

Abstract
To evaluate the effects of different gamma irradiation doses on PEGd,lPLA and PEG-PLGA multiblock copolymers. The behaviour of the multiblock copolymers to irradiation was compared to that of PLA, PLGA polymers. PEGd,lPLA, PEG-PLGA, PLA and PLGA polymers were irradiated by using a 60Co irradiation source at 5, 15, 25 and 50 kGy total dose. Characterization was performed on all samples before and after irradiation, by nuclear magnetic resonance (NMR), infrared absorption spectrophotometry (FTIR) and gel permeation chromatography (GPC). The effect of gamma irradiation on polymer stability was also evaluated. Results of NMR and FTIR suggest an increase in -OH and -COOH groups, attributed to scission reactions induced by irradiation treatment. Data of GPC analysis showed that the weight average molecular weight (Mw) of polymer samples decreased with increasing irradiation dose. The extent of Mw degradation expressed as percentage of Mw reduction was more prominent for polymers with high molecular weight as PEGd,lPLA and PLA. The dominant effect of gamma-irradiation on both polymer samples was chain scission. The multiblock copolymer PEGd,lPLA presented higher sensitivity to irradiation treatment with respect to PLA, likely due to the presence of PEG in the matrix. The effect of gamma irradiation continues over a much longer period of time after gamma irradiation has been performed. It is suggested that the material reacts with oxygen to form peroxyl free radicals, which may further undergo degradation reactions during storage after irradiation.
Key words: gamma-radiations, G(s) and G(x), multiblock copolymers, polymer stability
INTRODUCTION
Gamma and electron-beam irradiation are the most popular and well established methods for sterilizing polymer-based medical devices. Indeed, these methods are the best choice for the sterilization of polymeric drug delivery systems such as micro, nano-particles and implants intended for parenteral administration (1). Prior to using gamma or electron beam radiations for sterilization of healthcare products, it is essential to determine whether the radiation treatment may have any effects on the materials. Because each polymer reacts differently to ionizing radiation, it is important to verify that the maximum dose administered during the sterilization process does not adversely affect the quality, the safety and the performance of the product throughout its shelf life. It is well-known that radiosterilization introduces structural changes that may be detrimental to the integrity and performance of the polymer or polymeric system, limiting their useful working life (2–4). High-energy radiations generally produce radicals, which in turn may produce more radicals through chain scission, chain transfer quench radicals recombination, or cross linking (5–7). The stabilization process occurs during, immediately after, or even days, weeks, or months after irradiations and often results in physical and chemical cross linking or chain scission reactions (8).
The influence of radiations on the properties and performances of a polymer differs according to the polymer behaviour in terms of degradation or cross linking, and these phenomena are finally dependent on the specific sensitivity of the material to gamma radiations (9–10). In order to maintain its desirable properties, each polymer presents a fixed maximum dose of irradiation which is mainly dependent on its own chemical structure (11).
25 kGy represents the minimum absorbed dose considered adequate for the purpose of sterilizing pharmaceutical products without providing any biological validation (12–13). Lower doses can be used if a validation study has been carried out. Irradiation doses higher than 25 kGy may be used for research purposes to exasperate chain scission or cross linking phenomena. By gaining sufficient knowledge about these radiation-induced effects, manufacturers can make thoughtful choices regarding polymers to be used in sterile medical products and ensure that critical properties of materials and product performance are not compromised even after their irradiation sterilization.
Since poly α-hydroxyacids are commonly used to formulate injectable dug delivery systems, several studies have been published on the effect of gamma irradiation on these polymers. However, to the best of our knowledge no paper can be found in the literature concerning the effect of gamma irradiation on PEG-PLA, PEG-PLGA, PLA and PLGA and on drug delivery systems made of these polymers.
The present study was aimed to investigate the impact of gamma-irradiation doses on the physical–chemical properties of Poly(ethylene glycol)-co-Poly(d,l lactide) (PEGd,lPLA) and Poly(ethylene glycol)-co-Poly(d,l lactic and glycolic acid) (PEG-PLGA) multiblock copolymers. The block copolymers, consisting of biodegradable polyester and hydrophilic PEG segments, present several advantages in the preparation of drug carriers (micro-nanoparticles and implants), since the PLA segments provide rigidity, while the PEG segments confer a certain grade of hydrophilicity to the polymer that can be useful to prepare polymeric carriers designed for hydrophilic drugs (14). Moreover, the PEG segments confer stealth behaviour to the micro- or nano-particulate drug delivery systems. Stealth particles have the ability to evade the immune system and circulate after injection for longer periods of time without any clearance from immune system, increasing their effective lifespan (15–16). Aim of this work was to submit the polymers to irradiation doses up to 50 kGy, to evaluate the effect of irradiation treatment on multiblock copolymers (PEGd,lPLA and PEG-PLGA) and to compare the behaviour of the multiblock copolymers to that of PLA and PLGA polymers, respectively. The stability of the irradiated polymer, during storage in refrigerator at +4°C, 40% RH for 30, 60, 90 and 120 days was also evaluated.
MATERIALS AND METHODS
Materials
The copolymers PEGd,lPLA (Mw 130 kDa), PEG-PLGA (7525 DLG 3C-PEG 6000, Mw 22 kDa), PLA (Mw 78 kDa) and PLGA (7525 DLG 3E, 34 kDa) were purchased from Lakeshore Biomaterials, Birmingham, USA. Tetrahydrofuran (THF) and methylene chloride (CH2Cl2), analytical grade, were from Sigma Aldrich (Milan, Italy). All the reagents were of analytical grade.
Methods
The study was performed on polymers raw materials and on polymers films. The films were prepared to perform FTIR characterization.
Preparation of Polymeric Films
Polymer films were prepared by casting method as described in the literature (17). Briefly, 125 mg of polymer were solubilized in 4 mL of methylene chloride (CH2Cl2) in order to obtain a homogeneous solution (3.125% w/v). The solution was cast into Teflon moulds of 44 mm diameter. Homogeneous films were obtained after evaporation of CH2Cl2 at near 0°C. The casting at low temperature was performed in order to obtain flexible films in which the polymer was close to its amorphous state. The films were stored in desiccators for 72 h before performing their characterization.
Morphological Characterization of Polymeric Films
The polymer films were analysed for thickness and surface characteristics by scanning electron microscopy (SEM) and atomic force microscopy (AFM). An electron microscope (Jeol-Cx, Temscan, Tokyo, Japan) and Auto probe CP Research scanning probe microscope (Thermo Microscope, Sunnyvale, CA, USA) were used for SEM and AFM, respectively. AFM analyses were performed in air and under constant applied force condition (non-contact mode) with a cantilever resonant frequency of ~90 kHz and theoretical spring contact k: 3.2 Nm−1. Film samples were fixed on a silicon support, which had been mounted onto a steel disc to enable magnetic fixation under the scanning tip. Images were processed and analysed using the Image Processing Data Analysis 2.0 software provided by Thermo Microscope.
γ-Irradiation
Raw polymers and film samples were irradiated by using 60Co as irradiation source (Applied Nuclear Energy Laboratory (L.E.N.A.), University of Pavia) at 1 kGy/h dose rate. 400 mg of the polymer samples were placed in a glass container, closed with rubber stopper, and irradiated at 5, 15, 25 and 50 kGy total dose, in presence of air and at room temperature. It was checked by thermometric control that sample temperature did not significantly increase above the room temperature during the irradiation.
Nuclear Magnetic Resonance
The polymer samples were analyzed by 1H-NMR spectroscopy using a 400 MHz Bruker spectrometer with CDCl3 as the solvent. Samples of at least 10 mg were placed into glass NMR sample tubes with 1 ml of deuterated chloroformic solution (Sigma-Aldrich, Milano, Italy).
The proton NMR spectra were acquired on a NMR spectrometer (Bruker ADVANCE 400 spectrometer operating at 400 MHz, the temperature was regulated at 25°C). 1H NMR experimental parameters were as follows: an 8,012.82 Hz spectra window, a 2.0444 s acquisition time, a 6 μs (90°) pulse width, 16 transients and 30 s pulse intervals. The hydrogen of the -CH group of lactic acid unit resonates at 5.2 ppm while those of -CH3 group appears at 1.8 ppm and the hydrogens of methylene group of PEG homopolymer appears at 3.6 ppm (6). The areas under the peaks were integrated to determine the polymer composition. All spectra were performed in triplicate to estimate relative standard deviation of peak area. All data were processed using XWIN-NMR 3.1 software.
Molecular Weight Determination
The molecular weights of polymers were determined by Gel Permeation Chromatography (GPC). The GPC system consisted of three Ultrastyragel columns connected in series (7.7 × 250 mm each, with different pore diameters: 104 Å, 103 Å and 500 Å), a pump (Varian 9010 (MI), Italy), a detector Prostar 355 RI (Varian (MI), Italy), and software for computing molecular weight distribution (Galaxie Ws, ver. 1.8 Single-Instrument, Varian (MI), Italy). Sample solutions in tetrahydrofuran (THF) at a concentration of 20 mg/mL were filtered through a 0.45 µm filter (Millipore, USA) before injection into the GPC system, and were eluted with THF at 1 mL/min flow rate. The weight-average molecular weight (Mw) of each sample was calculated using monodisperse polystyrene standards (Mw 1,000–150,000 Da). The data were processed as weight average molecular weight (Mw), average molecular number (Mn).
Infrared Absorption Spectrophotometry (IR)
FTIR analyses were performed by FT-IR spectrophotometer (Perkin Elmer, UK). 20 scans for irradiated and non irradiated samples were collected for each spectrum in order to obtain an adequate signal-to-noise ratio. Measurements were performed at room temperature at a resolution of 2 cm−1. The range of acquisition was 400–4,000 cm−1.
Stability Study
The stability studies were performed over 120 days on raw polymer samples irradiated at 5, 10, 25 and 50 kGy total dose, room temperature in presence of air and at 25 kGy under vacuum. Samples of non irradiated polymers were used as controls.
All samples were stored at +4°C, with 40% RH in glass vials for all storage time. Changes in polymer molecular weight were monitored by GPC for a period of 120 days.
RESULTS AND DISCUSSION
Effects of Irradiation Doses
Figure 1 shows the characteristics of film surfaces analysed by AFM before irradiation. The films prepared with multiblock copolymers PEGd,lPLA appeared to have rough surface and some formations could be detected (Fig. 1a). PEG-PLGA film surface had similar characteristics to PEGd,lPLA film surface (image not reported). This could be attributed to the presence of hydrophilic domains based on PEG. PLA films seemed to have a crystalline matrix, as shown in Fig. 1b, whereas PLGA films presented a smooth, non porous surface and very compact structure (Fig. 1c). Film thickness resulted to be homogeneous within the range of 60–110 μm, as observed by SEM (Figures not reported).
Fig. 1.

AFM images of polymer films surface based on a PEGd,lPLA multiblock copolymer, b PLA polymer and c PLGA polymer
Polymeric films were irradiated at 5, 10, 25 and 50 kGy at RT in presence of air with 60Co source. The films prepared with PEGd,lPLA and PEG-PLGA multiblock copolymers became brittle to touch with increasing radiation dose. The embrittlement process, which can be found in irradiated films, could be attributed to their degradation as result of gamma-radiation treatment (17, 18). PLA and PLGA films maintain their texture after irradiation at all doses tested.
Visual inspection of the films samples revealed that no discrete changes in colour were observed increasing the irradiation dose. The multiblock copolymer PEG-PLGA exhibited post-irradiation a particular odour: this phenomenon could be due presumably to the high percentage of PEG in the polymeric matrix. As reported in literature, polymers that mainly exhibit post-irradiation odour are polyethilene, PVC and polyurethane (9).
NMR spectrum of PEGd,lPLA before irradiation (Fig. 2red line) showed typical signals of PEG at 3.6 ppm (-CH2-CH2-), and PLA units at 5.2 ppm (-CH-) and at 1.6 ppm (-CH3). The quartets at about 5.0 ppm and 1.7 ppm are related to the un-reacted lactic acid. From peak area ratio between PEG and PLA, the content of PEG in PEG-PLA resulted in 5 mol%. After irradiation, the peaks related to PLA are unchanged (Fig. 2blue line), while the peak related to PEG at 3.6 ppm is significantly modified in terms of shape and of area reduction. As shown in Fig. 2 and in its insert, a new peak at about 3.66 ppm and a broad peak between 3.72–3.78 ppm appear after irradiation. The peaks can be ascribed to PEG fragmentation.
Fig. 2.

NMR spectra of PEGd,lPLA multiblock copolymer before irradiation (red line) and after irradiation (blue line) at 25 kGy, at RT and in presence of air. The insert shows the magnification of NMR spectra in the range 4–3.5 ppm of PEGd,l-PLA not irradiated (red line) and irradiated (blue line) at 25 kGy
NMR spectra of PEG-PLGA obviously showed a further signal at 4.7–4.9 ppm due to glycolic acid units. The NMR spectra of the irradiated polymer showed reduced integration of signal corresponding to PEG and PGA fragments (spectra not shown). From peak area ratio between PEG and PLGA, the content of PEG in PEG-PLGA resulted to be 60 mol %.
IR spectrum of PEGd,lPLA (Fig. 3) did not show significant changes after irradiation, whereas IR spectrum of PEG-PLGA (Fig. 3) showed remarkable variations in the spectra following irradiation at 25 and 50 kGy. PEG-PLGA IR spectrum (Fig. 3) shows the characteristic absorption bands in the 1,300–1,500 cm−1 region: a C-H bending vibration of methyl group at 1,400–1,450 cm−1 and an additional C-H vibration of methylene group at 1,430 cm−1.
Fig. 3.

FT-IR spectra of PEGd,lPLA and PEG-PLGA multiblock copolymers before irradiation (blue line) and after irradiation at 25 kGy (red line), 50 kGy (green line) at RT and in presence of air
Moreover, a significant increase of the band at 3,000–4,000 cm−1 related to the -O-H stretching of the hydroxyl and carboxy functions was observed. The remarkable increase could be attributed to scission reactions induced by irradiation treatment that lead to the generation of additional carboxylic and hydroxyl end-groups in the polymer chains. Radiation usually affects polymers in two manners: (1) chain scission mechanism, that is a random cleavage of bonds, which reduces the polymer molecular weight, and (2) cross-linking mechanism of polymer molecules, which results in the formation of large three-dimensional molecular networks. Most often, the mechanisms occur as polymers are submitted to ionizing radiation, but frequently one mechanism predominates within a polymer (6–8).
The scission yield G(s) and cross linking yield G(x) are important characteristics of polymer radiation sensitivity. The radiation chemical yields for chain scission G(s) and cross-linking G(x) are defined as the number of such reactions per 100 eV of absorbed energy. G(s) and G(x) determine the extent of chain scission or cross-linking during irradiation and can be calculated from the following equations (19–23):
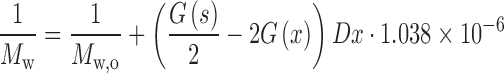 |
1 |
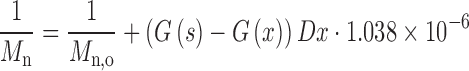 |
2 |
where Mw,0 and Mn,0 are the weight and number average molecular weights of non-irradiated samples. Mw and Mn are the corresponding values following exposure to irradiation dose, D. A ratio of G(s)/G(x) greater than 4 indicates that chain scission is the dominant mechanism within polymer, while values lower than 4 indicates that cross-linking mechanism prevails (22).
The values of G(s) and G(x) for PEGd,lPLA, PLA, PEG-PLGA and PLGA polymer samples are obtained using Equations (1) and (2), and tabulated in Table I. The results indicate that chain scission entity G(s) decreases with increasing dose, while G(s)/G(x) ratio appears to show dose independent behaviour. The G(s)/G(x) ratio >4 in Table I illustrates always the dominance of chain scission over cross-linking reactions in irradiated polymer samples irradiated at 5, 15, 25 and 50 kGy total dose.
Table I.
Chain Scission Yield G(s) and Cross Linking Yield G(x) of Polymer Samples Irradiated at 5, 15, 25 and 50 kGy
| Polymer | Doses (kGy) | G(s) | G(x) | G(s)/G(x) |
|---|---|---|---|---|
| PEGd,lPLA | 5 | 2.963 | 0.585 | 5.064 |
| 15 | 1.857 | 0.309 | 6.009 | |
| 25 | 0.606 | 0.080 | 7.575 | |
| 50 | 0.412 | 0.051 | 8.078 | |
| PLA | 5 | 1.802 | 0.236 | 7.635 |
| 15 | 1.406 | 0.168 | 8.369 | |
| 25 | 0.691 | 0.066 | 10.469 | |
| 50 | 0.683 | 0.096 | 7.114 | |
| PEG-PLGA | 5 | 10.216 | 0.621 | 16.450 |
| 15 | 5.200 | 0.322 | 16.149 | |
| 25 | 2.743 | 0.213 | 12.877 | |
| 50 | 1.487 | 0.129 | 11.527 | |
| PLGA | 5 | 6.291 | 0.671 | 9.375 |
| 15 | 3.016 | 0.280 | 10.77 | |
| 25 | 2.021 | 0.213 | 9.488 | |
| 50 | 0.863 | 0.073 | 11.82 |
Weight average molecular weights of the polymers tested, as determined by GPC analysis, before and after irradiation at 5, 15, 25 and 50 kGy total dose, are reported in Fig. 4. The values obtained for not treated polymers at time 0 were significantly lower than those given by the manufacturer because of the different solvent (THF instead of CHCl3) and calibration method (universal calibration method) used in GPC analysis (8). All polymers showed Mw reduction upon treatment, proportional to irradiation doses. The initial Mw of PEGd,lPLA and PLA polymer samples (130 and 78 kDa, respectively) seemed to be strongly influenced by the sensitivity of polymer towards gamma-irradiation (Fig. 4); in any case the Mw decay was higher for the multiblock copolymer PEGd,lPLA compared to the related homopolymer PLA. The higher sensitivity of the multiblock copolymer PEGd,lPLA with respect to PLA could be due to the presence of PEG in the matrix: at 50 kGy the Mw decays for PEGd,lPLA and PLA were 53.43 and 49.04%, respectively. The effect could be a consequence of increased polymer chain mobility due to the presence of PEG domains in the polymer chain. PEGs segments could imply a geometrical mismatch of the reacting centers minimizing cross-linking reactions.
Fig. 4.

Mw changes against irradiation dose (5, 15, 25 and 50 kGy) of PEGd,lPLA, PEG-PLGA multiblock copolymers and PLA, PLGA polymers
Average Mw decrease was of 26.66 and 28.8% for PEG-PLGA and PLGA respectively. GPC chromatogram related to PEG-PLGA and PLGA polymers did not show remarkable changes in the shape of peak after irradiation. Several authors described higher degradation, upon irradiation, for high molecular weight polymers (24, 25). This behaviour suggested that the degradation reactions induced by gamma irradiation process on a polymer depend on cleavage mechanism. Kissel et al. proposed both random chain scission and unzipping mechanism (26, 27). Volland et al. (26) observed that the content of monomers and oligomers significantly increased in irradiated polymers and that the number average molecular weight was more sensitive than weight average molecular weight towards gamma-irradiation. They hypothesized a cleavage mechanism which primary affected the terminal groups of polymer chains, hence the denomination of “unzipping mechanism”, causing a faster decay of the Mn compared to Mw (24). The chain flexibility of the polymer decreased with increasing of Mw and in these conditions the primary irradiation products could recombine with others or undergo further reactions depending on lifetime of the free radicals formed (24).
Figure 4 highlights also that the extent of weight average molecular weight decrease differs for samples irradiated below and above 25 kGy. The initial irradiation doses, up to 25 kGy, results in an important decrease in Mw, consequently, a more steady decay in molecular weight was detected for 50 kGy. This difference could be due to two different radiation-induced chain scission mechanisms below and above 25 kGy. The initial decay in Mw is due to the backbone main chains scission, where long polymeric backbone chains break into shorter chains. The energy from gamma-rays exceeds the attractive forces between atoms (22). This happens because the excited states dissipate part of the excess energy by bond scission, within both the amorphous and crystalline domains. However, at higher radiation doses (50 kGy), hydrogen abstractions become the main radiation-induced scission mechanism due to higher oxygen diffusivity in the amorphous regions. The alkyl free radicals in the amorphous domains react with oxygen to form peroxyl free radicals. The peroxyl radicals cause chain scissions, within the amorphous region and the crystal domain surface, through hydrogen abstraction (2).
Figure 5 plots GPC results expressed as percentages of Mw and Mn reduction of multiblock copolymers (PEGd,lPLA and PEG-PLGA), versus irradiation doses. At 50 kGy the Mw and Mn decay of PEGd,lPLA was 53.42 and 55.46%, while Mw and Mn decrease of PEG-PLGA was 26.66 and 31.47%, respectively. The results obtained from this study suggested that with shorter chain lengths the radicals lifetime increases and competitive reactions become more prominent.
Fig. 5.

Reduction (%) of Mw and Mn of PEGd,lPLA, PEG-PLGA multiblock copolymers irradiated at 25 kGy at RT and in presence of air
Since the Mn reduction (%) of the multiblock copolymers was bigger than the Mw reduction at all the irradiation doses, it could be possible to suppose that degradation reaction induced by gamma irradiation is based predominantly on unzipping mechanism.
Stability Study
Irradiated polymers are susceptible to continuous changes in physical and chemical properties for long after irradiation time. Therefore, it is highly probable that post-irradiation effects appear due to the entrapped radicals, influencing in vivo performance of the polymer.
To examine the stability of the polymer samples after irradiation in presence of air at 5, 15, 25 and 50 kGy dose, each polymer sample was subsequently stored for 4 months (120 days) under controlled temperature (+4°C) and relative environmental humidity (40% RH). The changes in molecular weight (ΔMw %), as a function of storage time, were detected by GPC on irradiated samples.
Figure 6 plots the GPC results obtained on irradiated polymer samples in terms of Mw reduction percentage. As shown in the four graphs (Fig. 6a,b,c and d), Mw always decayed along this storage time. The Mw decay entity after irradiation did not seem to be dependent on initial irradiation dose. Polymer samples irradiated at lower irradiation dose (5 kGy) after 30 days of storage presented Mw reduction % higher than the samples at time zero. A further increase of Mw reduction was detected only for PEG-PLGA multiblock copolymer, while for PEGd,lPLA, PLA and PLGA polymer samples the percentage values observed at 30th day were kept in the range of 15 and 40% up to day 120th (Fig. 6a).
Fig. 6.

Changes in weight average molecular weight (Mw reduction %) of PEGd,lPLA (♦), PEG-PLGA (●), PLA (▲) and PLGA (□) polymers irradiated at a 5, b 15, c 25 and d 50 kGy upon storage at +4°C and 40% RH for 120 days
The Mw decay calculated for polymer samples irradiated at 15 kGy was not remarkable for PEGd,lPLA, PLA and PLGA. PEG-PLGA showed an important increase of Mw decay after 30 and 60 days of storage. No variations were detectable up to day 120th (Fig. 6b). After 120 days of storage the Mw reduction was of about 40% and comparable to PEGd,lPLA Mw reduction.
At 25 kGy the Mw decay was low up to day 60th for PLA, PEGd,lPLA and PEG-PLGA than a faster decrease was observed between 60th and 90th day for PEGd,lPLA, PLA and PEG-PLGA. PLGA polymer sample showed a slight increase at day 60th, no variations of % Mw were observed up to 120th day (Fig. 6c).
PEGd,lPLA, PEG-PLGA and PLGA polymer samples irradiated at 50 kGy showed a gradual reduction of Mw during the storage time: PEGd,lPLA presented a Mw reduction of about 70% at 120th day, against 50% of Mw reduction for PEG-PLGA and PLGA. PLA homopolymer sample did not show a detectable variation of Mw during the storage time: a Mw reduction of 50% was observed up to 120th day (Fig. 6d).
The observations could be related to the number of free radicals formed during irradiation treatment, which are linearly related to the absorbed dose and to the oxidation reactions. The oxidation reaction kinetic depended to the diffusion of the oxygen among free radicals in the polymeric matrix (6).
The effect of ionizing energy continues over a much longer period of time after gamma irradiation treatment, allowing oxygen molecules to permeate into depleted areas of the material resulting in a greater degree of chain scission. The alkyl free radicals generated by irradiation process in the polymer matrix may react with oxygen to form peroxyl free radicals (22, 28).
The radicals may further undergo some reactions during storage after irradiation, resulting in remarkable alterations of the physical properties of irradiated polymers. The slow decrease observed in the stability study could be attributable to the time needed for permeation and diffusion of oxygen through polymer chains. Since oxygen concentration differed in the polymer matrix by location (i.e., higher percentage of oxygen is detectable near the external surface) during the stability study it should be observed an increase of the Mw reduction during storage time proportional to the penetration of oxygen inside the polymeric matrix.
CONCLUSIONS
The multiblock copolymers PEGd,lPLA and PEG-PLGA degrade through chain scission when exposed to radiations. Oxygen molecules which permeate the polymer matrix and free radicals formed by irradiation treatment can promote changes in the structure of the polymer, which continues with time. Since medical devices based on multiblock copolymers are intended for long-lasting applications in the body, the properties changes caused by γ irradiation must be kept in consideration.
References
- 1.Martinez-Sancho C., Herrero-Vanrell R., Negro S. Study of gamma-irradiation effects on acyclovir poly(D,L-lactic-co-glycolic)acid microspheres for intravitreal administration. J. Control Release. 2004;99:41–52. doi: 10.1016/j.jconrel.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 2.Sintzel M. B., Merkli A., Tabatabay C., Gurny R. Influence of irradiation sterilization on polymers used as drug carriers—a review. Drug Dev. Ind. Pharm. 1997;23(9):857–879. doi: 10.3109/03639049709148693. [DOI] [Google Scholar]
- 3.Hausberger A. G., Kenley R. A., DeLuca P. P. Gamma irradiation effects on molecular weight and in vitro degradation of poly(dl-lactide-co-glycolide) microparticles. Pharm. Res. 1995;12(6):851–856. doi: 10.1023/A:1016256903322. [DOI] [PubMed] [Google Scholar]
- 4.Montanari L., Cilurzo F., Selmin F., Conti B., Genta I., Poletti G., Orsini F., Valvo L. Poly(lactide-co-glycolide) microspheres containing bupivacaine: comparison between gamma and beta irradiation effects. J. Control Release. 2003;90(3):281–290. doi: 10.1016/S0168-3659(03)00153-6. [DOI] [PubMed] [Google Scholar]
- 5.Bushell J. A., Claybourn M., Williams H. E., Murphy D. M. An EPR and ENDOR study of γ and β-radiation sterilization in poly(lactide-co-glycolide) polymers and microspheres. J. Control Release. 2005;110:49–57. doi: 10.1016/j.jconrel.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 6.Dorati R., Genta I., Montanari L., Buttafava A., Faucitano A., Conti B. The effect of γ-irradiation on PLGA/PEG microspheres containing ovalbumin. J. Control Release. 2005;107:78–90. doi: 10.1016/j.jconrel.2005.05.029. [DOI] [PubMed] [Google Scholar]
- 7.Faucitano A., Buttafava A., Montanari L., Cilurzo F., Conti B., Genta I., Valvo L. Radiation induced free radical reactions in polymer/drug systems for controlled release: an EPR investigation. Radiat. Phys. Chem. 2003;67:61–72. doi: 10.1016/S0969-806X(02)00404-8. [DOI] [Google Scholar]
- 8.Montanari L., Costantini M., Signoretti E. C., Valvo L., Santucci M., Bortolomei M., Fattibene P., Onori S., Faucitano A., Conti B., Genta I. Gamma-irradiations effects on poly(d,l-lactide-co-glycolide) microspheres. J. Control Release. 1998;56(2):219–229. doi: 10.1016/S0168-3659(98)00082-0. [DOI] [PubMed] [Google Scholar]
- 9.K. J. Hemmerich. Radiation sterilization—polymer materials selection for radiation-sterilized products. MDDI Feb. 2000: p. 78.
- 10.Haugen H. J., Brunner M., Pellkofer F., Aigner J., Will J., Wintermantel E. Effect of different γ-irradiation doses on cytotoxicity and material properties of porous polyether-urethane polymer. J. Biomed. Mater. Res. B Appl. Biomater. 2006;80B(2):415–423. doi: 10.1002/jbm.b.30612. [DOI] [PubMed] [Google Scholar]
- 11.Claybourn M., Gray H., Murphy D. M., Purnell I. J., Rowlands C. C. Electron magnetic resonance study of gamma-irradiated poly(lactide-co-glycolide) microspheres. J. Control Release. 2003;91:431–438. doi: 10.1016/S0168-3659(03)00269-4. [DOI] [PubMed] [Google Scholar]
- 12.The use of ionizing radiation in the manufacture of medicinal products European Guidelines 3AQ4a.
- 13.Gèze A., Venier-Julienne M. C., Cottin J., Faisant N., Benoit J. P. PLGA microsphere bioburden evaluation for radiosterilization dose selection. J. Microencapsul. 2001;18(5):627–636. doi: 10.1080/02652040010019424. [DOI] [PubMed] [Google Scholar]
- 14.Y. Y. Huang, and T. W. Chung. Microencapsulation of gentamicin in biodegradable PLA and/or PLA/PEG copolymer. J. Microencapsul. 18(4):457–465 (2001). [DOI] [PubMed]
- 15.Quesnel R., Hildgen P. Synthesis of PLA-b-PEG multiblock copolymers for stealth drug carrier preparation. Molecules. 2005;10:98–104. doi: 10.3390/10010098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Panoyan T., Quesnel R., Hildgen P. Injectable nanospheres from a novel multiblock copolymer: cytocompatibility, degradation and in vitro release studies. J. Microencapsul. 2003;20(6):745–758. doi: 10.1080/02652040310001599760. [DOI] [PubMed] [Google Scholar]
- 17.Fayolle B., Audouin L., Verdu J. Radiation induced embrittlement of PTFE. Polymer. 2003;44:2773–2780. doi: 10.1016/S0032-3861(03)00116-2. [DOI] [Google Scholar]
- 18.Fayolle B., Colin X., Audouin L., Verdu J. Mechanism of degradation induced embrittlement in polyethylene. Polym. Degrad. Stab. 2007;92:231–238. doi: 10.1016/j.polymdegradstab.2006.11.012. [DOI] [Google Scholar]
- 19.Nakane K., Hata Y., Morita K., Ogihara T., Ogata N. Porous poly(L-lactic acid)/poly(ethylene glycol) blend films. J. Appl. Polym. Sci. 2004;94(3):965–970. doi: 10.1002/app.20959. [DOI] [Google Scholar]
- 20.Cleek R. L., Ting K. C., Eskin S. G., Mikos A. G. Microparticles of poly(d,l-lactic-co-glycolic acid)/poly(ethyleneglycol) blends for controlled drug delivery. J. Control Release. 1997;48:259–268. doi: 10.1016/S0168-3659(97)00052-7. [DOI] [Google Scholar]
- 21.Moad C. L., Winzor D. J. Quantitative characterization of radiation degradation in polymers by evaluation of scission and cross-linking yields. Prog. Polym. Sci. 1998;23:759–813. doi: 10.1016/S0079-6700(97)00041-5. [DOI] [Google Scholar]
- 22.Devasahayam S., Hill D. J. T., Whittaker A. K. G values for scission and crosslinking on γ-radiolysis of Ultem at 303K. High Perform Polym. 2003;15:259–267. doi: 10.1177/0954008303015003003. [DOI] [Google Scholar]
- 23.Nugroho P., Mitomo H., Yoshii F., Kume T. Degradation of poly(L-lactic acid) by γ-irradiation. Polym. Degrad. Stab. 2001;72:337–343. doi: 10.1016/S0141-3910(01)00030-1. [DOI] [Google Scholar]
- 24.Loo J. S. C., Ooi C. P., C Boey F. Y. Degradation of poly(lactide-co-glycolide) (PLGA) and poly(L-lactide) (PLLA) by electron beam radiation. Biomaterials. 2005;26:1359–1367. doi: 10.1016/j.biomaterials.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 25.Sen M., Uzun C., Kantoglu Ö., Erdogan S. M., Deniz V., Guven O. Effect of gamma irradiation conditions on the radiation-induced degradation of isobutylene-isoprene rubber. Nucl. Instrum. Methods Phys. Res. B. 2008;203:480–484. [Google Scholar]
- 26.Volland C., Wolff M., Kissel T. The influence of terminal gamma sterilization on captopril containing poly(D,L-lactide-co-glycolide) microspheres. J. Control. Release. 1994;31:293–305. doi: 10.1016/0168-3659(94)90012-4. [DOI] [Google Scholar]
- 27.Mohr D., Wolff M., Kissel T. Gamma irradiation for terminal sterilization of 17β-estradiol loaded poly(D,L-lactide-co-glycolide) microparticles. J. Control. Release. 1999;61:203–217. doi: 10.1016/S0168-3659(99)00118-2. [DOI] [PubMed] [Google Scholar]
- 28.Montanari L., Cilurzo F., Valvo L., Faucitano A., Buttafava A., Groppo A., Genta I., Conti B. Gamma irradiation effects on stability of poly(lactide-co-glycolide) microspheres containing clonazepam. J. Control. Release. 2001;75:317–330. doi: 10.1016/S0168-3659(01)00401-1. [DOI] [PubMed] [Google Scholar]