

Abstract
The objective of present investigation was to formulate self-microemulsifying drug delivery systems (SMEDDS) of tacrolimus (FK 506), a poorly water soluble immunosuppressant that exhibits low and erratic bioavailability. Solubility of FK 506 in various oils, surfactants cosurfactants and buffers was determined. Phase diagrams were constructed at different ratios of surfactant/cosurfactant (Km) to determine microemulsion existence region. The effect of oil content, pH of aqueous phase, dilution, and incorporation of drug on mean globule size of resulting microemulsions was studied. The optimized SMEDDS formulation was evaluated for in vitro dissolution profile in comparison to pure drug and marketed formulation (Pangraf capsules). The in vivo immunosuppressant activity of FK 506 SMEDDS was evaluated in comparison to Pangraf capsules. Area of o/w microemulsion region in phase diagram was increased with increase in Km. The SMEDDS yielded microemulsion with globule size less than 25 nm which was not affected by the pH of dilution medium. The SMEDDS was robust to dilution and did not show any phase separation and drug precipitation even after 24 h. Optimized SMEDDS exhibited superior in vitro dissolution profile as compared to pure drug and Pangraf capsules. Furthermore, FK 506 SMEDDS exhibited significantly higher immunosuppressant activity in mice as compared to Pangraf capsules.
Key words: emulsification efficiency, poorly water soluble, SMEDDS, Tacrolimus (FK506)
INTRODUCTION
Tacrolimus (FK 506), a 23-member macrolide lactone isolated from Streptomyces tsukubaensi is a potent immunosuppressant and has been clinically used for preventing rejection of liver, heart, kidney, pancreas, lung and bone marrow after transplantation (1–3). It is more potent and efficacious than cyclosporin A (CyA) in reducing acute and chronic rejections. It has been proved to be effective as a ‘rescue’ drug for patients in whom cyclosporin A and steroid based regimen fails to prevent persistent refractory rejections. These advantages of FK 506 coupled with its long-term tolerability led to improved graft and patient survival indicating its clinical potential (3). However, its efficacy is strongly limited due to its poor water solubility (5–8 μg/ml) (4), presystemic metabolism by combination of gastrointestinal (GI) cytochrome P450 3A isoenzymes and P-glycoprotein (P-gp) mediated efflux (3–6), all of which are responsible for its low oral bioavailability. Additionally, FK 506 shows large intra- and inter-individual pharmacokinetic variability (3). Several approaches such as oily solution (7), solid dispersions (8), complexation with cyclodextrins (9), and liposomes (10) have been investigated to improve oral delivery of FK 506. Amongst these, solid dispersions succeeded in improving its delivery leading to commercialization (Prograf® Capsules, Fujisawa). However, it requires about 2 h for complete drug release and exhibits 25% bioavailability (3). As per Biopharmaceutical Classification System; FK 506 is a class II drug (5). For such drugs dissolution in GI lumen is rate-controlling step for absorption. Improved absorption can be achieved by use of delivery systems, which can enhance drug dissolution from its dosage form and maintains drug in dissolved state in GI fluids. Therefore, current scenario demands a need for a delivery strategy that can improve its therapeutic efficacy. Self-microemulsifying drug delivery systems (SMEDDS) recently have gained great interest in drug delivery research for its potential in improving oral bioavailability of poorly water soluble drugs. SMEDDS are defined as isotropic mixtures of oil, surfactant, cosurfactant and drug that rapidly form o/w microemulsion when exposed to aqueous media under conditions of gentle agitation or digestive motility that would be encountered in GI tract. SMEDDS presents the drug in nanosized droplets offering large interfacial area for drug diffusion (11–13). Furthermore, SMEDDS offer various advantages such as reduction in inter- and intra subject pharmacokinetic variability, improvement in lymphatic transport and GI permeability and reversal of P-gp efflux, all of which help in improving bioavailability of hydrophobic drugs (11–13). Considering these advantages, SMEDDS could be a valuable strategy in overcoming problems associated with delivery of FK 506. Utility of SMEDDS has been successfully established in improving oral bioavailability of CyA (14,15), which further justifies rationale of the study. The objective of study was to design and evaluate FK 506 SMEDDS for improving its solubility, dissolution rate and therapeutic efficacy. The present study, for the first time, reports a successful development of FK 506 SMEDDS that can improve its delivery.
MATERIALS AND METHODS
Materials
FK 506 was a gift from Glenmark Pharmaceuticals Ltd. (Nashik, India). Commercially available formulation of FK 506, Pangraf™ capsules, 5 mg (Panacea Biotec Ltd., New Delhi, India) was purchased. The inactive ingredients include lactose, hydroxypropyl methylcellulose, croscarmellose sodium and magnesium stearate. Cremophor EL, Lutrol F 68, Lutrol F 127, Solutol HS 15 (BASF, Mumbai, India), Capryol 90, Gelucire 44/14, Gelucire 55/13, Labrafil M 1944 S, Labrafac CC, Labrasol, Lauroglycol 90, Maisine 35-1, Peceol, Plurol Oleique CC497 (Colorcon Asia Mumbai, India), Imwitor-742, Lipoid MCT, Miglyol 812 N (S. Zaveri, Mumbai, India), Akoline-MCM, Akomed-E (Karlshamns AB, Sweden), Capmul MCM C8, Capmul MCM C8/10 (Abitec Corporation, USA), hard gelatin capsules (Associated Capsules, Mumbai, India) were obtained as gift samples. Carbitol, propylene glycol, Tween 20 and Tween 80 were purchased (S.D. Fine Chemicals, Mumbai, India). All excipients and reagents were used as received. Freshly prepared double distilled water was used.
Analysis of FK 506
A high performance thin layer chromatography (HPTLC) method with was developed for analysis of FK 506 (16). HPTLC system (Camag, Switzerland) comprising of Limomat 5 sample applicator, Hamilton syringe (100 μl), twin trough chamber (10 × 10 cm), dipping chamber and TLC scanner 3 with winCATS software V1.1.3.0 was used. Silica gel 60 F254 TLC plates (E. Merck, Darmstadt, Germany) used as stationary phase. Samples were dissolved in methanol and spotted under drying stream of nitrogen. Mobile phase composed of toluene/acetonitrile/glacial acetic acid (6:4:0.1, v/v/v) was used. Prior to development, chamber was saturated with mobile phase for 20 min. FK 506 was visualized and quantified 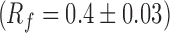 at 675 nm using anisaldehyde-sulfuric acid reagent. Minimum detectable amount was 28.90 ng per spot with limit of quantitation 97.04 ng per spot. Assay was linear (r2 = 0.998) in range 100–800 ng per spot. The method was accurate, precise and robust as percent relative standard deviation was consistently <5%.
at 675 nm using anisaldehyde-sulfuric acid reagent. Minimum detectable amount was 28.90 ng per spot with limit of quantitation 97.04 ng per spot. Assay was linear (r2 = 0.998) in range 100–800 ng per spot. The method was accurate, precise and robust as percent relative standard deviation was consistently <5%.
Solubility Studies
Equilibrium solubility of FK 506 in various excipients and buffers was determined by shake flask method. An excess of FK 506 was added to vial containing 1 g excipient or buffer. Mixtures were vortexed for 10 min and shaken for 48 h in reciprocating water bath shaker (Remi Equipments, Mumbai, India) at 25 ± 2 °C. After 48 h, vials were centrifuged at 5,000 rpm for 5 min, and mixtures were filtered through 0.45 μ filters (Pall Life Sciences, Mumbai, India). FK 506 in filtrate was quantified by a validated HPTLC method (16).
Emulsification Studies
Self-emulsification ability of surfactants was assessed to select the best surfactant from large pool of surfactants. Selected oil and surfactant were mixed in 1:3 (w/w), heated at 40–50 °C and vortexed to form homogenous mixture. Ratio of oil to surfactant was decided on the basis of requirements stated by Pouton (13) for spontaneously emulsifying systems and represents Type III system. Oil-surfactant mixture, 500 mg dispersed into 500 ml of double distilled water in a glass beaker with gentle stirring. Visual test was used to assess self-emulsification of surfactants in terms of dispersability, ease of emulsification and final appearance using grading system (Table I) (16). Various cosurfactants were screened by mixing surfactants with selected cosurfactants in 2:1 w/w ratios. Oily phase was added to this mixture in 1:3 (w/w), heated and vortexed gently to form homogenous mixture. They were evaluated using a visual test as explained in Table I.
Table I.
Visual Assessment of Efficiency of Self-Microemulsification
| Dispersability and Appearance | Time of Self-Emulsification (min) | Grade |
|---|---|---|
| Formulation spreads rapidly in water forming clear and transparent microemulsion | <1 | MEa |
| Formulation formed transparent, gel like intermediate structure prior to dispersing completely but could form microemulsion | 3–5 | MEb |
| Formulation droplets spread in water to form turbid emulsion | >5 | E |
| Formulation exhibits poor emulsification with coalescence of oil droplets | NA | NE |
Construction of Pseudo-Ternary Phase Diagrams
Pseudo-ternary phase diagrams of oil, surfactant, cosurfactant and water were developed using titration method at 25 ± 2 °C (17). Phase behavior of systems was studied at various ratios of surfactant to cosurfactant (Km) viz. 1:0.5, 1:1 and 1:2. Mixtures of surfactant and cosurfactant (at a specific Km) with water were prepared at ratios (w/w) of 10:0, 9:1, 8:2, 7:3, 6:4, 5:5, 4:6, 3:7, 2:8, 1:9, 0:10. A small amount of oil in 0.5% w/w increment was added in vials; vortexed and allowed to equilibrate. Resulting mixtures were evaluated visually for transparency and flow properties; and by polarizing microscope for optical isotropy. Endpoint of titration was the point, where mixture became turbid or phase separation was observed. At this point, amount of water, oil, surfactant and cosurfactant added was noted. Monophasic, clear, low viscous and non-birefringent systems were considered as microemulsion (ME) and shown as ME region.
Effect of Oily Phase Content on Mean Globule Size
A series of blank SMEDDS were prepared with varying oil content to study effect of oil content on mean globule size. Ratio of surfactant to cosurfactant was selected on the basis of phase diagrams and was maintained at 2:1 Table (II). Briefly, oil, surfactant and cosurfactant were weighed into glass vials, mixed by stirring, and heated (40–50 °C) to affect homogenous mixture. Oil–surfactant mixture, 500 mg was dispersed in 500 ml of various aqueous phases viz. double distilled water, buffer pH 1.2 (hydrochloric acid–potassium chloride buffer) and buffer pH 6.8 (phosphate buffer) with gentle stirring. Globule size was determined immediately after dilution.
Table II.
Compositions of SMEDDS Formulations Varying in Oil Content
| Ingredients | Formulation (mg) | ||||||
|---|---|---|---|---|---|---|---|
| S1 | S2 | S3 | S4 | S5 | S6 | S7 | |
| Cremophore-EL + Carbitol (At ratio of 2:1) | 425 | 412 | 400 | 387 | 375 | 362 | 350 |
| Capmul MCM C8 | 75 | 88 | 100 | 113 | 125 | 138 | 150 |
Preparation of FK 506 SMEDDS
FK 506 SMEDDS were prepared by dissolving drug into cosurfactant in glass vials. Oil and surfactant were accurately weighed in to glass vial. Components were mixed and heated (40–50 °C) to form a homogenous mixture and stored at room temperature until used.
Effect of Dilution and pH of Dilution Media on FK 506 SMEDDS
Robustness of SMEDDS to dilution and effect of pH of dilution medium was studied by diluting 500 mg SMEDDS with 500 ml of various aqueous phases mentioned above. Visual observations were made immediately after dilution for self-microemulsification efficiency and transparency. Resulting microemulsions were stored at room temperature for 24 h to assess phase separation and drug precipitation. Globule size was determined immediately and after 24 h.
Globule Size Analysis
Mean globule size and polydispersity index (P.I.) of microemulsions were determined by photon correlation spectroscopy (PCS) at 25 °C, using Beckman coulter N5 plus submicron particle size analyzer (Coulter Corporation, USA). Light scattering was monitored at 90° to incident beam on samples. Microemulsions were diluted suitably to ensure the intensity of light scattering was within the instrument’s sensitivity range. Samples were placed in transparent polystyrene cuvettes (1 cm2) and loaded in thermostated sample chamber.
Effect of FK 506 Loading
Effect of FK 506 loading on physical stability of microemulsions was studied using optimized composition i.e. formulation S3. Accordingly, a series of SMEDDS was prepared with varying amount of FK 506 (0 to 3%). SMEDDS, 500 mg was diluted with 500 ml of different media. Mean globule size and P.I. of microemulsions were determined by PCS.
In Vitro Dissolution Profile
FK 506 SMEDDS was filled in size ‘2’ hard gelatin capsules and evaluated for in vitro release using USP XXIII apparatus I at 37 ± 0.5 °C, at 100 rpm in buffer pH 1.2 and 6.8, 500 ml. During study, 2 ml of aliquots removed at 5, 10, 15, 20, 30, 60, 90 and 120 min and replaced with fresh buffer. Amount of drug released was determined using developed HPTLC method. In vitro release of Pangraf capsules and plain FK 506 was also determined.
Stability Studies
Chemical and physical stability of FK 506 SMEDDS was assessed at 40 ± 2 °C/75 ± 5% RH as per ICH Guidelines. SMEDDS equivalent to 5 mg FK 506 was filled in size ‘2’ hard gelatin capsules, packed in aluminum strips and stored for 3 months. Samples were analyzed at 0, 15, 30, 60 and 90 days for drug content, mean globule size and in vitro dissolution profile.
In Vivo Studies: Pharmacodynamic Evaluation in Mice
In vivo efficacy of FK 506 SMEDDS was evaluated by using method proposed by Kim et al. (18) and compared with commercial FK 506 formulation. Swiss albino mice weighing 20–25 g, were procured from Nicholas Piramal Research Centre (Mumbai, India) and housed in animal house of Bombay College of Pharmacy (Mumbai, India). Animals were given free access food, water was made available ad libitum and were acclimatized for >7 days before use. Experimental protocol was approved by Animal Ethical Committee of Bombay College of Pharmacy. Animals were divided into four groups of six animals each. Group 1—administered distilled water, group 2—treated with placebo SMEDDS, group 3—treated with Pangraf capsules, group 4—treated with FK 506 SMEDDS. Mice were administered formulations equivalent to 3.69 mg kg−1 day−1 of FK 506 by oral gavages for 10 days daily. Blood samples were collected in heparin containing tubes by retroorbital puncture under light ether anesthesia on zeroth, fifth and tenth days. Differential white blood cell (WBC) count was determined by standard method using Leishmann stain within 24 h after collection. Percent lymphocyte count was expressed as mean ± SD and data obtained was analyzed using a two-tail paired t-test to ascertain their statistical significance P < 0.05.
RESULTS AND DISCUSSION
Solubility Studies
Solubility of FK 506 in various excipients and buffers is shown in Table III. As it is important to achieve optimum drug loading (11,13), solubility study was aimed to identify suitable SMEDDS components that possess good solubilizing capacity for selected drug. FK 506 exhibited good solubility in Capryol 90, Capmul MCM C8, Capmul MCM C8/10 and Lauroglycol 90. Among surfactants, Cremophore EL solubilize maximum amount of FK 506; whereas in cosurfactants, Carbitol and propylene glycol exhibited good solubility for FK 506. Selection of surfactants and cosurfactants was governed by their emulsification efficiency for selected oily phases rather than their ability to solublize FK 506. Solubility study also indicated that FK 506 has poor aqueous solubility and was independent of pH of the medium.
Table III.
Solubility of FK 506 in Various Excipients and Buffers
| Oily phases | Solubilitya | Surfactants and Cosurfactants | Solubility | Buffer | Solubilityc |
|---|---|---|---|---|---|
| Akomed E | 12.28 ± 1.15 | Cremophor EL | 25.02 ± 1.16 | pH 1.2 | 6.81 ± 3.26 |
| Akoline MCM | 25.45 ± 2.21 | Gelucire 44/14b | 12.41 ± 0.35 | pH 4.5 | 5.72 ± 2.11 |
| Capmul MCM C8 | 32.19 ± 1.02 | Gelucire 55/13b | 11.22 ± 0.23 | pH 6.8 | 6.80 ± 3.14 |
| Capmul MCM C8/10 | 28.69 ± 2.38 | Labrasol | 9.63 ± 0.82 | pH 7.4 | 7.11 ± 4.32 |
| Capryol 90 | 34.44 ± 0.74 | Lutrol F 68b | 13.45 ± 1.21 | ||
| Imwitor 742 | 15.69 ± 1.75 | Lutrol F 127b | 9.41 ± 0.41 | ||
| Labrafac CC | 4.23 ± 1.01 | MYS-40b | 11.35 ± 0.96 | ||
| Lauroglycol 90 | 33.45 ± 1.12 | Solutol HS 15 | 9.60 ± 0.89 | ||
| Lipoid MCT | 2.16 ± 0.21 | Tween 20 | 12.76 ± 1.04 | ||
| Maisine 35-1 | 6.97 ± 1.49 | Tween 80 | 14.45 ± 2.19 | ||
| Miglyol 812 N | 2.10 ± 0.56 | Carbitol | 58.69 ± 3.94 | ||
| Oleic acid | 5.83 ± 0.78 | Labrafil M 1944 S | 6.77 ± 0.69 | ||
| Peceol | 7.79 ± 1.34 | Plurol Oleique CC497 | 5.08 ± 0.69 | ||
| Propylene glycol | 37.88 ± 4. 21 | ||||
| Triacetin | 17.17 ± 2.21 |
aData expressed as milligram per gram, mean ± SD, n = 3
b10% w/w surfactant solution
cData expressed as microgram per milliliter, mean ± SD, n = 3
Emulsification Studies
Grading for relative emulsification of surfactants is shown in Table IV which clearly distinguished ability of surfactants to emulsify selected oily phases, viz., Capryol 90, Capmul MCM C8, Capmul MCM C8/10 and Lauroglycol 90. The study indicated that Cremophor EL, Gelucire 44/14, MYS-40 and Tween 20 had very good ability to emulsify Capryol 90 and Capmul MCM C8 whereas Gelucire 55/13, Labrasol, Lutrol F 68, Lutrol F 127, Solutol HS 15 and Tween 80 appeared to be poor emulsifiers. None of the surfactant could emulsify Capmul MCM C8/10 and Lauroglycol 90. Although, HLB values of surfactants used in study were >10, there were considerable differences in their ability to emulsify oils. Results obtained indicated that apart from HLB value, other factors such as structure and relative length of hydrophobic chains of surfactants had influence on microemulsification. These results are in conformation with results reported in literature (19–21). Cremophor EL and Tween 20 rendered effective microemulsification and were selected for further study.
Table IV.
Emulsification Efficiency of Various Surfactants
| Surfactant | Oily Phases | |||
|---|---|---|---|---|
| Capryol 90 | Capmul MCM C8 | Capmul MCM C8/10 | Lauroglycol 90 | |
| Cremophor EL | MEa, c | MEa | E | E |
| Gelucire 44/14 | MEb | MEb | E | NE |
| Gelucire 55/13 | E | NE | NE | NE |
| Labrasol | E | NE | NE | NE |
| Lutrol F 68 | E | NE | NE | NE |
| Lutrol F 127 | E | NE | NE | NE |
| MYS-40 | MEb | MEb | E | NE |
| Solutol HS 15 | E | NE | NE | NE |
| Tween 20 | MEa | E | E | E |
| Tween 80 | E | NE | NE | NE |
cMeaning of abbreviations explained in Table I
Among the oils, Lauroglycol 90 was difficult to emulsify followed by Capmul MCM C8/10 whereas Capryol 90 and capmul MCM C8 were emulsified easily. This is explained by fact that ease of emulsification of oil and amount incorporated in microemulsion is affected by its molecular volume. As number and length of hydrophobic alkyl chains increases, molecular volume increases. Lauroglycol 90 and Capryol 90 are propylene glycol mono- and diester of lauric acid and caprylic acid, respectively. Though both oils are monoesters of respective fatty acids, lauric acid has longer chain length than caprylic acid which limits emulsification of Lauroglycol 90. Capmul MCM C8 and capmul MCM C8/10 both are mono and di-glycerides of medium chain fatty acids but differ in alkyl chain distribution. Capmul MCM C8 is a glycerin mono- and diester exclusively of caprylic acid about 97% and that of capric acid ~3% whereas Capmul MCM C8/10 is glycerin mono and diester of caprylic acid, ~82% and of capric acid ~17%. Capric acid has longer alkyl chain than that of caprylic acid. Higher capric acid content of Capmul MCM C8/10 limits its emulsification. The observations are in line with studies reported by Malcolmson et al. (20) and Warisnoicharoen et al. (21) Capryol 90 and Capmul MCM C8 were selected as oily phases for further study due to their relative ease of self-microemulsification.
Table V shows relative efficacy of cosurfactants to improve emulsification of surfactants. Carbitol and propylene glycol, hydrophilic cosurfactants increased spontaneity of microemulsion formation. Lipophilic cosurfactants Labrafil M 1944 CS, Plurol olique CC 497 and Triacetin were less effective as they could not improve emulsification of surfactants. As ratio of surfactant to cosurfactant is constant, study clearly distinguished ability of cosurfactants to improve emulsification of surfactants. Furthermore, as cosurfactants improve emulsification of surfactants by penetrating interfacial surfactant monolayer, their performance is affected by their structure and chain length (20,21). Labrafil M 1944 CS and Plurol olique CC 497 have oleate/linoleate and oleate backbones, whereas Triacetin have three alkyl chains. This increases molecular volume and affects penetration at interface. In contrast, Carbitol and propylene glycol, short chain amphiphiles, can penetrate perfectly at interface. Thus, the study gave an insight into relative emulsification properties of SMEDDS components and forms the basis of their selection.
Table V.
Emulsification Studies on Surfactant–Co-surfactant Combinations
| Surfactant + Cosurfactant | Oily Phases | |
|---|---|---|
| Capryol 90 | Capmul MCM C8 | |
| Cremophor EL + Carbitol | Ec | MEa |
| Cremophor EL + Propylene glycol | MEa | MEa |
| Cremophor EL + Labrafil M 1944 CS | E | E |
| Cremophor EL + Plurol olique CC 497 | NE | NE |
| Cremophor EL + Triacetin | NE | NE |
| Tween 20 + Carbitol | MEa | E |
| Tween 20 + Propylene glycol | E | E |
| Tween 20 + Labrafil M 1944 CS | E | NE |
| Tween 20 + Plurol olique CC 497 | NE | NE |
| Tween 20 + Triacetin | NE | NE |
| Gelucire 44/14 + Carbitol | MEb | MEb |
| Gelucire 44/14 + Propylene glycol | MEb | MEb |
| Gelucire 44/14 + Labrafil M 1944 CS | E | E |
| Gelucire 44/14 + Plurol olique CC 497 | NE | NE |
| Gelucire 44/14 + Triacetin | NE | NE |
| MYS-40 44/14 + Carbitol | MEb | MEb |
| MYS-40 44/14 + Propylene glycol | MEb | MEb |
| MYS-40 44/14 + Labrafil M 1944 CS | E | NE |
| MYS-40 44/14 + Plurol olique CC 497 | NE | NE |
| MYS-40 44/14 + Triacetin | NE | NE |
cMeaning of abbreviations explained in Table I
Carbitol and propylene glycol were equally effective as cosurfactant however; propylene glycol could not form isotropic mixture with oils and surfactants. It is an important prerequisite for SMEDDS, as by definition they are isotropic mixtures of oil, surfactant, and cosurfactant. Furthermore, due to its less solubilizing potential was not used further. Carbitol due to its superior solubilizing potential for FK 506 was selected. Accordingly, Cremophor EL-Carbitol-Capmul MCM C8 and Tween 20-Carbitol-Capryol 90 systems were selected for further study.
Construction of Pseudo-Ternary Phase Diagrams
Figures 1a–c and 2a–c show ternary phase diagrams for Cremophor EL-Carbitol-Capmul MCM C8 and Tween 20-Carbitol-Capryol 90 systems, respectively. Microemulsion formation area was increased with an increase in Km and was highest at Km = 2. Hence, surfactant to cosurfactant ratio was maintained at 2:1. The size of microemulsion region was compared; larger the size, greater is the self-microemulsification efficiency. From Figs. 1 and 2, it is evident that Cremophor EL-Carbitol-Capmul MCM C8 system has larger microemulsification region compared to Tween 20-Carbitol-Capryol 90. Cremophor EL-Carbitol-Capmul MCM C8 system yielded as high as 35% w/w of oil incorporation (Fig. 1c) whereas Tween 20-Carbitol-Capryol 90 system can incorporate 25% w/w of oil (Fig. 2c). Therefore, due to larger microemulsification area and greater capacity for oil incorporation, which is desirable to improve drug loading, Cremophor EL-Carbitol-Capmul MCM C8 system was selected for further studies.
Fig. 1.

Phase diagrams of Cremophor EL-Carbitol-Capmul MCM C8 system indicating o/w microemulsion existence region (ME) at a K m = 0.5, b K m = 1 and c K m = 2
Fig. 2.

Phase diagrams of Tween 20-Carbitol-Capryol 90 system indicating o/w microemulsion existence region (ME) at a K m = 0.5, b K m = 1 and c K m = 2
In conclusion, the study helped to identify microemulsion formation area, effect of ratio of surfactant to cosurfactant on it and maximum oil incorporation. It also helped to determine a suitable Km and concentration rage of various components for formation of SMEDDS.
Effect of Oily Phase Content on Mean Globule Size
Effect of oil concentration and pH of dilution media on mean globule size are shown in Fig. 3. SMEDDS formulations S1 to S4 were found robust to dilution and did not show any phase separation after 24 h. From phase behaviour, it was evident that Cremophor EL-Carbitol-Capmul MCM C8 system can incorporate 35% w/w of oily phase. However, compositions with such high oil content may not be necessarily stable. Hence, SMEDDS with varying oil content at Km = 2 were formulated to identify optimum oil concentration that can yield microemulsions of desired globule size. It is evident that up to 20% w/w concentration of Capmul MCM C8 (formulations S1, S2 and S3), mean globule size was <25 nm irrespective of pH of dilution medium. However, increase in oil content above 20%; mean globule size increased considerably. Formulations S5, S6 and S7 that contained oily phase 25% and above exhibited globule size greater than 150 nm (which is greater than globule size described by classical definition of microemulsion) and were opalescent. These observations are in line with inferences drawn from phase behavior studies. Based on this study, formulation S3 that contained 20% w/w oil was selected and is a classical example of type III-B systems as described by Pouton (13).
Fig. 3.

Effect of oil content on mean globule size (n = 3, relative standard deviation <10%) and polydispersity index (mean, n = 3)
Selection of Optimum Composition
Optimized composition was selected based on drug loading efficiency, larger micro-emulsification area with high amount of oil incorporation, fast dispersion and minimum effect of pH and dilution on mean globule size. Accordingly, formulation S3 was selected for further experiments and its composition is shown in Table VI.
Table VI.
Composition of Optimized FK 506 SMEDDS
| Ingredient | Quantity (mg per capsule) |
|---|---|
| Cremophor EL | 160 |
| Carbitol | 80 |
| Capmul MCM C8 | 60 |
| Tacrolimus | 5 |
| Total | 305 |
Effect of Dilution and pH of Dilution Media on FK 506 SMEDDS
Effect of dilution and pH of dilution media on SMEDDS containing FK 506 is shown in Table VII.
Table VII.
Globule Sizea and Polydispersity Indexb of FK 506 SMEDDS at Different pH Conditions
| Dilution Medium | Buffer pH 1.2 | Water | Buffer pH 6.8 | |||
|---|---|---|---|---|---|---|
| Immediately | After 24 h | Immediately | After 24 h | Immediately | After 24 h | |
| Blank SMEDDS | 19.8 (0.34c) | 21.8 (0.32) | 20.9 (0.48) | 19.3 (0.51) | 20.6 (0.33) | 21.4 (0.43) |
| FK 506 SMEDDS | 19.9 (0.24) | 18.2 (0.44) | 19.7 (0.39) | 19.1 (0.58) | 21.4 (0.44) | 24.4 (0.64) |
aData expressed in nanometer as mean, n = 3, where relative standard deviation <10%
bData expressed as mean, n = 3.
cValues in parentheses represent polydispersity index
Physical integrity of microemulsion formed and drug solublization capacity after dilution of SMEDDS must be assessed and ensured as it gives an idea about its performance in vivo (22,23). In view of this, FK 506 SMEDDS were diluted with aqueous phases differing in pH. FK 506 SMEDDS dispersed effectively and formed microemulsion in a minute without precipitation of drug. Microemulsions exhibited globule size <25 nm with narrow distribution irrespective of pH of dilution medium. Furthermore, they were robust to dilution as they did not show any phase separation, increase in globule size and drug precipitation even after 24 h of storage.
Effect of FK 506 Loading
Effect of FK 506 loading on mean globule size is shown in Fig. 4. Drug Incorporation can have significant influence on mean globule size and needs to be investigated (22,23). Because of hydrophobic nature of FK 506, it may affect globule size of microemulsions or may precipitate upon dilution. Globule size experiments showed that incorporation of FK 506 in SMEDDS does not have any impact on globule size when its concentration was up to 1.5% w/w. Furthermore, FK 506 did not precipitated after dilution at a concentration of 1.5% w/w. However, mean globule size increased significantly when concentration of FK 506 was 2% w/w and above. At this drug loading, resulting microemulsions appeared turbid and drug precipitated out in 30 min after dilution irrespective of pH of aqueous phase.
Fig. 4.

Effect of FK 506 loading on mean globule size (n = 3, relative standard deviation <10%) and polydispersity index (mean, n = 3)
In Vitro Dissolution Study
In vitro dissolution profile of FK 506 SMEDDS in comparison to its commercial formulation and plain drug in buffer pH 1.2 and 6.8 is shown in Fig. 5. SMEDDS release 100% FK 506 in 15 min in both dissolution media. Pangraf capsules required 2 h to release 100% of FK 506 and less than 10% of drug was released in 2 h from plain FK 506. Dramatic increase in the rate of release of FK 506 from SMEDDS compared to Pangraf capsules can be attributed to its quick dispersability and ability to keep drug in solubilized state. It was also evident that release of FK 506 from SMEDDS was independent of pH dissolution medium.
Fig. 5.

Dissolution profiles of FK 506 from various formulations, data expressed as mean ± SD, n = 3
Stability Studies
No change in the physical parameters such as homogeneity and clarity was observed during the stability studies. Interestingly, no decline in the FK 506 content was observed at the end of 3 months indicating that FK 506 remained chemically stable in SMEDDS (Table VIII). Furthermore, no change in other parameters such as dissolution profile, globule size and self-microemulsion efficiency was observed for the FK 506 SMEDDS (data not shown).
Table VIII.
Stability Study of FK 506 SMEDDS at 40 ± 2 °C/75 ± 5% RH
| Time (Days) | % Drug Contenta | Globule Sizeb (nm) | Polydispersity Indexb | ||||
|---|---|---|---|---|---|---|---|
| Water | Buffer pH 1.2 | Buffer pH 6.8 | Water | Buffer pH 1.2 | Buffer pH 6.8 | ||
| 15 | 99.89 ± 0.88 | 18.9 | 19.9 | 21.5 | 0.43 | 0.43 | 0.37 |
| 30 | 99.67 ± 1.08 | 21.2 | 18.6 | 20.1 | 0.32 | 0.43 | 0.56 |
| 45 | 100.06 ± 0.94 | 19.1 | 21.2 | 19.6 | 0.21 | 0.51 | 0.53 |
| 60 | 99.99 ± 1.21 | 22.8 | 21.6 | 22.6 | 0.33 | 0.63 | 0.48 |
| 90 | 98.87 ± 0.62 | 19.8 | 20.9 | 20.7 | 0.51 | 0.23 | 0.18 |
aData expressed as mean ± SD, n = 3
bData expressed as mean, n = 3, where relative standard deviation <10%
In Vivo Efficacy Studies: Pharmacodynamic Evaluation In Mice
Immunosuppressant effect of FK 506 is due to its selective and reversible inhibition of T lymophocytes, preventing formation of interleukins that are responsible for T-cell activation and proliferation. In addition, it has an inhibitory effect on humoral immunity i.e. inhibition of B-cell proliferation. Therefore, immunosuppressive activity of FK 506 would be clinically evident by reduction in T and B cell lymphocytes in peripheral blood and can be used as pharmacodynamic marker for FK 506 (1,3). Results of in vivo studies are shown in Fig. 6. Comparison of percent lymphocyte count between zeroth day and fifth day revealed a statistically significant difference for FK 506 SMEDDS. However, when a similar comparison was made for Pangraf capsules, statistically significant difference was not obtained on fifth day. When study was continued up to 10 days, wherein percent lymphocyte count obtained on tenth day was compared to that of baseline. Statistically significant difference was obtained for both i.e. FK 506 SMEDDS and Pangraf capsules. However, magnitude of reduction of lymphocyte was much greater for SMEDDS. Further, FK 506 SMEDDS and Pangraf capsules were compared for relative efficacy, where significant difference was not observed on the fifth day. But on tenth day, magnitude of reduction of lymphocyte showed by FK 506 SMEDDS was significantly greater than Pangraf capsules. Thus, there was a significant and profound effect of FK 506 SMEDDS on lowering lymphocytes in peripheral blood and was achieved much earlier as compared to Pangraf capsules. This clearly indicates the superiority of FK 506 SMEDDS and validates the rationale behind this study.
Fig. 6.

Percent peripheral lymphocyte count (mean ± SD, n = 6). Asterisk Significant reduction in percent lymphocyte count on fifth day for FK 506 SMEDDS, dagger percent lymphocyte count on tenth day was significantly less for FK 506 SMEDDS compared to commercial formulation
The improved therapeutic efficacy could be primarily because of increased solubility and dissolution rate of FK 506 leading to its rapid and efficient dispersion in GI tract. In addition, microemulsion formed was sufficiently stable and had very small globule size which provides a large interfacial surface area for drug diffusion. Superior performance of FK 506 SMEDDS may also be attributed although not confirmed, to inhibition of P-gp mediated efflux by Cremophor-EL and possible saturation of first-pass metabolism in GI wall. Thus, it was thought that, combination of above factors may result in an increase in rate and extent of absorption of FK 506 leading to its improved therapeutic efficacy over commercial formulation.
CONCLUSION
SMEDDS appeared to be an interesting approach to improve problems associated with oral delivery of FK 506. FK 506 SMEDDS formulation was superior to commercial formulation with respect to in vitro dissolution profile and in vivo immunosuppressant activity. Thus, SMEDDS can be regarded as novel and commercially feasible alternative to current FK 506 formulations.
ACKNOWLEDGEMENTS
Authors are thankful to Glenmark Pharmaceuticals, India; Colorcon Asia, India; BASF India Ltd, India; Karlshamns AB, Sweden; S. Zaveri & Co., India; Abitec Corporation USA and, Associated Capsules, India for providing gift samples. Authors are thankful to All India Council for Technical Education (AICTE), Delhi, India for providing the financial assistance for project.
References
- 1.Spencer C. M., Goa K. L., Gillis J. C. Tacrolimus: an update of its pharmacology and clinical efficacy in the management of organ transplantation. Drugs. 1997;54:925–975. doi: 10.2165/00003495-199754060-00009. [DOI] [PubMed] [Google Scholar]
- 2.Shapiro R. Tacrolimus in solid organ transplantation: an update. Transpl. Proc. 1999;31:2203–2205. doi: 10.1016/S0041-1345(99)00306-1. [DOI] [PubMed] [Google Scholar]
- 3.Christine E., Susan E. Clinical pharmacokinetics and pharmacodynamics of FK 506 in solid organ transplantation. Clin. Pharmacokinet. 2004;43:623–653. doi: 10.2165/00003088-200443100-00001. [DOI] [PubMed] [Google Scholar]
- 4.Kino T., Hatanaka H., Hashimoto M., Nishiyama M., Goto T., Okuhara M., Kohsaka M. FK 506, a novel immunosuppressant isolated from a Streptomyces I. Fermentation, isolation and physico-chemical and biological characteristics. J. Antibiot. 1987;42:1249–1255. doi: 10.7164/antibiotics.40.1249. [DOI] [PubMed] [Google Scholar]
- 5.Shigeki T., Atsuo O., Rinta I., Gordon L. Tacrolimus is a class II low-solubility high permeability drug: The effect of P-glycoprotein efflux on regional permeability of tacrolimus in rats. J. Pharm. Sci. 2002;91:719–729. doi: 10.1002/jps.10041. [DOI] [PubMed] [Google Scholar]
- 6.Shimomura M., Masuda S., Saito H., Sakamoto S., Uemoto S., Tanaka K., Inui K. Roles of the Jejunum and ileum in the first-pass effect as absorptive barriers for orally administered tacrolimus. J. Surg. Res. 2002;103:215–222. doi: 10.1006/jsre.2002.6359. [DOI] [PubMed] [Google Scholar]
- 7.Honbo T., Kobayashi M., Hane K., Hata T., Ueda Y. The oral dosage form of FK 506. Transpl. Proc. 1987;19:17–22. [PubMed] [Google Scholar]
- 8.Yamashita K., Nakate T., Okimoto K., Higaki K., Kimura T. Establishment of new preparation method for solid dispersion formulation of tacrolimus. Int. J. Pharm. 2003;267:79–91. doi: 10.1016/j.ijpharm.2003.07.010. [DOI] [PubMed] [Google Scholar]
- 9.Arima H., Yunomae K., Miyake K., Irie T., Hirayama F., Uekama K. Comparative studies of the enhancing effects of cyclodextrins on the solubility and oral bioavailability of tacrolimus in rats. J. Pharm. Sci. 2001;90:690–701. doi: 10.1002/jps.1025. [DOI] [PubMed] [Google Scholar]
- 10.McAlister V. C., Keshavamurthy M., Lee T. D. G. Oral delivery of liposomal tacrolimus: increased efficacy and reduced toxicity. Transplant. Proc. 1999;31:1110–1113. doi: 10.1016/S0041-1345(98)01923-X. [DOI] [PubMed] [Google Scholar]
- 11.Gursoy R. N., Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed. Pharmacother. 2004;58:173–182. doi: 10.1016/j.biopha.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 12.Lawrence M. J., Rees G. D. Microemulsion-based media as novel drug delivery systems. Adv. Drug Deliv. Rev. 2000;45:89–121. doi: 10.1016/S0169-409X(00)00103-4. [DOI] [PubMed] [Google Scholar]
- 13.Pouton C. W. Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur. J. Pharm. Sci. 2000;11(Supplement):S93–S98. doi: 10.1016/S0928-0987(00)00167-6. [DOI] [PubMed] [Google Scholar]
- 14.Vonderscher J., Meinzer A. Rationale for the development of Sandimmune Neoral. Transplant Proc. 1994;26:2925–2927. [PubMed] [Google Scholar]
- 15.Gershanika T., Benita S. Self-dispersing lipid formulations for improving oral absorption of lipophilic drugs. Eur. J. Pharm. Biopharm. 2000;50:179–188. doi: 10.1016/S0939-6411(00)00089-8. [DOI] [PubMed] [Google Scholar]
- 16.V. B. Borhade. Study of specialized emulsions for drug delivery (Masters thesis). University of Mumbai, India, 2006.
- 17.Corswant C. V., Söderman O. Microemulsions based on soybean phosphotidylcholine and triglycerides: Phase behavior and microstructure. Langmuir. 1997;13:5061–5070. doi: 10.1021/la9702897. [DOI] [Google Scholar]
- 18.Kim S. J., Choi H. K., Lee Y. B. Pharmacokinetic and pharmacodynamic evaluation of cyclosporin A o/w emulsion in rats. Int. J. Pharm. 2002;249:149–156. doi: 10.1016/S0378-5173(02)00490-8. [DOI] [PubMed] [Google Scholar]
- 19.Kawakami K., Yoshikawa T., Moroto Y., Kanaoka E., Takahashi K., Nishihara Y., Masuda K. Microemulsion formulation for enhanced absorption of poorly soluble drugs I. Prescription design. J. Control. Rel. 2002;81:65–74. doi: 10.1016/S0168-3659(02)00049-4. [DOI] [PubMed] [Google Scholar]
- 20.Malcolmson C., Sidhu A., Satra C., Kantaria S., Lawrence M. J. Effect of the nature of oil on the incorporation of testosterone propionate into non ionic oil-in-water microemulsions. J. Pharm. Sci. 1998;87:109–116. doi: 10.1021/js9700863. [DOI] [PubMed] [Google Scholar]
- 21.Warisnoicharoen W., Lansley A. B., Lawrence M. J. Nonionic oil-in-water microemulsions: effect of oil type on phase behavior. Int. J. Pharm. 2000;198:7–27. doi: 10.1016/S0378-5173(99)00406-8. [DOI] [PubMed] [Google Scholar]
- 22.Li P., Ghosh A., Wagner R. F., Krill S., Joshi Y. M. Effect of combined use of nonionic surfactant on formation of oil-in-water microemulsions. Int. J. Pharm. 2004;288:27–34. doi: 10.1016/j.ijpharm.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 23.Kanga B. K., Lee J. S., Chona S. K., Jeong S. Y., Khanga G., Lee H. B., Choc S. H. Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int. J. Pharm. 2004;274:65–73. doi: 10.1016/j.ijpharm.2003.12.028. [DOI] [PubMed] [Google Scholar]