2<248::AID-IJC16>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 15.Athauda G, Giubellino A, Coleman JA, et al. c-Met ectodomain shedding rate correlates with malignant potential. Clin Cancer Res. 2006;12:4154–62. doi: 10.1158/1078-0432.CCR-06-0250. [DOI] [PubMed] [Google Scholar]
- 16.Lal B, Xia S, Abounader R, et al. Targeting the c-Met pathway potentiates glioblastoma responses to {gamma}-radiation. Clin Cancer Res. 2005;11:4479–86. doi: 10.1158/1078-0432.CCR-05-0166. [DOI] [PubMed] [Google Scholar]
- 17.Sheng-hua C, Zhi-an Z, Xian-hou Y, et al. In vitro and in vivo potentiating the cytotoxic effect of radiation on human U251 gliomas by the c-Met antisense oligodeoxynucleotides. J Neurooncol. 2006;80:143–9. doi: 10.1007/s11060-006-9174-5. [DOI] [PubMed] [Google Scholar]
- 18.Burgess T, Coxon A, Meyer S, et al. Fully human monoclonal antibodies to hepatocyte growth factor with therapeutic potential against hepatocyte growth factor/c-Met-dependent human tumors. Cancer Res. 2006;66:1721–9. doi: 10.1158/0008-5472.CAN-05-3329. [DOI] [PubMed] [Google Scholar]
- 19.Burgess TL, Sun J, Meyer S, et al. Biochemical characterization of AMG 102: a neutralizing, fully human monoclonal antibody to human and nonhuman primate hepatocyte growth factor. Mol Cancer Ther. 2010;9:400–9. doi: 10.1158/1535-7163.MCT-09-0824. [DOI] [PubMed] [Google Scholar]
- 20.Jun HT, Sun J, Rex K, et al. AMG 102, a fully human anti-hepatocyte growth factor/scatter factor neutralizing antibody, enhances the efficacy of temozolomide or docetaxel in U-87 MG cells and xenografts. Clin Cancer Res. 2007;13:6735–42. doi: 10.1158/1078-0432.CCR-06-2969. [DOI] [PubMed] [Google Scholar]
- 21.Gao CF, Xie Q, Zhang YW, et al. Therapeutic potential of hepatocyte growth factor/scatter factor neutralizing antibodies: inhibition of tumor growth in both autocrine and paracrine hepatocyte growth factor/scatter factor:c-Met-driven models of leiomyosarcoma. Mol Cancer Ther. 2009;8:2803–10. doi: 10.1158/1535-7163.MCT-09-0125. [DOI] [PubMed] [Google Scholar]
- 22.Davis IJ, McFadden AW, Zhang Y, et al. Identification of the receptor tyrosine kinase c-Met and its ligand, hepatocyte growth factor, as therapeutic targets in clear cell sarcoma. Cancer Res. 2010;70:639–45. doi: 10.1158/0008-5472.CAN-09-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu B, Kim S-T, Lim D-S, et al. Two molecularly distinct G2/M checkpoints are induced by ionizing irradiation. Mol Cell Biol. 2002;22:1049–59. doi: 10.1128/MCB.22.4.1049-1059.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qian L-W, Mizumoto K, Inadome N, et al. Radiation stimulates HGF receptor/c-Met expression that leads to amplifying cellular response to HGF stimulation via upregulated receptor tyrosine phosphorylation and MAP kinase activity in pancreatic cancer cells. Int J Cancer. 2003;104:542–9. doi: 10.1002/ijc.10997. [DOI] [PubMed] [Google Scholar]
- 25.Schweigerer L, Rave-Fränk M, Schmidberger H, et al. Sublethal irradiation promotes invasiveness of neuroblastoma cells. Biochem Biophys Res Commun. 2005;330:982–8. doi: 10.1016/j.bbrc.2005.03.068. [DOI] [PubMed] [Google Scholar]
- 26.Pawlik TM, Keyomarsi K. Role of cell cycle in mediating sensitivity to radiotherapy. In J Radiat OncolBiolPhys. 2004;59:928–42. doi: 10.1016/j.ijrobp.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 27.Cilby W, Roberts C, Cimprich K, et al. Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J. 1998;17:159–69. doi: 10.1093/emboj/17.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y, Li J, Booher RN, et al. Radiosensitization of p53 Mutant Cells by PD0166285, a Novel G2 Checkpoint Abrogator. Cancer Res. 2001;61:8211–7. [PubMed] [Google Scholar]
- 29.Broker LE, Kruyt FAE, Giaccone G. Cell death independent of caspases: a review. Clin Cancer Res. 2005;11:3155–62. doi: 10.1158/1078-0432.CCR-04-2223. [DOI] [PubMed] [Google Scholar]
- 30.Olive PL. The role of DNA single- and double-strand breaks in cell killing by ionizing radiation. Radiat Res. 1998;150:S42–52. [PubMed] [Google Scholar]
- 31.Banath JP, Olive PL. Expression of phosphorylated histone H2AX as a surrogate of cell killing by drugs that create DNA double-strand breaks. Cancer Res. 2003;63:4347–50. [PubMed] [Google Scholar]
- 32.Bonner WM, Redon CE, Dickey JS, et al. [gamma]H2AX and cancer. Nat Rev Cancer. 2008;8:957–67. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rogakou EP, Pilch DR, Orr AH, et al. DNA Double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 34.Olive PL, Banath JP. The comet assay: a method to measure DNA damage in individual cells. Nat Protocols. 2006;1:23–9. doi: 10.1038/nprot.2006.5. [DOI] [PubMed] [Google Scholar]
- 35.CBTRUS. Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2004–2005. Hinsdale, IL: Central Brain Tumor Registry of the United States; 2009. [Google Scholar]
- 36.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 37.Gordon MS, Sweeney CS, Mendelson DS, et al. Safety, pharmacokinetics, and pharmacodynamics of AMG 102, a fully human hepatocyte growth factor-neutralizing monoclonal antibody, in a first-in-human study of patients with advanced solid tumors. Clin Cancer Res. 2010;16:699–710. doi: 10.1158/1078-0432.CCR-09-1365. [DOI] [PubMed] [Google Scholar]
- 38.Gao C-F, Xie Q, Su Y-L, et al. Proliferation and invasion: plasticity in tumor cells. Proc Nat Acad Sci USA. 2005;102:10528–33. doi: 10.1073/pnas.0504367102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Toulany M, Dittmann K, Fehrenbacher B, et al. PI3K-Akt signaling regulates basal, but MAP-kinase signaling regulates radiation-induced XRCC1 expression in human tumor cells in vitro. DNA Repair. 2008;7:1746–56. doi: 10.1016/j.dnarep.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 40.Kim M-J, Byun J-Y, Yun C-H, et al. c-Src-p38 mitogen-activated protein kinase signaling is required for Akt activation in response to ionizing radiation. Mol Cancer Res. 2008;6:1872–80. doi: 10.1158/1541-7786.MCR-08-0084. [DOI] [PubMed] [Google Scholar]
- 41.Fan S, Gao M, Meng Q, et al. Role of NF-[kappa]B signaling in hepatocyte growth factor//scatter factor-mediated cell protection. Oncogene. 2005;24:1749–66. doi: 10.1038/sj.onc.1208327. [DOI] [PubMed] [Google Scholar]
- 42.Miller AC, Luo L, Chin WK, et al. Proto-oncogene expression: a predictive assay for radiation biodosimetry applications. Radiat Prot Dosimetry. 2002;99:295–302. doi: 10.1093/oxfordjournals.rpd.a006789. [DOI] [PubMed] [Google Scholar]
- 43.Fan S, Ma Y, Wang J, et al. The cytokine hepatocyte growth factor/scatter factor inhibits apoptosis and enhances DNA repair by a common mechanism involving signaling through phosphatidyl inositol 3’ kinase. Oncogene. 2000;19:2212–23. doi: 10.1038/sj.onc.1203566. [DOI] [PubMed] [Google Scholar]
- 44.Ganapathipillai SS, Medova M, Aebersold DM, et al. Coupling of mutated Met variants to DNA repair via Abl and Rad51. Cancer Res. 2008;68:5769–77. doi: 10.1158/0008-5472.CAN-08-1269. [DOI] [PubMed] [Google Scholar]
- 45.Welsh J, Mahadevan D, Ellsworth R, et al. The c-Met receptor tyrosine kinase inhibitor MP470 radiosensitizes glioblastoma cells. Radiat Oncol. 2009;4:69. doi: 10.1186/1748-717X-4-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim KJ, Wang L, Su Y-C, et al. Systemic anti-hepatocyte growth factor monoclonal antibody therapy induces the regression of intracranial glioma xenografts. Clin Cancer Res. 2006;12:1292–8. doi: 10.1158/1078-0432.CCR-05-1793. [DOI] [PubMed] [Google Scholar]
- 47.Zou HY, Li Q, Lee JH, et al. An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007;67:4408–17. doi: 10.1158/0008-5472.CAN-06-4443. [DOI] [PubMed] [Google Scholar]
- 48.Eder JP, Vande Woude GF, Boerner SA, et al. Novel therapeutic inhibitors of the c-Met signaling pathway in cancer. Clin Cancer Res. 2009;15:2207–14. doi: 10.1158/1078-0432.CCR-08-1306. [DOI] [PubMed] [Google Scholar]
- 49.Scott AM, Lee F-T, Tebbutt N, et al. A phase I clinical trial with monoclonal antibody ch806 targeting transitional state and mutant epidermal growth factor receptors. Proc Natl Acad Sci. 2007;104:4071–6. doi: 10.1073/pnas.0611693104. [DOI] [PMC free article] [PubMed] [Google Scholar]