

Abstract
The ability of self-emulsifying drug delivery systems (SEDDS) to improve solubility, dissolution rate and bioavailability of a poorly water-soluble calcium channel blocker, nimodipine (NM) was evaluated in the present investigation. Solubility of NM in various oils, surfactants and cosurfactants was determined. The influence of the ratio of oil to surfactant + cosurfactant, pH of aqueous phase on mean globule size of resulting emulsions was studied by means of photon correlation spectroscopy. The NM loaded SEDDS selected for the in vitro and in vivo studies exhibited globule size less than 180 nm. In vitro dissolution studies indicated that NM loaded SEDDS could release complete amount of NM irrespective of the pH of the dissolution media. Pharmacokinetics of NM suspension, NM oily solution, NM micellar solution and NM SEDDS were evaluated and compared in rabbits. Relative bioavailability of NM in SEDDS was significantly higher than all the other formulations. NM loaded SEDDS were subjected to various conditions of storage as per ICH guidelines for 3 months. NM SEDDS successfully withstood the stability testing.
Key words: in vitro drug release, low oral bioavailability, nimodipine, pharmacokinetics, self-emulsifying drug delivery systems (SEDDS)
INTRODUCTION
Nimodipine (NM), isopropyl-2-methoxyethyl-1,4-dihydro-2,6-dimethyl-4-(3-nitrophenyl)-3,5-pyridine dicarboxylate, is a dihydropyridine calcium antagonist with therapeutic indications for cerebrovascular spasm, stroke and migraine (1,2). Recently, NM has been shown to be effective in ameliorating memory degeneration and preventing senile dementia in the old age (3,4). However, the clinical efficacy of NM is strongly limited by its poor water solubility (2.30 μg/ml) and low oral bioavailability (∼13%) (5,6). Apart from the poor water solubility, extensive first-pass metabolism by Cytochrome P450-3A4 (CYP3A4) isoenzymes and P-glycoprotein (P-gp) mediated efflux are also responsible for the low oral bioavailability of the NM (7). The current therapeutic scenario demands a strong need for a delivery strategy that can improve the therapeutic efficacy of NM by means of increasing its solubility or by reducing its first pass metabolism or P-gp mediated efflux.
Self-emulsifying drug delivery systems (SEDDS) have gained great importance as a promising technology to improve the bioavailability of poorly water-soluble drugs. A substantial amount of individual reports about the potential SEDDS formulations has been published during the last decade and the number is continuously increasing per year. Furthermore, successful commercialization of the products Fortovase® (Saquinavir) and Norvir® (Ritonavir) (all based on SEDDS technology) is sufficient to establish the commercial viability of this delivery strategy (8).
SEDDS are defined as isotropic mixtures of lipid, surfactant, co-surfactant and drug substance that rapidly form a fine oil-in-water (O/W) emulsion when exposed to aqueous media under conditions of gentle agitation or digestive motility that would be encountered in the gastrointestinal tract (9,10). The spontaneous formation of emulsion advantageously presents the drug in a dissolved form, and the resultant small droplet size provides a large interfacial surface area. These characteristics result in faster drug release from emulsion in a reproducible manner, which can be designed further to make the release characteristics independent of the gastro intestinal physiology and the fed/fasted state of the patient (9,10).
Oral absorption of several drugs has been enhanced by SEDDS employing single or combined mechanism (10). Additionally, the specific components of SEDDS promote the intestinal lymphatic transport of drugs which would be very useful in reducing the first pass of the drugs such as NM. Main mechanisms include increasing membrane fluidity to facilitate transcellular absorption, opening tight junction to allow paracellular transport, inhibiting P-gp and/or Cytochrome P450 (CYP450) enzymes to increase intracellular concentration and residence time by surfactants, and stimulating lipoprotein/chylomicron production by lipid (11–14). Recently, SEDDS have been shown to improve the bioavailability of Carvedilol, a poorly water-soluble β-blocker (15) that also undergoes extensive first pass metabolism like NM. The objectives of the present investigation were to design SEDDS for NM and to evaluate its potential to improve oral bioavailability of NM by carrying out in vivo studies in rabbits. The stability of NM in developed SEDDS has also been investigated in the present investigation.
MATERIALS AND METHODS
Materials
Nimodipine (NM) was a generous gift from Nivedita Chemicals Pvt. Ltd., (Mumbai, India). Transcutol (Diethylene glycol monoethyl ether), Plurol Oleique CC 497 (Polyglyceryl-6dioleate), Capryol 90 (Propylene glycol monocaprylate), Labrafil 2125 CS (Linoleoyl macroglycerides), Labrafac CC (Medium chain triglycerides), Labrasol (Caprylocaproyl macrogolglycerides), Peceol (Glyceryl monooleate), Gelucire 44/14 (Lauroyl macrogol-32 glycerides), Gelucire 33/01 (Glycerol esters of fatty acids), Gelucire 50/13 (Stearoyl macrogol-32 glycerides), hydroxypropyl methylcellulose (Colorcon Asia Pvt. Ltd., Mumbai, India); Miglyol 812 (S. Zaveri & Co., Mumbai, India), and Hard gelatin capsules (Associated Capsules, Mumbai, India) were obtained as gift samples. Tween 80, Brij 35, ammonium phosphate (AR grade) and methanol (HPLC grade) were purchased from s.d. fine chemicals (Mumbai, India). All the excipients and reagents were used as received. Double distilled water was prepared freshly whenever required.
Solubility Studies
The solubility of NM in various modified oils, surfactants and cosurfactants was determined by using shake flask method. Briefly, an excess amount of NM was added to each vial containing 1 mL of the selected vehicle i.e. either oil, surfactant or solubilizer. After sealing, the mixture was vortexed using a cyclomixer for 10 min in order to facilitate proper mixing of NM with the vehicles. Mixtures were then shaken for 72 h in an isothermal shaker (Remi, Mumbai, India) maintained at 37 ± 1°C. Mixtures were centrifuged at 5,000 rpm for 15 min, followed by filtration through membrane filter (0.22 μm, 13 mm, Pall Life sciences, Mumbai, India). The concentrations of NM were then determined by high-performance liquid chromatography (HPLC) method. No interference was observed from the excipients used to solubilize NM.
HPLC Analysis of NM
The solubility of NM in various excipients was determined by a validated reverse-phase HPLC method developed in house. The HPLC apparatus consisted of Jasco PU-2080 Plus Intelligent HPLC pump (Jasco, Japan) equipped with a Jasco UV-2075 Intelligent UV/VIS detector (Jasco, Japan), a Rheodyne 7725 injector (Rheodyne, USA), a Jasco Borwin Chromatography Software (version 1.50) integrator software and a Spherisorb ODS 2 RP-18 (4.6 mm × 250 mm and 5 μm particle size) column. The mobile phase consisted of a mixture of methanol and ammonium phosphate (0.05M) buffer pH 6.5 (60:40 v/v) at a flow rate of 1.0 mL/min that led to retention time of 8.86 min when detection was carried out at 238 nm. The assay was linear (r2 = 0.99) in the concentration range 0.1–40 μg/mL with the lowest detection limit of 10 ng/mL of NM. The method was validated with respect to accuracy and inter- and intra-day precision as per ICH guidelines and the relative standard deviation was less than 2% in both the cases.
Preparation of SEDDS
A series of SEDDS was prepared (Table I) with varying ratio of oil to surfactant + cosurfactant mixture. The ratio of oil (Gelucire 44/14) to surfactant + cosurfactant (Labrasol containing 80% Transcutol P + Plurol Oleique CC 497) was varied from 9:1 to 1:9. The ratio of surfactant to cosurfactant was maintained at 5:1 and the concentration of NM was also kept constant in all the formulations (30 mg). Briefly, NM was dissolved in the surfactant in glass vials. Oil and cosurfactant were accurately weighed into glass vials. Then, the components were mixed by gentle stirring and vortex mixing, and heated at 37°C in an incubator to obtain a homogenous isotropic mixture. The SEDDS formulations were stored at room temperature until used. Various SEDDS were assessed for robustness to dilution by diluting them 50, 100 and 1,000 times with various dissolution media namely water and buffer pH 1.2. The resulting fine emulsions were stored for 24 h and observed for any signs of phase separation and drug precipitation. Furthermore, the effect of the ratio of oil to surfactant + cosurfactant (O:SC) and pH of the dilution medium on the globule size of the various SEDDS formulations was evaluated in order to arrive at optimum formulation.
Table I.
Composition of Various SEDDS
| Ingredients | Formulation | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| S1 | S2 | S3 | S4 | S5 | S6 | S7 | S8 | S9 | |
| LTCPa + P 497b (at ratio of 5:1) | 42 | 84 | 126 | 168 | 210 | 252 | 294 | 336 | 378 |
| Gelucire 44/14 | 378 | 336 | 294 | 252 | 210 | 168 | 126 | 84 | 42 |
| NM | 30 | 30 | 30 | 30 | 30 | 30 | 30 | 30 | 30 |
All values are expressed in milligrams
aLCTP: Labrasol containing 80% Transcutol-P
bP 497: Plurol Oleique CC 497
Globule Size Analysis
SEDDS formulation, 100 mg, was diluted to 100 mL with double distilled water and buffer pH 1.2. Visual observations were made immediately after dilution for assessment of self-emulsification efficiency, phase separation, and precipitation of drug. The mean globule size and polydispersity index of the resulting emulsions was determined by photon correlation spectroscopy (Beckman N4 Coulter, Wipro, India). Measurements were obtained in duplicate at an angle of 90°. Emulsions were diluted with respective vehicles to ensure that the light scattering intensity was within the instrument’s sensitivity range. The resultant emulsions were also allowed to stand for 12 h at room temperature to assess dilution stability.
In-vitro Dissolution Test
Formulation S4 (Table I; SEDDS of NM containing 30 mg of NM) was filled in size ‘2’ hard gelatin capsules. In-vitro release profiles of SEDDS of NM were studied using USP XXIII apparatus I (Electrolab India, Mumbai, India) at 37 ± 0.5°C with a rotating speed of 100 rpm in buffer pH 1.2 and distilled water as the dissolution media (900 mL) so as to evaluate the effect of pH on in vitro dissolution. During the study, 2 mL of aliquots were removed at predetermined time intervals (10, 20, 30, 45 and 60 min) from the dissolution medium and replaced with fresh media. The amount of NM released in the dissolution medium was determined by UV spectrophotometer at λmax = 238 nm.
Bioavailability Studies
Bioavailability of NM in SEDDS was compared with NM suspension, NM oily solution and NM micellar solution. NM suspension was prepared by milling NM powder with a small amount of 2.5% (w/v) hydroxymethylcellulose (Colorcon Asia Pvt. Ltd, Mumbai, India) solution and diluted to a definite volume to yield NM concentration of 10 mg/mL. The oily solution of NM (10 mg/mL) was prepared by dissolving it in Gelucire 44/14 with the help of gentle heating. The micellar solution of NM (10 mg/mL) was prepared by dissolving it in appropriate amount of Labrasol® containing 80% Transcutol® P (LCTP). The SEDDS was prepared as per the aforementioned procedure. The chemical stability of NM in all the formulations was assessed prior to and during the course of the study by a validated HPLC method described earlier.
Animal care and handling throughout the experimental procedure was performed in accordance to the CPCSEA (Committee for the purpose of the control and supervision on experiments on animals) guidelines. The experimental protocol was approved by the Animal Ethical Committee of University Institute of Chemical Technology. Male rabbits weighing (2.5 ± 0.3) kg were used in the study. They were fasted for 24 h prior to the experiment and water was available ad lib. Rabbits (n = 5) were administered NM suspension, NM oily solution, NM micellar solution and NM SEDDS (equivalent to a 5 mg/kg of NM) by oral gavage in a crossover design. According to half-life of NM, the wash-out periods between the doses were kept for 7 days. Blood samples (4 mL) were collected from peripheral ear vein into heparinized tubes at 0, 0.16 0.33, 0.66, 1, 2, 3, 4, 5, and 6 h. Blood samples were centrifuged at 5,000 rpm for 10 min using a high speed centrifuging machine (Remi, Mumbai, India). Supernatant (plasma) in all the tubes was separated and stored at −18°C. Frozen plasma samples were thawed in the dark just prior to extraction. Extraction of NM was carried out as per the procedure described by Qian and Gallo (16). Briefly, in a 15 mL polypropylene screw-capped conical tube was added 0.5 mL of plasma followed by 0.25 mL of 0.1 N HCl. After vortex mixing for 30 s, the acidified plasma was mixed well with 0.25 mL methanol for 5 min. This mixture was then vortexed with 1 mL mixture of diethyl ether: n-hexane (1:1) for 15 min. After centrifuging at 5,000 rpm for 15 min, the organic layer was transferred to another tube and evaporated under a light stream of nitrogen. The residue was dissolved in 150 μL of mobile phase and 100 μL was injected into the chromatographic system.
HPLC Analysis of NM in Plasma
The concentration of NM in plasma was determined by a validated reverse-phase HPLC method developed in house. The details of the HPLC apparatus have already been mentioned earlier. The mobile phase consisted of a mixture of methanol:water (60:40 v/v) at a flow rate of 0.8 mL/min that led to retention time of 9.22 min when detection was carried out at 238 nm. The assay was linear (r2 = 0.99) in the concentration range 0.01–1.5 μg/mL. The method was validated with respect to accuracy and inter- and intra-day precision as per ICH guidelines and the relative standard deviation was less than 2% in both the cases.
Pharmacokinetic Data Analysis
Pharmacokinetic parameters were determined using WinNonLin Professional version 3.3 (Pharsight Corp., Mountain View, CA, USA). The area under the drug concentration–time curve from zero to 6 h (AUC0–6 h) was calculated using the trapezoidal rule. The maximal plasma concentration of drug (Cmax) and the time to reach maximum plasma concentration (Tmax) were directly obtained from plasma data. The relative bioavailability (BA) of various NM formulations to the NM suspension was calculated using the following equation:
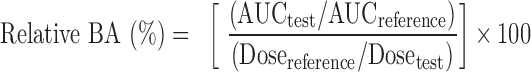 |
1 |
Statistics
The data from different formulations was compared for statistical significance by using a non-parametric Kruskal–Wallis one-way analysis of variance (ANOVA) at a level of significance P < 0.05 for the calculated parameters. All results were expressed as mean ± SD.
Stability Studies
Chemical and physical stability of NM loaded SEDDS (formulation S4) was assessed under various storage conditions namely room temperature (R. T.), 30 ± 2°C/65 ± 5% RH and 40 ± 2°C/75 ± 5% RH as per ICH guidelines. SEDDS equivalent to 30 mg of NM was filled in size ‘2’ hard gelatin capsules. Ten such capsules were packed in Alu–Alu strip packs and stored at various aforementioned storage conditions up to 3 months. Samples were removed at 0, 30, 60 and 90 days of interval and checked for NM content (by HPLC) and dissolution efficiency at 15 min (DE15%). The % dissolution efficiency at 15 min was calculated as follows
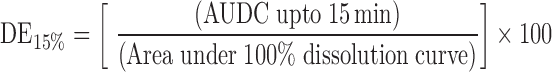 |
2 |
where AUDC = Area under dissolution curve.
RESULTS AND DISCUSSION
Solubility Studies
Solubility studies were performed to identify suitable oil, surfactant and cosurfactant that possess good solubilizing capacity for NM. Identifying the suitable oil, surfactant/co-surfactant having maximal solubilizing potential for drug under investigation is very important to achieve optimum drug loading and also to minimize the final volume of SEDDS (9,10). Solubility of NM in various oils, surfactants and surfactants is shown in Figs. 1 and 2. It is evident that NM exhibited highest solubility in Gelucire 44/14 whereas least solubility was observed in Gelucire 33/01 (Fig. 1). The dramatic difference in the solubility can be attributed to the chemical nature and HLB of the oily phases. Gelucire 44/14 is obtained by PEGylation of lauroyl glycerides and has a higher HLB value of 14. It has surfactant like properties and is well known for its good solubilization potential.
Fig. 1.

Solubility of NM in various oils; data expressed as mean ± SD (n = 3)
Fig. 2.

Solubility of NM in various co-surfactants and surfactants; data expressed as mean ± SD (n = 3). Double asterisk Cosurfactant; asterisk surfactant
Gelucire 33/01 is a mixture of glycerides of saturated C8–C18 fatty acids and has a HLB value of 1. It is usually used as oily carrier for soft or hard gelatin capsule formulations and considerably less solubilization potential. However, it is noteworthy that there was considerable difference in the solubilizing potential of Gelucire 50/13 and Gelucire 44/14 which differ by HLB value only of 1. Gelucire 50/13 is obtained by PEGylation of stearoyl glycerides and has considerably higher molecular volume as compared to Gelucire 44/14 which may be the reason for less solubilization potential. Gelucire 44/14 was selected as an oil phase for further investigation due to its solubilizing potential for NM and also due to its well established bioavailability enhancing properties (17,18) which would be helpful in case of NM. Amongst various surfactants that were screened, Labrasol exhibited highest solubilizing potential for NM whereas amongst various co-surfactants, Transcutol P and Plurol oleique CC 497 (P 497) exhibited good solubilization potential for NM (Fig. 2). For further investigations, Labrasol containing 80% transcutol P (LTCP) was chosen as a surfactant. The blending of transcutol P with the labrasol mainly helps in improving the emulsification ability of labrasol and this approach has already been employed by Kim et al. (19) Furthermore, both Labrasol and Transcutol P are known bioavailability enhancers. Plurol oleique CC 497 (P 497) was chosen as a cosurfactant due to its solubilizing potential for NM and also due to its long chain fatty acid backbone which might help in improving lymphatic transport of the NM thereby increasing its bioavailability.
Evaluation of NM Loaded SEDDS
All the SEDDS formulations (S1 to S9) were robust to the 1,000-time dilution irrespective of the dissolution media used namely water and buffer pH 1.2 and did not show any phase separation and drug precipitation after 24 h. Fig. 3 shows the effect of the ratio of oil to surfactant + cosurfactant (O:SC) and the pH of the dilution media on the mean globule size of the various SEDDS. The mean globule size remained fairly constant when the ratio of oil to surfactant + cosurfactant was between 4:6 to 7:3. The pH of the dilution medium showed negligible effect on the mean globule size of emulsions for all the SEDDS formulations. The polydispersity index values of the SEDDS formulations were less than 0.6 in all the cases except S1 (1.21) and S2 (1.34). This could be due to the high concentration of oily phase in the systems S1 and S2.
Fig. 3.

Effect of O:SC ratio and pH of dilution medium on the mean globule size of various SEDDS (Mean globule size expressed as mean, n = 3, where relative standard deviation was <5%)
Based on this study, S4 (O:SC = 6:4) was chosen for the further experiments. The formulation S4 exhibits mean globule size <175 nm, polydispersity index of 0.45 and is a classical example of type III-A systems as described by Pouton (20).
In-vitro Dissolution Study
The dissolution profile of NM loaded SEDDS in pH 1.2 and water is shown in Fig. 4. The SEDDS was found to release more than 80% of NM within first 10 min in both the dissolution media. It is evident from Fig. 4 that the release profile of NM from SEDDS was unaffected by the pH of dissolution medium.
Fig. 4.

Dissolution profiles SEDDS of NM in various dissolution media (Data expressed as mean, n = 3, where relative standard deviation was <5%)
Bioavailability Study
Pharmacokinetic parameters of NM loaded SEDDS, oily solution, micellar solution and suspension were compared in rabbits. Mean plasma NM concentration was plotted as a function of time and is shown in Fig. 5. It is evident that plasma concentration profiles of NM loaded SEDDS were considerably higher than all the other NM formulations indicating significantly greater improvement of drug absorption than the other formulations. Pharmacokinetic parameters of SEDDS, micellar solution, oily solution and suspension are shown in Table II. Calculated on AUC0–6 h, the mean relative bioavailability of NM loaded SEDDS was 460.74%, 191.03% and 153.85% as compared to NM suspension, NM oily solution and NM micellar solution respectively (P < 0.05). This clearly demonstrates the advantage of SEDDS systems over other formulations. Interestingly, NM micellar solution and oily solution showed mean relative bioavailability of 299.42% and 241.14% as compared to NM suspension (P < 0.05). This increase in the bioavailability can be attributed to the absorption enhancing properties of Gelucire 44/14 (used in oily solution), Labrasol and Transcutol P (used in micellar solution) which are well established (17, 18, 21). Furthermore, Labrasol has recently been shown to improve the absorption of digoxin which is a CYP3A substrate like NM (14). This could be the reason behind higher bioavailability of NM in micellar solution as compared to that in oily solution. Interestingly, SEDDS showed even higher bioavailability than micellar solution. This could be due to the combined effect of Gelucire 44/14, Labrasol and Transcutol P. Furthermore, the presence of P 497, (a lipophile with long chain fatty acid backbone) in the SEDDS might promote lymphatic transport of NM. In short, we believe that the improvement in bioavailability of NM when administered as SEDDS is a result of several mechanisms.
Fig. 5.

Plasma concentrations of NM after oral administration of various NM formulations to rabbits (n = 5). ** P < 0.05 when compared to NM suspension
Table II.
Pharmacokinetic Parameters of Various NM Formulations in Rabbits (n = 5)
| Parameter | NM Suspension | Oily Solution | Micellar Solution | SEDDS |
|---|---|---|---|---|
| C max (ng/mL) | 66.97 ± 4.51 | 150.96 ± 10.62 | 182.83 ± 11.02 | 282.61 ± 18.04 |
| T max (h) | 0.82 ± 0.07 | 0.91 ± 0.07 | 0.90 ± 0.06 | 0.89 ± 0.07 |
| T 1/2 (h) | 1.21 ± 0.05 | 1.11 ± 0.06 | 1.10 ± 0.08 | 1.10 ± 0.09 |
| AUC0–6 (ng h/mL) | 149.09 ± 16.24 | 359.52 ± 39.13 | 446.41 ± 41.36 | 686.82 ± 67.59 |
Stability Studies
No change in the physical parameters such as homogeneity and clarity was observed during the stability studies. No decline in the NM content (Table III) was observed at the end of 3 months indicating that NM remained chemically stable in SEDDS. Furthermore, no change in dissolution efficiency (DE15%) was observed for the NM loaded SEDDS (Table IV).
Table III.
Effect of Various Storage Conditions on NM Content in SEDDS
| Month | Storage Condition | ||
|---|---|---|---|
| Room Temperature (25 ± 3°C) | 30°C/65% RH | 40°C/75% RH | |
| 0 | 100.77 ± 0.18 | 100.77 ± 0.18 | 101.06 ± 1.29 |
| 1 | 100.31 ± 0.77 | 100.31 ± 0.77 | 99.93 ± 0.58 |
| 2 | 100.01 ± 0.39 | 100.01 ± 0.39 | 100.25 ± 0.09 |
| 3 | 100.15 ± 0.86 | 100.15 ± 0.86 | 99.51 ± 1.04 |
Data expressed as mean ± SD, n = 3
Table IV.
Effect of Various Storage Conditions on Dissolution Efficiency (DE15%) of NM SEDDS
| Month | Storage Condition | ||
|---|---|---|---|
| Room Temperature (25 ± 3°C) | 30°C/65% RH | 40°C/75% RH | |
| 0 | 94.77 ± 0.18 | 92.24 ± 0.95 | 93.57 ± 0.19 |
| 1 | 93.31 ± 0.71 | 92.58 ± 0.78 | 92.23 ± 0.28 |
| 2 | 94.01 ± 0.30 | 92.45 ± 0.18 | 92.25 ± 0.21 |
| 3 | 92.21 ± 0.26 | 93.9 ± 0.15 | 93.51 ± 1.4 |
Data expressed as mean ± SD, n = 3
CONCLUSION
Self-emulsifying drug delivery systems (SEDDS) could successfully improve the in vitro and in vivo performance of nimodipine, a P-gp substrate exhibiting poor water-solubility and high-first pass metabolism. The chemical stability of nimodipine was preserved in the investigated SEDDS. Thus, SEDDS is a novel, effective and commercially viable alternative to the currently existing nimodipine formulations.
Acknowledgements
Authors are thankful to Nivedita Chemicals Ltd., India; Colorcon Asia Pvt. Ltd., India; Gattefosse, France; S. Zaveri & Co., India; Associated Capsules, India and Abitec Corporation USA, for providing gift samples. Authors are also thankful to Mr. Abhijit Date for assisting in manuscript preparation, revision and for participating in technical discussions related to the paper.
References
- 1.Gelmers H. J. Calcium-channel blockers in the treatment of migraine. Am. J. Cardiol. 1985;55:139B–143B. doi: 10.1016/0002-9149(85)90622-8. [DOI] [PubMed] [Google Scholar]
- 2.Langley M. S., Sorkin E. M. Nimodipine. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic. Drugs. 1989;37:669–699. doi: 10.2165/00003495-198937050-00004. [DOI] [PubMed] [Google Scholar]
- 3.Zhang J. T. Altered calcium homeostasis in brain aging and senile dementia, new approach to treat Alzheimer’s disease. Yao Xue Xue Bao. 1993;28:641–646. [PubMed] [Google Scholar]
- 4.Pantoni L., Rossi R., Inzitari D., Bianchi C., Beneke M., Erkinjuntti T., Wallin T. Efficacy and safety of nimodipine in subcortical vascular dementia: a subgroup analysis of the Scandinavian Multi-Infarct Dementia Trial. J. Neurol. Sci. 2000;175:124–134. doi: 10.1016/S0022-510X(00)00300-2. [DOI] [PubMed] [Google Scholar]
- 5.Muck W., Breuel H. P., Kuhlmann J. The influence of age on the pharmacokinetics of nimodipine. Int. J. Clin. Pharmacol. 1996;34:293–298. [PubMed] [Google Scholar]
- 6.Blardi P., Urso R., De Lalla A., Volpi L., Perri T. D., Auteri A. Nimodipine: drug pharmacokinetics and plasma adenosine levels in patients affected by cerebral ischemia. Clin. Pharmacol. Ther. 2002;72:556–561. doi: 10.1067/mcp.2002.128127. [DOI] [PubMed] [Google Scholar]
- 7.Yang D., Zhu J., Zheng Y., Ge L. Preparation, characterization, and pharmacokinetics of sterically stabilized nimodipine-containing liposomes. Drug Dev. Ind. Pharm. 2006;32:219–227. doi: 10.1080/03639040500466270. [DOI] [PubMed] [Google Scholar]
- 8.Strickley R. G. Solubilizing excipients in oral and injectable formulations. Pharm. Res. 2004;21:201–230. doi: 10.1023/B:PHAM.0000016235.32639.23. [DOI] [PubMed] [Google Scholar]
- 9.Lawrence M. J., Rees G. D. Microemulsion-based media as novel drug delivery systems. Adv. Drug Deliv. Rev. 2000;45:89–121. doi: 10.1016/S0169-409X(00)00103-4. [DOI] [PubMed] [Google Scholar]
- 10.Gursoy R. N., Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed. Pharmacother. 2004;58:173–182. doi: 10.1016/j.biopha.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 11.Humberstone A. J., Charman W. N. Lipid-based vehicles for the oral delivery of poorly water soluble drugs. Adv. Drug Deliv. Rev. 1997;25:103–128. doi: 10.1016/S0169-409X(96)00494-2. [DOI] [Google Scholar]
- 12.O’Driscoll C. M. Lipid-based formulations for intestinal lymphatic delivery. Eur. J. Pharm. Sci. 2002;15:405–415. doi: 10.1016/S0928-0987(02)00051-9. [DOI] [PubMed] [Google Scholar]
- 13.Cornaire G., Woodley J., Hermann P., Cloarec A., Arellano C., Houin G. Impact of excipients on the absorption of P-glycoprotein substrates in vitro and in vivo. Int. J. Pharm. 2004;278:119–131. doi: 10.1016/j.ijpharm.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 14.Sha X., Yan G., Wu Y., Li J., Fang X. Effect of self-microemulsifying drug delivery systems containing Labrasol on tight junctions in Caco-2 cells. Eur. J. Pharm. Sci. 2005;24:477–486. doi: 10.1016/j.ejps.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 15.Wei L., Sun P., Nie S., Pan W. Preparation and evaluation of SEDDS and SMEDDS containing carvedilol. Drug Dev. Ind. Pharm. 2005;31:785–794. doi: 10.1080/03639040500216428. [DOI] [PubMed] [Google Scholar]
- 16.Qian M., Gallo J. HPLC determination of calcium channel blocker nimodipine in monkey plasma. J. Chromatogr. 1992;578:316–320. doi: 10.1016/0378-4347(92)80432-P. [DOI] [PubMed] [Google Scholar]
- 17.Barker S. A., Yap S. P., Yuen K. H., McCoy C. P., Murphy J. R., M Craig D. Q. An investigation into the structure and bioavailability of α-tocopherol dispersions in Gelucire 44/14. J. Control Rel. 2003;91:477–488. doi: 10.1016/S0168-3659(03)00261-X. [DOI] [PubMed] [Google Scholar]
- 18.Yuksel N., Karatas A., Ozkan Y., Savaser A., Ozkan S., Baykara T. Enhanced bioavailability of piroxicam using Gelucire 44/14 and Labrasol: in vitro and in vivo evaluation. Eur. J. Pharm. Biopharm. 2003;56:453–459. doi: 10.1016/S0939-6411(03)00142-5. [DOI] [PubMed] [Google Scholar]
- 19.Kim H. J., Yoon K. A., Hahn M., Park E. S., Chi S. C. Preparation and in vitro evaluation of self-microemulsifying drug delivery systems containing idebenone. Drug Dev. Ind. Pharm. 2000;26:523–529. doi: 10.1081/DDC-100101263. [DOI] [PubMed] [Google Scholar]
- 20.Pouton C. W. Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur. J. Pharm. Sci. 2000;11(Suppl. 2):S93–S98. doi: 10.1016/S0928-0987(00)00167-6. [DOI] [PubMed] [Google Scholar]
- 21.Hu Z., Tawa R., Konishi T., Shibata N., Takada K. A novel emulsifier, Labrasol, enhances gastrointestinal absorption of gentamicin. Life Sci. 2001;69:2899–2910. doi: 10.1016/S0024-3205(01)01375-3. [DOI] [PubMed] [Google Scholar]