

Abstract
The aim of this study was to investigate a newly synthesized dextrin–zidovudine (AZT) conjugate designed as a sustained release prodrug of AZT for parenteral administration. AZT was first reacted with succinic anhydride to form a succinoylated AZT which was subsequently coupled with dextrin to yield the dextrin–AZT conjugate. The structure of the conjugate was characterized by FT-IR and 1H-NMR spectroscopy. The drug content of the conjugate was 18.9 wt.%. The release in vitro of free AZT and succinoylated AZT was investigated in buffer solutions at pH 5.5 and 7.4 and in human plasma. AZT and succinoylated AZT release from the conjugate was 1.4% (pH 5.5), 41.7% (pH 7.4) and 78.4% in human plasma after 24 h. Release was complete in human plasma after 48 h. A pharmacokinetic study in rats following intravenous administration of the conjugate showed prolonged plasma levels of AZT compared to free AZT. The use of the conjugate extended the plasma half-life of AZT from 1.3 to 19.3 h and the mean residence time from 0.4 to 23.6 h. Furthermore, the conjugate provided a significant greater area under the plasma concentration-time curve and reduced the systemic clearance of AZT. This study suggested the potential of this novel dextrin–AZT conjugate as a new intravenous preparation of AZT.
Key words: AZT, dextrin, polymeric prodrug, sustained release, zidovudine
INTRODUCTION
Zidovudine (AZT) was the first clinically approved nucleoside reverse transcriptase inhibitor widely used for the treatment of acquired immunodeficiency syndrome (AIDS) caused by the human immunodeficiency virus (HIV). It has been used in many anti-HIV combination therapies including the suppression of HIV transmission from mother to fetus and the post-exposure prophylaxis of healthcare workers following exposure to HIV (1–8).
The clinical limitations of AZT are its short plasma half-life (approximately 1 h), and its dose-dependent toxicities (9,10), especially granulocytopenia and anemia, often found in AZT-treated patients (11–13). Its short plasma half-life demands frequent administration of high doses to maintain therapeutic drug levels. Current oral and intravenous (I.V.) AZT dosage forms lead to fluctuations of plasma drug concentration with an initial high concentration that increases the risk of hematological toxicities, and subsequent low drug levels that are below the therapeutic threshold. Oral dosage forms have the disadvantage that AZT undergoes extensive first pass metabolism thus only 64% of the dose reaches blood circulation (13,14) and the absorption of AZT from gastrointestinal tract is variable patient to patient (15). I.V. therapy is used in patients that cannot receive oral medication, such as pregnant women during labor and delivery (16). The available I.V. preparation is in the form of solution for infusion. It is infused into the vein over 1 h six times a day (every 4 h) at a dose of 1–2 mg/kg body weight. After injection, it is rapidly excreted in the urine as metabolized and unchanged forms (16). There is no sustained release I.V. preparation of AZT available on the market. In our research, a sustained release I.V. dosage form of AZT was formed to reduce the frequency of administration. The sustained release of AZT may allow dose reduction and smaller fluctuations in drug levels, using here the strategy of a polymer–drug conjugate (a polymeric AZT prodrug).
Polymer–drug conjugates employ a water soluble, polymeric carrier as a platform to covalently bind drugs via a spacer which is designed to allow drug release through simple hydrolysis or enzymatic cleavage (17–21). Ideal polymeric carriers are water-soluble, non-toxic, non-immunogenic, easily chemical manipulated and available at low cost (22). Dextrin is a promising polymer as it has these characteristics. It is biodegradable so it does not accumulate in long-term use. It has been already used clinically as a peritoneal dialysis solution (23–25). Dextrin is a natural, water-soluble, linear polymeric carrier containing primarily α-1,4 linkages with small amounts of branching α-1,6 linkages prepared by acid or enzymatic hydrolysis of starch (23,26). Its chemical structure is also suitable for conjugation with drugs as it has hydroxyl groups, providing reaction sites that can be used to attach bioactive molecules. Dextrin modified by attachment of sulfate groups at its hydroxyl groups (sulfated dextrin) has anti-HIV activity and anti-coagulant activity which results in thrombocytopenia, an increase in bleeding time and a decrease in platelet count, toxicities that limit the use of sulfated dextrin (and other sulfated polysaccharides) by the I.V. route (27–29). Studies in HIV-infected patients showed the failure of I.V. sulfated dextran to reduce viral titer (serum p24 levels) and the high toxicity was unacceptable (29). As a consequence we used non-sulfated dextrin for conjugation with AZT.
Our aim was to investigate a new dextrin–AZT conjugate for prolonging AZT blood levels and improving the pharmacokinetics of AZT. A dextrin–AZT conjugate was synthesized and characterized. The in vitro drug release was determined in buffer solutions at pH 5.5 and 7.4 chosen to mimic the intracellular and extracellular pH, and in human plasma. The hemolytic activity of the dextrin–AZT conjugate was examined to judge the safety of the conjugate. The pharmacokinetic study in rats compared the dextrin–AZT conjugate and free drug following I.V. administration.
MATERIALS AND METHODS
Materials
AZT originated from Samchully Pharmaceuticals (Seoul, South Korea) and was obtained from The Government Pharmaceutical Organization, Bangkok, Thailand. Dextrin (weight-average molecular weight of 6,600 g/mol with the weight-average molecular weight to number-average molecular weight ratio being 1.8, determined by gel permeation chromatography), 4-dimethylaminopyridine (DMAP), dry tetrahydrofuran (THF), poly(ethyleneimine) (PEI), dextran and phosphate buffered saline (PBS) were purchased from Sigma Chemical (St. Louis, MO, USA). Succinic anhydride was purchased from Aldrich Chemical (Milwaukee, Wl, USA). N,N′-dicyclohexylcarbodiimide (DCC) was from Merck (Hohenbrunn, Germany). 1-Hydroxybenzotriazole (HOBt) was from Epochem (Shanghai, China). Triethylamine was from Carlo Erba Reagent (Rodano, MI). Dry N,N′-dimethylformamide (DMF) and dry dimethylsulfoxide (DMSO) were from Sigma-Aldrich Chemicals (Steinheim, Germany). 2, 5-Dihydroxybenzoic acid (DHB) was from Bruker Daltonics (Leipzig, Germany). Methanol (HPLC grade) and acetonitrile (HPLC grade) were from Fisher Scientific UK (Leicestershire, UK). Citric acid and di-sodium hydrogen orthophosphate (Na2HPO4) were from Ajax Finechem (Auckland, New Zealand). SpectraPor membranes (MWCO = 2,000) were obtained from Spectrum Laboratories (Dominguez, CA, USA). Sephadex LH-20 was from Amersham Biosciences (Uppsala, Sweden). All other chemicals and solvents were analytical grade. Human blood and plasma of healthy volunteer blood donors were supplied by the Thai Red Cross Society (Bangkok, Thailand). The protocol was approved by the Ethics Committee of Chulalongkorn University.
Synthesis of Succinoylated AZT
Succinoylated AZT was synthesized by a modification of the reported method (30). Briefly, AZT (1.976 mmol) was reacted with succinic anhydride (3.163 mmol) in the presence of triethylamine (2.570 mmol) in 25 ml of dry THF at 69°C. The progress of reaction was followed by thin layer chromatography (TLC) (chloroform/methanol, 9:1, vol/vol) and after 6 h, succinic anhydride (0.988 mmol) and triethylamine (1.383 mmol) were added again and the reaction was left at 69°C for 6 h. After reaction, the solvent was evaporated and the residue was dissolved in distilled water (50 ml) and then adjusted to pH 2 by 1M HCl. The solution was transferred to a separating funnel and extracted with ethyl acetate several times. The ethyl acetate phase was collected and dried over anhydrous sodium sulfate overnight. The organic phase was filtered and evaporated. The residue was purified by flash column packed with Sephadex LH-20 and eluted with mixed solvent (chloroform/methanol, 9:1, vol/vol) (yield ∼80%). The white residue was characterized by Fourier transform-infrared spectroscopy (FT-IR) (Perkin Elmer FT-IR Spectrometer Spectrum 2000) and 1H nuclear magnetic resonance spectroscopy (1H-NMR) (Avance DPX-400 at 400 MHz spectrometer).
Synthesis of the Dextrin–AZT Conjugate
Dextrin (0.976 mmol) was dissolved in 17 ml of dry DMF and 3 ml of dry DMSO at 50°C with agitation overnight. The solution of succinoylated AZT (0.391 mmol) and DCC (1.562 mmol) in 10 ml of dry DMF was stirred for 15 min under nitrogen. The solution of HOBt (1.172 mmol) in 4 ml of dry DMF was added and the reaction was left for 30 min. The overnight-prepared dextrin solution and DMAP (0.976 mmol) were added to the reaction mixture. The reaction was left at room temperature, with stirring under nitrogen for 48 h. The precipitate of dicyclohexylurea was then filtered, the filtrate collected and the solvent removed and the residue precipitated in vigorously stirring diethyl ether. The precipitate was collected and dissolved in distilled water (50 ml). The impurity was successively extracted with ethyl acetate (4 × 40 ml) and chloroform (4 × 40 ml). The aqueous phase was then dialyzed against distilled water. The dextrin–AZT conjugate solution was dried using a freeze-drying technique (yield 87%). The white residue was characterized by FT-IR, 1H-NMR and matrix-assisted laser desorption/ionization time-of-flight mass spectroscopy (MALDI-TOF-MS). The percentage molar content of linked AZT was estimated by UV and 1H-NMR. For UV analysis, the calibration curve of AZT was used to calculate AZT loading. For 1H-NMR analysis, the integrated signals of anomeric protons of dextrin at 4.60 to 5.45 ppm (as internal reference signal) were compared with the signals of protons of AZT at 1.66 ppm (CH3–C5), 5.94 ppm (H–C1′) and 7.28 ppm (H–C6). One unit of dextrin monomer contains one anomeric proton, therefore the ratio of anomeric proton of dextrin to observed protons of AZT is 1:5. The calculation from 1H-NMR analysis follows (Eq. 1):
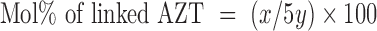 |
1 |
where x is the sum of the integrated areas of protons of AZT at 1.66 ppm (CH3–C5), 5.94 ppm (H–C1′) and 7.28 ppm (H–C6) and y is the integrated area of anomeric protons of dextrin at 4.60 to 5.45 ppm.
MALDI-TOF-MS Analysis
Dextrin or the dextrin–AZT conjugate (0.5 mg) was dissolved in 1 ml of nanopure water as a sample solution. A saturated solution of 2, 5-dihydroxybenzoic acid (DHB) prepared in acetonitrile: water (1:1, vol/vol) was used as a matrix solution. The sample solution (1 μl) was mixed with the matrix solution (5 μl). An aliquot of the mixture solution (1 μl) was then deposited on an MTP AnchorChip target (Bruker Daltonics) and air-dried. Samples were analyzed using a Reflex IV MALDI-TOF mass spectrometer (Bruker Daltonics) equipped with delayed extraction, a 2 GHz LeCroy digitizer and a 337 nm nitrogen laser. The instrument was operated in linear and negative ion mode. The accelerating potential was 20 kV.
HPLC Analysis
High-performance liquid chromatography (HPLC) analysis was conducted with a Shimadzu instrument (Model LC-10AD VP), equipped with a Shimadzu (Model SPD-10A VP) variable-wavelength UV detector. The analytical column was a reversed phase C-18 (ODS-3, 250 × 4.6 mm i.d., particle size 5 μm, from Inersil) connected to a C-18 guard column.
The mobile phase system for quantification of AZT and succinoylated AZT consisted of components A and B. Mobile phase A was 0.01% (vol/vol) ortho-phosphoric acid in water and mobile phase B was a mixture of acetonitrile: methanol (1:1, vol/vol). A gradient elution program was used, at 0 to 14 min, mobile phase A was fixed at 93% with a linear increase in flow rate from 1.5 to 2 ml/min. At 14th min, the mobile phase B was increased immediately from 7% to 22% and then linearly increased up to 40% at the 28th min. After 28 min, mobile phase A was returned to 93% with a flow rate of 1.5 ml/min for 7 min to re-equilibrate the column. A UV detector was used and fixed at 266 nm. Stavudine (165 μg/ml) was used as an internal standard.
Calibration curves for AZT were prepared using a concentration range of 0.12–37.17 μg/ml. The stavudine standard (60 μl) was added into the AZT standard solutions (720 μl) with final AZT concentrations of 0.12–37.17 μg/ml. Aliquots of these solutions (10 μl) were injected onto the HPLC, and the peak area ratio of AZT/stavudine determined. The calibration curves for succinoylated AZT were created using the same method over a concentration range of 0.11–549.23 μg/ml.
Calibration curves for AZT and succinoylated AZT were also prepared in plasma. Human plasma (650 μl) in a microcentrifuge tube was spiked with AZT standard solutions (100 μl) and the stavudine internal standard (75 μl). The mixture was mixed, and then 20% (wt/vol) of trichloroacetic acid (150 μl) was added. The tubes were vortexed for 30 s and then centrifuged at 12,000 rpm for 7 min to obtain a clear supernatant. The final concentrations of AZT ranged from 0.02 to 40.40 μg/ml. An aliquot of the supernatant (10 μl) was then injected onto the HPLC. For succinoylated AZT, a calibration curve was prepared using the same method but over the concentration range of 0.03–103.08 μg/ml.
The HPLC method for AZT and succinoylated AZT was validated following ICH (Q2B) guidelines (31). AZT and succinoylated AZT in plasma were prepared in the range of 0.02–40.40 μg/ml and 0.03–103.08 μg/ml, respectively. Linearity was evaluated by linear regression analysis of the plot between the peak area ratios of AZT/stavudine or succinoylated AZT/stavudine and sample concentrations. Accuracy was determined as percent recovery. The percent recovery of AZT from plasma was performed using three replicates of three different concentrations of 6.73, 13.47 and 40.40 μg/ml and succinoylated AZT was assayed at concentrations of 2.58, 20.62 and 82.46 μg/ml. The percent recovery was determined by comparing observed concentrations with actual concentrations. Intra-day precision of AZT or succinoylated AZT was determined by analyzing six replicates of three different concentrations of AZT (8.08, 20.20 and 40.40 μg/ml) or succinoylated AZT (5.15, 34.36 and 82.46 μg/ml) in plasma within the same day. For inter-day precision, three replicates of corresponding concentrations of AZT or succinoylated AZT were analyzed for 3 days. The quantification limit and the detection limit of AZT or succinoylated AZT were determined by reduction of AZT (0.40 μg/ml) or succinoylated AZT (0.41 μg/ml) concentration to a minimum concentration that provided signal-to-noise ratios of 10:1 and 3:1, respectively.
In Vitro Drug Release
Buffer Solutions at pH 5.5 and 7.4
The release of AZT and succinoylated AZT from the dextrin–AZT conjugate was investigated in citrate–phosphate buffer solution at pH 5.5 and PBS at pH 7.4. An aliquot of the dextrin–AZT conjugate (1 ml, 7.23 μmol) was added into 5 ml of preheated buffer (37 ± 0.1°C) under continuous stirring at 420 rpm using a magnetic stirrer. Paraffin film closures were used to prevent evaporation. Samples (300 μl) were taken at various time intervals and the stavudine internal standard (25 μl, 165 μg/ml) was added. The amounts of AZT and succinoylated AZT released were determined by HPLC analysis.
Human Plasma
An aliquot of the dextrin–AZT conjugate (1 ml, 7.23 μmol) was added into 5 ml of preheated plasma (37 ± 0.1°C) under continuous stirring at 420 rpm using a magnetic stirrer. Paraffin film closures were used to prevent evaporation. Samples (300 μl) were taken at different time intervals. The stavudine internal standard (30 μl, 165 μg/ml) was added, and thoroughly mixed into the collected sample. Trichloroacetic acid (60 μl, 20% wt/vol) was then added to precipitate plasma proteins. The mixture was vortexed for 30 s and centrifuged at 12,000 rpm for 7 min. The supernatant was analyzed by HPLC to determine the amounts of AZT and succinoylated AZT released from the dextrin–AZT conjugate.
Hemolysis Assay
Human red blood cells were separated and washed three times with cold PBS. The red blood cells (RBC) were then diluted in cold PBS to obtain 2% (wt/vol) of RBC suspension. The solutions (100 μl) of various concentrations of free AZT, the conjugate (calculated equivalent to AZT loading), dextrin, dextran or poly(ethyleneimine) (PEI) in PBS were incubated with RBC suspension (100 μl) at 37°C for 4 h. The incubated samples were centrifuged at 1,000×g for 10 min to remove the intact RBC. The supernatant (100 μl) was transferred to 96-well plate. The absorbance was measured at 550 nm using a microtitre plate reader. The percentage of hemolysis was calculated relative to 100% hemolysis obtained from incubation with a 1% vol/vol solution of Triton X-100 (32).
In Vivo Pharmacokinetic Study
Male Sprague Dawley rats weighing 250–300 g were obtained from the National Laboratory Animal Centre (Nakhon Pathom, Thailand). The experimental protocol was approved by the Institutional Animals Ethics Committee of Chulalongkorn University. Three rats were used in each group to investigate the pharmacokinetics of the dextrin–AZT conjugate and AZT following I.V. administration. A freshly prepared solution of either free AZT or the dextrin–AZT conjugate (AZT equivalent dose of 8.46 mg/kg body weight) in sterile normal saline was injected as an I.V. bolus into the tail vein. Blood samples (150 μl) were taken from the tail vein at different time intervals up to 30 h, and collected in microcentrifuge tubes. The plasma was separated by centrifugation at 12,000 rpm for 10 min, and the resultant supernatants were processed for analysis by HPLC. The plasma half-life (t1/2) of AZT was calculated using Eq. 2:
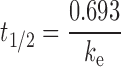 |
2 |
where ke was the elimination rate constant of the terminal elimination phase. The area under the plasma concentration-time curve (AUC0→α) and the area under the first moment of the plasma concentration-time curve (AUMC0→α) were calculated using the trapezoidal rule. The mean residence time (MRT), the apparent distribution volume (Vd) and the systemic clearance (CL) were calculated following Eqs. 3, 4 and 5, respectively:
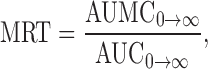 |
3 |
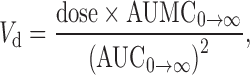 |
4 |
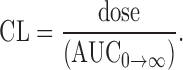 |
5 |
Statistic Analysis
Statistical significance of differences was assessed using Student’s t test and analysis of variance (ANOVA) with post-hoc Turkey’s comparison. The P-value of <0.05 was considered statistically significant.
RESULTS AND DISCUSSION
Synthesis and Characterization of the Dextrin–AZT Conjugate
Synthesis of the dextrin–AZT conjugate involved two steps (Fig. 1). Firstly, AZT was succinoylated to introduce a carboxylic functional group by reacting with succinic anhydride in the presence of triethylamine. Then, the succinoylated AZT was conjugated with dextrin by using DCC and HOBt as coupling agents. In brief, AZT reacted with succinic anhydride to form succinoylated AZT, which was purified by solvent extraction and then by flash column chromatography (Sephadex LH-20). The synthetic process provided succinoylated AZT of purity >97%, determined by HPLC. Its FT-IR spectrum (Fig. 2) exhibited a broad band of O-H stretching in the range of 2,700–3,660 cm−1 centered at 3,190 cm−1. The azido group (–N3) appeared at 2,109 cm−1. The carbonyl stretching of the ester of carboxylic acid showed at 1,735 cm−1. The C=O band was a broad band due to the overlap of two esters of the carboxylic acid and of 6 pie-position owing to the incorporation of succinic groups together with carbonyls of –CO–NH–CO– of AZT. The 1H-NMR in CDCl3 (Fig. 3) showed the disappearance of the proton of hydroxyl group at 8.55 ppm, the presence of the protons of succinic group at 2.62 ppm and of the carboxylic group at 10.62 ppm. The conjugation of succinoylated AZT and dextrin was carried out in the presence of DCC and HOBt. The structure of the dextrin–AZT conjugate was confirmed by FT-IR and 1H-NMR. The FT-IR spectrum (Fig. 2) showed the absorption of the –N3 group of succinoylated AZT at 2,113 cm−1. The ester of succinic spacer absorbed at 1,734 cm−1 whereas the amide I and amide II absorptions of succinoylated AZT occurred at 1,648 and 1,568 cm−1, respectively. The remaining unreacted hydroxyl groups of dextrin appeared as a broad band centered at 3,401 cm−1. The CH, CH2 stretching of dextrin exhibited at 2,930 cm−1 and the absorption of O–CH2 of dextrin was shown at 1,025 cm−1.
Fig. 1.

Synthesis of succinoylated a AZT and b the dextrin–AZT conjugate
Fig. 2.

FT-IR spectra of a AZT, b succinoylated AZT, c dextrin and d the dextrin–AZT conjugate
Fig. 3.

1H-NMR spectra of a AZT in CDCl3, b succinoylated AZT in CDCl3, c dextrin in D2O and d the dextrin–AZT conjugate in D2O
The 1H-NMR in D2O of the dextrin–AZT conjugate (Fig. 3) showed a peak at 1.66 ppm due to the methyl protons of succinoylated AZT. The conjugation caused a 0.22 ppm upfield shift of the methyl protons of succinoylated AZT. The signals between 2.31 and 2.53 ppm were from the protons of C2′ of sugar ring of succinoylated AZT and those of the succinic spacer. The peak at 5.94 ppm corresponded to the proton of C1′ of succinoylated AZT and the peak at 7.28 ppm was due to the proton of C6 on the pyrimidine ring of succinoylated AZT. A small downfield shift of 0.01 ppm for the proton of C1′ of succinoylated AZT and an upfield shift of 0.16 ppm for the proton of C6 of succinoylated AZT was observed after conjugation. The broad bands from 2.94 to 5.45 ppm were due to the protons of the dextrin polymer. The MALDI-TOF mass spectrum of the dextrin–AZT conjugate was compared with that of dextrin as shown in Fig. 4. Dextrin had average molecular weight of around 2,264.6 Da. After conjugation, the dextrin–AZT conjugate had a higher average molecular weight of about 2,980.9 Da indicating the success of conjugation. The absence of free AZT or succinoylated AZT in the dextrin–AZT conjugate was confirmed by HPLC analysis. Drug loading on the polymeric backbone was estimated by UV spectroscopy and found to be 18.9% wt (15.24 mol%). The average AZT content was also confirmed by 1H-NMR analysis and was in good agreement with that from UV analysis (15.03 mol%).
Fig. 4.

MALDI-TOF mass spectra of a dextrin and b the dextrin–AZT conjugate
In Vitro Drug Release
First, the HPLC conditions for quantitation of AZT and succinoylated AZT in plasma were developed and validated. Figure 5 shows the HPLC chromatograms of AZT and succinoylated AZT in plasma. Retention times for stavudine internal standard, AZT and succinoylated AZT were 11.9, 20.2 and 26.7 min, respectively. The analytes’ peaks were not interfered with endogenous peaks of plasma matrix showing the specificity of the HPLC method. For AZT, the quantitation limit was 0.017 μg/ml and the detection limit was found to be 0.006 μg/ml. The coefficient of variation for intra-day precision was between 0.5% and 2.5%, and for inter-day precision was between 1.6% and 3.2%. The calibration curve of peak area ratio of AZT/stavudine against the AZT concentration showed good linearity (r2 of 0.9997). For succinoylated AZT, the quantitation limit was 0.034 μg/ml and the detection limit was 0.008 μg/ml. In this case, the coefficient of variation for intra-day precision and inter-day precision were 0.8–1.7% and 0.9–2.4%, respectively, and the linearity was good (r2 of 0.9998). The percent recovery of AZT and succinoylated AZT from plasma was found to be 99.3% and 102.1%, respectively.
Fig. 5.

HPLC chromatograms of stavudine, AZT and succinoylated AZT in human plasma
The drug release profiles of the dextrin–AZT conjugate were investigated in buffer solutions at pH 5.5 (equivalent to the pH of the endosomal compartment) and 7.4 (equivalent to that of extracellular fluids). Theoretically hydrolysis can occur at two sites in the succinic spacer resulting in the release of either free AZT or succinoylated AZT. Figure 6 showed 1.4% of the drug was released from the conjugate at pH 5.5 after 24 h. This release was corresponded to 0.7% AZT and 0.7% succinoylated AZT. At pH 7.4, 41.7% of the drug was released as free drug and succinoylated drug (14.7% and 27.0% respectively). The release profiles of both were almost linear over time with r2 values of 0.9948 and 0.9897, respectively. These release profiles followed zero order kinetics, the succinoylated AZT release being greater than that of AZT release (P < .05) indicating that the ester linkage of the succinic spacer bonded to dextrin was more susceptible to hydrolysis than the bond to AZT. The dextrin–AZT conjugate was stable at pH 5.5 but hydrolyzed to release drug at pH 7.4.
Fig. 6.

Release of AZT and succinoylated AZT from the dextrin–AZT conjugate in buffer solutions at pH 5.5 and pH 7.4, at 37 ± 0.1°C. Mean±SD, n = 3
In human plasma (Fig. 7), approximately 78% of the bound drug (42.3% AZT and 36.1% succinoylated AZT) was released within 24 h. Initially succinoylated AZT release was greater than for AZT (P < 0.05), however, after 24 h AZT was released from both the dextrin–AZT conjugate and succinoylated AZT. AZT and succinoylated AZT release from the dextrin–AZT conjugate was complete within 48 h and the appearance of AZT was nearly linear (r2 of 0.9874) with time over 48 h following zero order release. In plasma, the rate of the release of AZT and succinoylated AZT from the conjugate was faster than in buffer at pH 7.4 indicating that the succinic spacer allows the release of drug through both hydrolytic and enzymatic mechanisms. Due to the ester type of the succinic spacer, succinoylated AZT would be cleaved by esterases in the blood resulting in free AZT that exerts its anti-HIV activity. Free succinate (spacer) will be metabolized to fumarate by succinate dehydrogenase (33–35) hence the free spacer would not accumulate and would thus be safe for long-term use of the dextrin–AZT conjugate. In AIDS compromised patients, the free spacer is metabolized as the same process as in healthy humans.
Fig. 7.

Release of AZT and succinoylated AZT from the dextrin–AZT conjugate in human plasma, at 37 ± 0.1°C. Mean±SD, n = 3
Hemolysis Study
As the dextrin–AZT conjugate is used for I.V. administration, an in vitro hemolysis assay was used as a simple measure of safety determination (36). The degree of hemolysis obtained for the dextrin–AZT conjugate was compared with that of the parent polymer (dextrin), dextran and poly(ethyleneimine) (PEI) as reference controls. Dextran is used as a plasma volume expander in the form of I.V. infusion solutions (37–39). PEI was used as positive control because it has high hemolytic activity. The hemolytic activities of the dextrin–AZT conjugate, dextrin, dextran and PEI are shown in Fig. 8a. The dextrin–AZT conjugate showed a low hemolytic activity that was equivalent to dextrin, dextran (P > 0.05) and to AZT (P > 0.05) (Fig. 8b). The hemolytic activity of AZT did not increase when it was covalently linked to dextrin or physically mixed with dextrin (P > 0.05).
Fig. 8.

Hemolytic effect of a dextrin, dextran, poly(ethyleneimine) (PEI) and the dextrin–AZT conjugate and b those of the dextrin–AZT conjugate, AZT and combination of AZT and dextrin. Mean±SD, n = 3
In Vivo Pharmacokinetic Study
The in vivo pharmacokinetic study was conducted to compare the pharmacokinetics of the conjugate with free AZT. The plasma concentrations of AZT over time following I.V. administration of the conjugate and free drug are shown in Fig. 9, with the corresponding pharmacokinetic parameters in Table I. After administration of the conjugate, an initial AZT level of 4 μg/ml was observed and after 3 h the AZT concentration was maintained at ∼0.10 μg/ml up to 30 h (range, 0.06–0.17 μg/ml), values in the IC50 range (0.03–0.13 μg/ml) for suppression of viral replication (14). Whereas the administration of free AZT showed initial AZT concentration of 24 μg/ml which was six times greater than that obtained from the conjugate but AZT level decreased dramatically to undetectable level after 5 h. This AZT plasma profile following I.V. administration of the dextrin–AZT conjugate providing more stable drug level may result in a decreased risk of AZT toxicity. With appropriate dose adjustment, the dextrin–AZT conjugate would provide therapeutic levels and it would need less frequent administration.
Fig. 9.

Free AZT plasma concentrations versus time following I.V. administration of the dextrin–AZT conjugate and free AZT to rats. Mean±SD, n = 3
Table I.
Pharmacokinetic Parameters of AZT Following I.V. Administration of Free AZT and Dextrin–AZT Conjugate, Dose of 8.46 mg/kg in Rats, Mean±SD, n = 3
| Pharmacokinetic parameters | AZT | Dextrin–AZT conjugate |
|---|---|---|
| k e (h−1) | 0.527 ± 0.035 | 0.037 ± 0.008 |
| t 1/2 (h) | 1.32 ± 0.09 | 19.34 ± 4.93 |
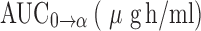
|
5.14 ± 0.48 | 6.09 ± 0.09 |
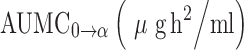
|
1.78 ± 0.12 | 142.31 ± 17.70 |
| MRT (h) | 0.35 ± 0.02 | 23.63 ± 3.04 |
| V d (l) | 0.16 ± 0.02 | 9.30 ± 1.25 |
| CL (l/h) | 0.47 ± 0.04 | 0.39 ± 0.01 |
The pharmacokinetic parameters obtained from administration of the conjugate (Table I) indicated that it improved the pharmacokinetic profile of AZT. The half-life of AZT was increased from 1.3 to 19.3 h (P < 0.05). The mean residence time (MRT) was calculated to be 23.6 h (P < 0.05). The area under the plasma concentration-time curve (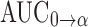 ), the area under the first moment of the plasma concentration-time curve (
), the area under the first moment of the plasma concentration-time curve (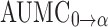 ) and the apparent distribution volume (Vd) obtained following the administration of the dextrin–AZT conjugate were significantly increased (P < 0.05). Moreover, the conjugate exhibited a reduced systemic clearance (CL) (P < 0.05) which could be explained by the higher molecular weight of the conjugate reducing its renal filtration. The in vivo study indicated that the dextrin–AZT conjugate has potential as a polymeric prodrug producing sustained release of AZT after I.V. administration. This novel conjugate could be helpful in the treatment of HIV infection providing the required constant drug levels to successfully suppress viral replication.
) and the apparent distribution volume (Vd) obtained following the administration of the dextrin–AZT conjugate were significantly increased (P < 0.05). Moreover, the conjugate exhibited a reduced systemic clearance (CL) (P < 0.05) which could be explained by the higher molecular weight of the conjugate reducing its renal filtration. The in vivo study indicated that the dextrin–AZT conjugate has potential as a polymeric prodrug producing sustained release of AZT after I.V. administration. This novel conjugate could be helpful in the treatment of HIV infection providing the required constant drug levels to successfully suppress viral replication.
CONCLUSION
The synthesis of a novel dextrin–AZT conjugate has been reported. AZT was covalently attached to the dextrin backbone via ester bonds by succinoylation, and the conjugate formed was stable at pH 5.5 and drug released at pH 7.4, both in buffer and in human plasma in vitro. An in vivo investigation in rats showed that I.V. administration of the dextrin–AZT conjugate led to a sustained release of AZT over at least 30 h, the pharmacokinetics of AZT were improved and a maintenance plasma level was achieved. The more stable plasma levels obtained using the dextrin–AZT conjugate may also be expected to decrease the fluctuations in drug levels that can lead to toxicity.
Acknowledgments
This work was financially supported by the Thailand Research Fund through the Royal Golden Jubilee Ph.D. Program (Grant No. PHD/0110/2544) to SW and UN. Authors would like to thank Professor Ruth Duncan and Dr Maria Vicent for their advice and they are grateful to Professor Alexander T. Florence for his excellent suggestions.
References
- 1.Yarchoan R., Klecker R. W., Weinhold K. J., et al. Administration of 3¢-azido-3¢-deoxythymidine, an inhibitor of HTLV-III/LAV replication, to patients with AIDS or AIDS-related complex. Lancet. 1986;327:575–580. doi: 10.1016/S0140-6736(86)92808-4. [DOI] [PubMed] [Google Scholar]
- 2.D’Alessandro A. M., Rinaldi A. C., D’Andrea G., et al. Evidences that zidovudine (AZT) could not be directly responsible for iron overload in AZT-treated patients: an in vitro study. Clin Chim Acta. 2000;300:119–130. doi: 10.1016/S0009-8981(00)00314-4. [DOI] [PubMed] [Google Scholar]
- 3.G. K. McEvoy, J. Miller, E. K. Snow, O. H. Welsh, and K. Litvak (eds.), AHFS Drug information. American Society of Health-System Pharmacists, Bethesda, MD, 2004, pp. 731–745.
- 4.Chitnis S., Mondal D., Agrawal K. C. Zidovudine (AZT) treatment suppress granulocyte-monocyte colony stimulating factor receptor type alpha (GM-CSFR alpha) gene expression in murine bone marrow cells. Life Sci. 2002;71:967–978. doi: 10.1016/S0024-3205(02)01790-3. [DOI] [PubMed] [Google Scholar]
- 5.Fauci A. S. HIV and AIDS: 20 years of science. Nat Med. 2003;9:839–843. doi: 10.1038/nm0703-839. [DOI] [PubMed] [Google Scholar]
- 6.Narishetty S. T., Panchagnula R. Transdermal delivery of zidovudine: effect of terpenes and their mechanism of action. J Control Release. 2004;95:367–379. doi: 10.1016/j.jconrel.2003.11.022. [DOI] [PubMed] [Google Scholar]
- 7.Lynx M. D., Bentley A. T., McKee E. E. 3¢-Azido-3¢-deoxythymidine (AZT) inhibits thymidine phosphorylation in isolated rat liver mitochondria: a possible mechanism of AZT hepatotoxicity. Biochem Pharmacol. 2006;71:1342–1348. doi: 10.1016/j.bcp.2006.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Newell M. L. Current issues in the prevention of mother-to-child transmission of HIV-1 infection. Trans R Soc Trop Med Hyg. 2006;100:1–5. doi: 10.1016/j.trstmh.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 9.Klecker R. W., Jr., Collins J. M., Yarchoan R., et al. Plasma and cerebrospinal fluid pharmacokinetics of 3¢-azido-3¢-deoxythymidine: a novel pyrimidine analog with potential application for the treatment of patients with AIDS and related diseases. Clin Pharmacol Ther. 1987;41:407–412. doi: 10.1038/clpt.1987.49. [DOI] [PubMed] [Google Scholar]
- 10.Thomas N. S., Panchagnula R. Transdermal delivery of zidovudine: effect of vehicles on permeation across rat skin and their mechanism of action. Eur J Pharm Sci. 2003;18:71–79. doi: 10.1016/S0928-0987(02)00242-7. [DOI] [PubMed] [Google Scholar]
- 11.Richman D. D., Fischl M. A., Grieco M. H., et al. The toxicity of azidothymidine (AZT) in the treatment of patients with AIDS and AIDS-related complex. A double-blind, placebo-controlled trial. N Engl J Med. 1987;317:192–197. doi: 10.1056/NEJM198707233170402. [DOI] [PubMed] [Google Scholar]
- 12.Barry M., Howe J. L., Back D. J., et al. Zidovudine pharmacokinetics in zidovudine-induced bone marrow toxicity. Br J Clin Pharmacol. 1994;37:7–12. doi: 10.1111/j.1365-2125.1994.tb04231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flexner C. Antiretroviral agents and treatment of HIV infection. In: Brunton L. L., Lazo J. S., Parker K. L., editors. Goodman and Gilman’s The Pharmacological Basis of Therapeutics. 11. New York: McGraw-Hill; 2006. pp. 1273–1285. [Google Scholar]
- 14.Fletcher C. V., Kakuda T. N., Collier A. C. Human immunodeficiency virus infection. In: DiPiro J. T., Talbert R. L., Yee G. C., Matzke G. R., Wells B. G., Posey L. M., editors. Pharmacotherapy A Pathophysiologic Approach. 4. Connecticut: Appleton and Lange Stamford; 1999. pp. 1937–1938. [Google Scholar]
- 15.Hayden F. G. Antimicrobial agents. In: Hardman J. G., Limbird L. E., Molinoff P. B., Ruddon R. W., Gilman A. G., editors. Goodman and Gilman’s The Pharmacological Basis of Therapeutics. 9. New York: McGraw-Hill; 1996. pp. 1204–1207. [Google Scholar]
- 16.Lacy C. F., Armstrong L. L., Goldman M. P., Lance L. L. Drug Information Handbook. 15. Hudson, OH: Lexi-Comp; 2007. pp. 1812–1815. [Google Scholar]
- 17.Dumitriu S., Dumitriu M. Polymeric drug carriers. In: Dumitriu S., editor. Polymeric Biomaterials. New York: Marcel Dekker; 1994. pp. 474–475. [Google Scholar]
- 18.Hoste K., Winne K. D., Schacht E. Polymeric prodrugs. Int J Pharm. 2000;277:119–131. doi: 10.1016/j.ijpharm.2003.07.016. [DOI] [PubMed] [Google Scholar]
- 19.Lovrek M., Zorc B., Boneschans B., Butula I. Macromolecular prodrugs. VIII. synthesis of polymer-gemfibrozil conjugates. Int J Pharm. 2000;200:59–66. doi: 10.1016/S0378-5173(00)00340-9. [DOI] [PubMed] [Google Scholar]
- 20.Zovko M., Zorc B., Lovrek M., Boneschans B. Macromolecular prodrugs. IX. synthesis of polymer-fenoprofen conjugates. Int J Pharm. 2001;228:129–138. doi: 10.1016/S0378-5173(01)00822-5. [DOI] [PubMed] [Google Scholar]
- 21.Duncan R. The dawning era of polymer therapeutics. Nat Rev Drug Discov. 2003;2:347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 22.Matthews S. E., Pouton C. W., Threadgill M. D. Macromolecular systems for chemotherapy and magnetic resonance imaging. Adv Drug Deliv Rev. 1996;18:219–267. doi: 10.1016/0169-409X(95)00098-R. [DOI] [Google Scholar]
- 23.Hreczuk-Hirst D., German L., Duncan R. Dextrins as carriers for drug targeting: reproducible succinoylation as a means to introduce pendent groups. J Bioact Compat Polym. 2001;16:353–365. doi: 10.1106/QBKY-E3VM-19K4-3GA5. [DOI] [Google Scholar]
- 24.diZerega G. S., Verco S. J., Young P., et al. A randomized, controlled pilot study of the safety and efficacy of 4% icodextrin solution in the reduction of adhesions following laparoscopic gynaecological surgery. Hum Reprod. 2002;17:1031–1038. doi: 10.1093/humrep/17.4.1031. [DOI] [PubMed] [Google Scholar]
- 25.Davies S. J. Exploring new evidence of the clinical benefits of icodextrin solutions. Nephrol Dial Transplant. 2006;21(S2):ii47–ii50. doi: 10.1093/ndt/gfl190. [DOI] [PubMed] [Google Scholar]
- 26.Sivakama Sundari C., Raman B., Balasubramanian D. Artificial chaperoning of insulin, human carbonic anhydrase and hen egg lysozyme using linear dextrin chains-a sweet route to the native state of globular proteins. FEBS Lett. 1999;443:215–219. doi: 10.1016/S0014-5793(98)01720-7. [DOI] [PubMed] [Google Scholar]
- 27.D. S. Davies. Dextrin sulfates as anti HIV-1 agents and composition thereof. US Patent, 5439892 (1969) December 31.
- 28.R. K. Hershline. Antiviral composition. US Patent, 6821958 (2004) November 23.
- 29.Flexner C., Barditch-crovo P. A., Kornhauser D. M., et al. Pharmacokinetics, toxicity, and activity of intravenous dextran sulfate in human immunodeficiency virus infection. Antimicrob Agents Chemother. 1991;35:2544–2550. doi: 10.1128/aac.35.12.2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giammona G., Cavallaro G., Fontana G., Pitarresi G., Carlisi B. Coupling of the antiviral agent zidovudine to polyaspartamide and in vitro drug release studies. J Control Release. 1998;54:321–331. doi: 10.1016/S0168-3659(98)00020-0. [DOI] [PubMed] [Google Scholar]
- 31.International Conference on Harmonization (ICH). ICH topic Q2B validation of analytical procedures: methodology. November 6, 1996.
- 32.Carreno-Gomez B., Duncan R. Evaluation of the biological properties of soluble chitosan and chitosan microspheres. Int J Pharm. 1997;148:231–240. doi: 10.1016/S0378-5173(96)04847-8. [DOI] [Google Scholar]
- 33.P. Y. Bruice. Carbonyl compounds I. In J. Challice, P. F. Corey, C. Trueheart, and K. Schiaparelli (eds.), Organic Chemistry, 2nd ed, Prentice-Hall, Upper Saddle River, NJ, 1998, pp. 679.
- 34.Olson M. S. Bioenergetics and oxidative metabolism. In: Devlin T. M., editor. Textbook of Biochemistry with Clinical Correlations. 4. New York: Wiley-Liss; 1997. [Google Scholar]
- 35.R. H. Garrett and C. M. Grisham. Glycolysis. In J. Vondeling, S. Kiselica, and B. Bonnie (eds.), Principles of Biochemistry with a Human Focus, Brooks/Cole, London, UK, 1997.
- 36.Jumaa M., Furkert F. H., Muller B. W. A new lipid emulsion formulation with high antimicrobial efficacy using chitosan. Eur J Pharm Biopharm. 2002;53:115–123. doi: 10.1016/S0939-6411(01)00191-6. [DOI] [PubMed] [Google Scholar]
- 37.van Dijk-Wolthuis W. N., Tsang S. K. Y., Kettenes-van den Bosch J. J., Hennink W. E. A new class of polymerizable dextrans with hydrolyzable groups: hydroxyethyl methacrylated dextran with and without oligolactate spacer. Polymer. 1997;38:6235–6242. doi: 10.1016/S0032-3861(97)00189-4. [DOI] [Google Scholar]
- 38.Won C. Y., Chu C. C. Dextran-estrone conjugate: synthesis and in vitro release study. Carbohydr Polym. 1998;36:327–334. doi: 10.1016/S0144-8617(97)00125-2. [DOI] [Google Scholar]
- 39.British Pharmacopoeia, Vol. 1, Stationery Office, London, UK, 2002, pp. 567–569.