

Abstract
In the present study, the aim was to optimize an orodispersible formulation of indomethacin using a combined approach of subliming agent and superdisintegrant. The tablets were made by non-aqueous wet granulation technique with superdisintegrant incorporated both intragranularly and extragranularly. A 23 factorial design was used to investigate the effects amount of subliming agents namely camphor and ammonium bicarbonate and taste masking and soothening hydrophilic agent mannitol as independent variables and disintegration time and crushing strength as dependent responses. The volatilization time of eight hours at 50°C was optimized by conducting solid-state kinetic studies of optimized formulations. Optimized orodispersible tablets were evaluated for wetting time, water absorption ratio, porosity and in vitro and in vivo disintegration tests. Results show that higher levels of camphor and mannitol and a lower level of ammonium bicarbonate is desirable for orodispersion. Scanning electron microscopy (SEM) revealed the porous surface morphology and kinetic digital images substantiated the orodispersible property. Differential Scanning Calorimetry (DSC) studies exhibited physiochemical compatibility between indomethacin and various excipients used in the tablet formulation. Stability studies carried out as per ICH Q1 A guidelines suggested the stable formulations for the tested time period of 6 months. The systematic approach of using subliming and disintegrating agents helped in achieving a stable, optimized orodispersible formulation, which could be industrially viable.
Key words: 23 factorial design, orodispersible tablet, subliming agents, superdisintegrant
INTRODUCTION
Oral administration is a preferred route administration and children younger than 5 years of age have great difficulty with, or are unable to swallow a solid dosage form. Manufacturers have developed liquid formulations for many commonly used pediatric products. The liquid dosage forms are not free of problems and often unstable and have short expiration dates. Accurate measurement and administration of prescribed dose is also a problem. Chewable tablets and sprinkle capsule formulations are well received for use in children with full dentition. Chewable tablets rely on the chewing action for their bioavailability and the sprinkle capsule formulation and dispersible tablets need to be delivered with the aid of external help. Orodispersible formulations provide solutions to the above limitations as it can be administered without any external help and when placed in mouth disintegrate and dissolve rapidly in the saliva without the need of drinking water (1,2).
Clinically, nonsteroidal anti-inflammatory drugs (NSAIDs) are the most frequently prescribed class of drugs by physicians for inflammatory disorders. Indomethacin[1-(4-Chlorobenzoyl)-5-methoxy-2-methyl-1H-indole-3-acetic acid)], a nonsteroidal anti-inflammatory drug was chosen as a model drug for formulation into a pediatric orodispersible tablet. Indomethacin is rapidly and completely absorbed from the gut following oral administration. The plasma concentration required for its antiinflammatory activity is probably less than 1 μg/ml. It undergoes extensive hepatic metabolism and both the parent substance and metabolites take part in enterohepatic circulation. Indomethacin though not preferred as a routine analgesic-antipyretic, is proven to be useful as an antipyretic in Hodgkin’s disease when fever is refractory to other agents. It has analgesic properties independent of its antiinflammatory actions. As an antiinflammatory agent, indomethacin reduces pain; swelling and tenderness of joints, increasing grip strength and decreases the duration of morning stiffness. It is also used to treat uveitis and inflammation following eye surgery (3).
Most of the marketed formulations of indomethacin are available as capsules, suspensions and sustained release tablets that address to the needs of adults and present difficulty in administration to the pediatric patients. Therefore, an attempt was made in the present study to formulate orodispersible tablets of indomethacin by a combination of sublimation and superdisintegrant addition techniques. The aim was to optimize an orodispersible formulation by 23 factorial design thereby developing a dosage form that may enhance its bioavailability, as a significant portion of the dose may be dissolved in the saliva and get absorbed in the pregastric region thus improving the extent of bioabsorption.
MATERIAL AND METHODS
Materials
Indomethacin was obtained as gift sample from Siemen Laboratories, Haryana, India. Polyplasdone XL® was obtained as gift sample from ISP Technologies, New Jersey, USA. Camphor, ammonium bicarbonate, mannitol, colloidal silicone dioxide and spray dried lactose were obtained from Ranbaxy Fine Chemicals Ltd, New Delhi, India, magnesium stearate and sodium saccharine were purchased from S.D. Fine Chem Ltd., Mumbai, India. Polyvinylpyrrolidone (PVP) was procured from Qualigens Ltd., Mumbai, India. Solvents of reagent grade and all glass double distilled water was used throughout the study.
Methods
Preparation of Orodispersible Tablet (Preliminary Trials and Factorial Design)
Accurately weighed quantities of indomethacin (pediatric dose, 50 mg/tablet, comparable to marketed formulation), subliming agents namely camphor and ammonium bicarbonate, intragranular fraction of PolyplasdoneXL® and other excipients were passed through the sieve (#20) and mixed in a glass mortar. The above blend was granulated by 10% w/v alcoholic solution of PVP and passed through sieve (#18). The granules were air dried and mixed with extra granular fraction of polyplasdoneXL®. About 1% w/w colloidal silicone dioxide was incorporated and the granules were passed through sieve (#22), lubricated with magnesium stearate and compressed on electrically operated single punch tablet press (HICON, India). The prepared tablets were subjected to volatilization at 50°C for 8 h until constant weight was achieved (to ensure complete sublimation of camphor and ammonium bicarbonate). The composition of preliminary and factorial design batches is shown in Tables I and II respectively.
Table I.
Results of Disintegration Times and Hardness for the Preliminary Trail Batches (PF1–PF4)
| Trail Batches | Excipient Added | Percentage of Excipients Added | Disintegration Time (s)a | Hardness (kg)b |
|---|---|---|---|---|
| PF1 | Volatilizing agent | 14% w/w | 79.8 ± 0.9 | 2.66 ± 0.09 |
| PF2 | Superdisintegrant incorporated intragranularly | 4% w/w | 71.7 ± 0.8 | 3.50 ± 0.08 |
| PF3 | Superdisintegrant incorporated intragranularly (2% w/w) | 4% w/w | 40.4 ± 0.4 | 3.10 ± 0.08 |
| PF4 | Volatilizing agent + superdisintegrant | 14% w/w + 4% w/w | 38.3 ± 0.6 | 2.33 ± 0.34 |
aValue represents mean ± SD of 6 determinations.
bValue represents mean ± SD of 3 determinations.
Table II.
Experimental Design (23) Used to Optimize the Formulations
| Factor Combination | Camphor (% w/w) | Ammonium Bicarbonate (% w/w) | Mannitol (% w/w) |
|---|---|---|---|
| F1 | 10 | 14 | 29 |
| F2 | 14 | 10 | 29 |
| F3 | 10 | 14 | 29 |
| F4 | 14 | 14 | 29 |
| F5 | 10 | 10 | 45 |
| F6 | 14 | 10 | 45 |
| F7 | 10 | 14 | 45 |
| F8 | 14 | 14 | 45 |
Optimization of Volatilization Time
The optimized formulations based on disintegration time and hardness values were subjected to kinetic studies. Twelve tablets of the optimized formulation were stored in uncovered glass petri plates at 50°C. Periodic samples were withdrawn for 6 h and the residual levels of ammonium bicarbonate were analyzed quantitatively by titrimetric method (4). Residual levels of camphor were determined by measuring the weight on micro analytical balance (Shimadzu AY 220, Japan) until a constant weight was obtained.
Evaluation of Tablet properties
The disintegration time of orodispersible tablets was determined by employing a modified dissolution apparatus (JP XII paddle method) (5). Nine hundred milliliters of water, maintained at 37°C ± 0.5, stirred with paddle at 100 rpm was used as the disintegration fluid. The test tablet was placed in sinker basket suspended in disintegration fluid to a depth of 6 cm from the neck of dissolution vessel. Disintegration time (n = 6) was recorded till all the fragments of the disintegrated tablet passed through the screen of the basket. The crushing strength (n = 6) of tablets was determined using a Pfizer hardness tester (HICON Enterprises, Delhi, India). Based on disintegration time and crushing strength values, optimized formulations were selected and subjected to evaluation of orodispersible tablet properties.
Evaluation of Optimized Formulations
The porosity of the tablet was determined by liquid (ethanol, 95% v/v) displacement method (6) using modified pycnometer (Jindal Scientific Industries, Ambala, India). The wetting time of the tablet (n = 6) was measured by placing five circular tissue papers of 10 cm diameter in a glass petridish of 10 cm internal diameter. Ten milliliters of water containing methylene blue (0.1% w/v) was added to the petridish (7). A tablet was carefully placed on the surface of the tissue paper and the time required for the dye to reach the upper surface of the tablet was recorded. The measurements were done in triplicate. The wetting time was recorded and water absorption ratio was calculated using the following formula (8),
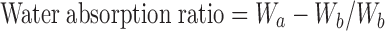 |
1 |
where, Wb is weight of tablet before absorption of water and Wa is weight of tablet after absorption of water.
In Vivo Disintegration Time
Six healthy volunteers aged between 20–24 years, from whom informed consent was obtained, were selected for determination of in vivo disintegration time. The study was duly approved by the Institutional Human Ethical Committee, K.D. Dental College, Mathura, India. One tablet was placed on the tongue of each volunteer and the time required for complete dispersion of the tablet, the mouth feel and grittiness were recorded (9).
Surface Morphology
The surface morphology of the optimized tablets before and after volatilization of camphor and ammonium bicarbonate from the tablet matrix, were studied using Jeol 6100 SEM (Jeol, Tokyo, Japan). The tablet surface was sputter coated for 10 minutes with gold by using fine coat ion sputter and examined under SEM.
Experimental Design
The 23 factorial design was implemented for the optimization of pediatric indomethacin orodispersible tablet. The dependent responses measured were disintegration time and crushing strength. Three independent factors namely concentration of camphor, concentration of ammonium bicarbonate and concentration of mannitol were set at two different levels (Table II).
Validation of the Experimental Design
In order to validate the experimental design using a polynomial equation, two parameters namely disintegration time and crushing strength were selected. A two level experimental design provides sufficient data to fit a polynomial equation, which is in the following form,
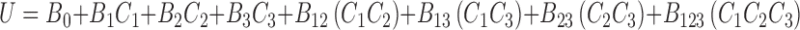 |
2 |
where, Y represents the experimental response, B0 the intercept and B1–B123 are the coefficients for the factors X1 (camphor), X2 (ammonium bicarbonate) and X3 (mannitol). The Student’s t test was conducted to examine the probability of each coefficient being equal to zero. All tests were performed at 95% level confidence level (P > 0.05). In the final model equation, only the significant factors were included. The polynomial equation was applied on the response parameters, disintegration time and crushing strength independently. The theoretical responses for disintegration time and crushing strength were calculated using software (New Statistica 10v, USA).
Drug Excipient Compatibility Study by DSC
The DSC profile of pure and physical mixtures of indomethacin and the excipients used in the formulation were recorded on Pyris Diamond DSC-4 (Perkin-Elmer, Wellesley, MA) in order to assess the compatibility of excipients used in the formulation with respect to the drug. The drug excipient ratios were selected on the basis of their ratios in experimental design and thermal behaviors were studied under normal conditions with perforated and sealed quartz pans with a nitrogen gas flow of 400 ml/min. The samples (11.47 mg for pure indomethacin, 5.45 mg for indomethacin and camphor, 4.42 mg for indomethacin and ammonium bicarbonate, and 5.80 mg for indomethacin and PolyplasdoneXL) were heated at 5°C/min over a temperature range of 40 to 260°C. The reference sample used in all the determinations was alumina with a weight of 10.5 mg.
Stability Studies
Stability studies were carried out as per ICH Q1A stability testing guidelines for zone III (10). The optimized formulations were stored in aluminum capped clear glass vials and were subjected to a storage condition of 40°C ± 2/75% RH ± 5 for 6 months. The samples were withdrawn at time intervals of 0, 1, 2, 3 and 6 months and evaluated for percentage drug content using UV spectrophotometer at 320 nm (Shimadzu-1700, Kyoto, Japan), crushing strength and in vitro disintegration time.
RESULTS AND DISCUSSION
Preliminary Trials Based on Experimental Design
Preliminary trial batches, PF1–PF4 were designed and the effects namely level of volatilizing agents, mode of superdisintegrant addition and combination thereof were studied. The trial batch PF1, comprising only volatilizing agents in the formulation showed a disintegration time of 79.8 s ± 0.9 (Table I), which does not comply with the limit of dispersion within 60 s for orodispersible tablet according to European Pharmacopoeia (11). The hardness of the prepared tablets was found 2.66 kg ± 0.09 which could present friability problems. This suggests that incorporation of volatilizing agents alone will not be sufficient for developing an acceptable orodispersible tablet. On the other hand, trial formulation PF2 with superdisintegrant alone, incorporated intragranularly exhibited a disintegration time of 71.7 s ± 0.8 and a hardness of 3.50 kg ± 0.08. The formulation exhibited improved hardness values but the disintegration time did not suffice the needs of orodispersible tablet. However, in trial formulation PF3, when the mode of superdisintegrant addition was modified [incorporated both intragranularly (2% w/w) and extragranularly (2% w/w)], the disintegration time reduced to 40.4 s ± 0.4 with a hardness of 3.10 kg ± 0.08. When compared to PF2, the hardness of PF3 remained largely unchanged with significant reduction in disintegration time. This may be due to the distribution of superdisintegrant both in the tablet as well as within the granules. Finally, the trial batch, PF4 with both subliming agent and superdisintegrant showed a disintegration time of 38.3 s ± 0.6 and a hardness of 2.33 kg ± 0.34. The least value for disintegration time observed suggested good dispersion, however, the hardness of the tablet presented friability problems. Hence, colloidal silicon dioxide, was incorporated extra granularly at a level of 1% w/w in an attempt to improve the hardness of the tablets. The effect of incorporation of colloidal silicon dioxide was evidenced by an increase in the crushing strength of the tablets from a value of 2.33 kg ± 0.34 in PF4 to 3.10 kg ± 0.08 without any change in the disintegration time. Colloidal silicon dioxide is known to restore the bonding property of the excipients thereby improving the hardness of tablets.
Evaluation of Orodispersible Tablet (Batch F1–F8)
All the formulations exhibited a disintegration time of less than 60 s except for formulation F8 that disintegrated in 62 s ± 1.7 (Table III). The crushing strength of formulated tablets varied within a wide range of 0.86–4.30 kg. For the purpose of handling, without friability problems, a hardness value of greater than 3.00 kg was selected as optimum value (Table III). Hence, the formulations F5, F6 and F8 with crushing strengths of 4.30 kg ± 0.35, 3.33 kg ± 0.18 and 3.36 kg ± 0.20 respectively, complied with the selected criteria. The formulation F8 with a disintegration time of 62 s ± 1.7 was rejected and formulations F5 and F6 with disintegration time values of 52 s ± 0.5 and 27 s ± 0.7 respectively, free from handling problems were selected as optimized formulations. The formulations F5 and F6, clearly demonstrate that with higher hardness values more time is required for disintegration. In both these formulations, mannitol, an orodispersion aid (13), is at a higher level and ammonium bicarbonate, one of the subliming agent is at a lower level. The difference in values of disintegration time for F5 and F6, can thus be attributed to the difference in the levels of camphor which is present at higher level in F6 and at lower level in F5, thus resulting in disintegration time of 27 s ± 0.7 as compared to 52 s ± 0.5 for F5.
Table III.
Hardness and Disintegration Time for F1–F8 Formulations of 23 Factorial Design of Indomethacin Orodispersible Tablets
| Formulation Code | Hardnessa (kg) | Disintegration Timea (s) |
|---|---|---|
| F1 | 2.33 ± 0.39 | 18.3 ± 0.6 |
| F2 | 1.26 ± 0.41 | 17.3 ± 0.9 |
| F3 | 2.30 ± 0.37 | 31.8 ± 0.1 |
| F4 | 0.86 ± 0.09 | 12.1 ± 0.7 |
| F5 | 4.30 ± 0.35 | 52.0 ± 0.5 |
| F6 | 3.33 ± 0.18 | 27.0 ± 0.7 |
| F7 | 1.66 ± 0.09 | 35.8 ± 1.5 |
| F8 | 3.36 ± 0.20 | 62.0 ± 1.7 |
aValue represents a mean ± SD of 6 determinations.
Optimization of Volatilization Time
It is important to optimize the time required for complete volatilization of subliming agents as any residual levels of camphor and ammonium bicarbonate in the tablets may make them unpalatable. Taste being an important criteria for acceptability of orodispersible formulation, it is essential to ensure zero levels of any component in the formulation that may contribute negatively to the organoleptic character and at the same time the component is not required in the final formulation. When percentage residual ammonium bicarbonate for F5 and F6 was plotted against time (Fig. 1), a straight line was obtained suggesting a zero order volatilization. The rate of volatilization of ammonium bicarbonate was calculated as 11.30 and 9.38 mg/h for F5 and F6 formulations respectively. The straight line when extrapolated to X-axis, where, the concentration of ammonium bicarbonate equals to zero, gives the time (TO%) required for complete volatilization of ammonium bicarbonate. The time (TO%) was determined to be 7.02 and 7.94 h for F5 and F6 formulations respectively. Similarly, for residual level determination studies of camphor, a constant weight was achieved by the optimized formulations after 6 hours of volatilization at 50°C (i.e. from 0.25 mg at the zeroth hour to 0.21 mg at the fourth hour till the seventh hour for F5 and 0.25 mg at the zeroth hour to 0.20 mg at the fifth hour till seventh hour). A constant weight value indicated complete removal of camphor, from the tablet. These kinetic studies justify an optimized volatilization period of 8 h to ensure complete removal of camphor and ammonium bicarbonate from the tablets at 50°C.
Fig. 1.

Decrease in the residual levels of ammonium bicarbonate in formulation F5 and formulation F6
Validation of the Experimental Design
The parameter disintegration time can be described by the model equation,
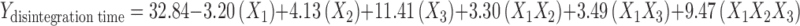 |
3 |
The negative coefficient for X1 in Eq. 3 indicates that the lower percentage of camphor contributes negatively to the disintegration time of the tablet. The theoretical response at Y50% was found to be 39 s (Table IV) and the experimental response of the extra design check point formulation was found to be 36.1 s ± 0.3 which was quite close to that of the theoretical response, indicating the validation of factorial design.
Table IV.
Predicted Response Values Calculated for Disintegration Time and Crushing Strength Using New Statistica 10 v Software
| B-Weight | Value | B-Weight Value | |
|---|---|---|---|
| Predicted values for disintegration time (seconds) | |||
| X1 | −3.20 | 0.50 | −1.60 |
| X2 | 4.13 | 0.50 | 2.06 |
| X3 | 11.41 | 0.50 | 5.70 |
| Intercept | 32.84 | Predicted | 39.01 |
| Predicted values for hardness (kg) | |||
| X1 | −0.22 | 0.50 | −0.11 |
| X2 | −0.38 | 0.50 | −0.19 |
| X3 | 0.73 | 0.50 | 0.36 |
| Intercept | 2.42 | Predicted | 3.09 |
The parameter, crushing strength can be described by the polynomial equation.
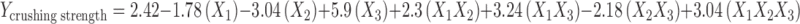 |
4 |
The Y50% theoretical was found to be 3.09 kg and the experimental response formulation showed a crushing strength value of 3.10 kg ± 0.115, which was significantly closer to the theoretical response at Y50%.
Evaluation of the Optimized Formulations
The first step in the disintegration process is the penetration of water into the tablet, consequently the tablet porosity has an important influence on disintegration. For orodispersible tablet, this factor is of extreme importance as the performance is based on rapid ingress of limited amount of saliva present in the oral cavity into the formulation, which in turn is dependent on the porous structure of the tablet matrix. Therefore, porosity of the formulated tablets was evaluated before and after volatilization of the subliming agents. An increase in porosity of up to 70.5 and 77.8% with respect to initial porosity for F5 and F6 respectively indicated a complete volatilization of subliming agents resulting in a porous matrix without friability problems (Table V).
Table V.
Wetting Time, Water Absorption Ratio, Percentage Increase in Porosity of Formulations F5 and F6
| Parameter | F5 | F6 |
|---|---|---|
| Wetting time (s) | 7.8 ± 0.3 | 7.8 ± 0.5 |
| Water absorption ratio | 1.25 ± 0.12 | 1.38 ± 0.08 |
| Percentage increase in porosity after volatilization | 70.5% | 77.8% |
The significantly lower wetting time(s) of F5 and F6, 7.8 s ± 0.3 and 7.8 s ± 0.5 can be attributed to the porous structure of the tablet and the hydrophilicity of the excipients resulting in faster water uptake. The water absorption ratio of the optimized tablets was found to be 1.25 and 1.38 for F5 and F6 respectively indicating that the formulated tablets could uptake water up to approximately 1.5 times more than there own weight, thus facilitating the process of orodispersion. The in vitro disintegration behavior without agitation of formulation F6 was recorded by digital camera (Kodak, 5.0 megapixels) and a complete disintegration was obtained within 44 s (Fig. 2). The difference in the numerical values of disintegration time observed, when compared with in vitro disintegration test may be attributed to the stirring element in the test causing faster disintegration (27 s) against the 44 s obtained without agitation.
Fig. 2.

Kinetic digital images of disintegration process of orodispersible tablet formulation F6
In Vivo Disintegration Time
Formulation F6 completely disintegrated in the saliva within 34.2 s ± 2.9 as compared to the in vitro disintegration time of 27 s ± 0.7. The in vitro disintegration time was of lower order as compared to in vivo due to the stirring element incorporated in the modified disintegration apparatus. Additionally, all the volunteers reported pleasant mouth feel and none reported grittiness.
Surface Morphology
SEM studies were carried out to estimate the extent of pore formation after volatilization of the camphor and ammonium bicarbonate from the tablet matrix. The images (Fig. 3a,b) show the formation of pores on the tablet surface that may have extended into the matrix after sublimation of the volatilizing agents, thus providing a sufficiently porous structure to facilitate rapid ingress of dispersing medium. This is evident from the magnified tablet surface images (Fig. 3c,d) of the orodispersible tablets before and after volatilization. This physical process however, did not affect the integrity and physical stability of the tablet.
Fig. 3.

SEM micrographs: prepared tablet a before volatilization (X1600), b after volatilization (X1500); tablet surface c before volatilization (×2,000), d after volatilization (×2,000)
Drug Excipient Compatability Study
DSC thermogram of pure drug exhibited a sharp endothermic peak at 155°C with a ΔH value 68.3 mJ/mg. In the presence of the excipients neither an appearance of a new peak nor a shift in DSC thermograms (Table VI), indicated absence of physical or chemical instabilities.
Table VI.
DSC Thermogram Endothermic Peaks of Indomethacin, Excipients and Combinations Thereof
| Sample | Endothermic Peaks Observed (°C) |
|---|---|
| Indomethacin | 155 (68.3 mJ/mg) |
| Camphor | 149 (179 mJ/mg) |
| Ammonium bicarbonate | 133 (1,277 mJ/mg) |
| Mannitol | 167 (237 mJ/mg) |
| PolyplasdoneXL | 65 (131 mJ/mg) |
| Magnesium stearate | 125 (7.63 mJ/mg), 106 (39.5 mJ/mg) |
| Sodium saccharine | 130 (144 mJ/mg) |
| Lactose | 213 (106 mJ/mg), 143 (66.3 mJ/mg) |
| Indomethacin +Camphor | 158 (86.9 mJ/mg) |
| Indomethacin + Ammonium bicarbonate | 158 (39.4 mJ/mg), 133 (600 mJ/mg) |
| Indomethacin + Mannitol | 166 (230 mJ/mg), 158 (66.8 mJ/mg) |
| Indomethacin + PolyplasdoneXL | 158 (68.0 mJ/mg), 66 (130 mJ/mg) |
| Indomethacin + Magnesium stearate | 157 (102 mJ/mg), 124 (7.88 mJ/mg), 106 (40.1 mJ/mg) |
| Indomethacin + Sodium saccharine | 157 (106 mJ/mg), 129 (140 mJ/mg), |
| Indomethacin + Lactose | 208 (23 mJ/mg), 158 (58.6 mJ/mg), 143 (12.3 mJ/mg) |
The values in parentheses indicate the ΔH.
Stability Studies
The optimized formulations were found to be stable, both chemically and physically (Table VII).
Table VII.
Stability Data for F5 and F6 Formulations
| Parameters | Time Interval (months) | |||||
|---|---|---|---|---|---|---|
| F5 | F6 | |||||
| 0 | 3 | 6 | 0 | 3 | 6 | |
| Disintegration time (s) | 52.0 ± 0.5 | 51.0 ± 0.4 | 50.9 ± 0.1 | 27.0 ± 0.7 | 26.0 ± 0.8 | 27.1 ± 0.3 |
| Hardness (kg) | 4.30 ± 0.35 | 4.29 ± 0.33 | 4.31 ± 0.15 | 3.33 ± 0.18 | 3.32 ± 0.29 | 3.32 ± 0.33 |
| Drug content (%) | 98.28 ± 0.29 | 98.21 ± 0.11 | 97.19 ± 0.29 | 98.13 ± 0.34 | 98.11 ± 0.12 | 97.88 ± 0.88 |
CONCLUSION
Formulations F5 and F6, formulated using a combined approach of subliming agents and superdisintegrants were identified as the optimized orodispersible formulations of indomethacin. The experimental design concluded that higher levels of both mannitol and camphor resulted in faster disintegration of the orodispersible tablets. Solid-state kinetic studies were used to optimize the volatilization time for complete removal of subliming agents.
Acknowledgement
The authors gratefully acknowledge the cooperation offered by IIT Roorkee, India for conducting SEM and DSC analysis of the formulations.
References
- 1.Banker G. S., Rhodes C. T. Modern Pharmaceutics. 2. New York, NY: Marcel Dekker; 1999. [Google Scholar]
- 2.Chang R., Guo X., Burnside B., Couch R. A review of fast dissolving tablets. Pharm. Tech. 2000;12:52–58. [Google Scholar]
- 3.Brar F. S. K. Essentials of Pharmacotherapeutics. 3. New Delhi, India: Chand; 2000. [Google Scholar]
- 4.Her Majesty’s Stationary Office . British Pharmacopoeia. London, UK: Stationary Office; 2004. [Google Scholar]
- 5.Bi Y., Sunada H., Yonezawa Y., Danjo K., Otsuka A. Preparation and evaluation of a compressed tablet rapidly disintegrating in the oral cavity. Chem. Pharm. Bull. 1996;44:2121–2127. doi: 10.1248/cpb.44.2121. [DOI] [PubMed] [Google Scholar]
- 6.Schmidt P. C., Schiermeier S. Fast dispersible ibuprofen tablets. Eur. J. Pharm. Sci. 2002;15:295–305. doi: 10.1016/S0928-0987(02)00011-8. [DOI] [PubMed] [Google Scholar]
- 7.Gohel M., Patel M., Amin A., Agrawal R., Dave R., Bariya N. Formulation design and optimization of mouth dissolve tablets of nimesulide using vacuum drying technique. AAPS Pharm. Sci. Tech. 2004;5:1–6. doi: 10.1208/pt050336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gohel M. C., Bhatt N. Formulation and evaluation of orodispersible taste masked tablets of famotidine. Pharm. BioWorld. 2005;4:75–78. [Google Scholar]
- 9.Kornblum S., Stoopak S. A new tablet disintegrating agent: crosslinked polyvinyl pyrollidone. J. Pharm. Sci. 1973;62:43–49. doi: 10.1002/jps.2600620107. [DOI] [PubMed] [Google Scholar]
- 10.ICH Q1A guidelines. Stability testing of existing drug and drug products. Available at http://www.ichguidelines.com. Accessed January 12, 2005.
- 11.Council of Europe . European pharmacopoeia. 5. Strasbourg Cedex, France: Directorate; 2005. [Google Scholar]
- 12.Bi Y., Sunada H., Danjo K., Yonezawa Y. Evaluation of rapidly disintegrating tablets prepared by a direct compression method. Drug Dev. Ind. Pharm. 1999;25:571–581. doi: 10.1081/DDC-100102211. [DOI] [PubMed] [Google Scholar]
- 13.Selection guide to excipients. http://www.signetchem.com. Accessed Feburary 14, 2005.