

Abstract
The aim of this study was to formulate extended release compression coated core tablets of fenoterol hydrobromide, a selective β2 adrenergic receptor agonist, in an attempt to prevent nocturnal asthma. Two hydrophilic polymers viz Kollidon® SR, Polyox® WSR 303 and a hydrophobic one (Precirol® ATO5) were employed. Compression coated tablets were formulated by preparing a core tablet containing 7.5 mg drug and various amounts of polymer and Emcompress® then compressed coated with the same polymeric materials. For comparison purpose different matrix tablets were also prepared employing the same polymers. In-vitro release studies were carried out at different pH (1.2 and 6.8). Pharmacokinetics of extended release tablets as well as commercially available immediate release tablets (Berotec®) were studied after oral administration to beagle dogs using a new developed LC-MS/MS method with a lower limit of quantification of 1 ng/ml. Fenoterol release from compression coated tablets was significantly lower than matrix tablets. The mechanism of release was changed with the nature and content of polymer. The release pattern of drug from F16 containing 40 mg Kollidon® SR divided in the core tablet (15 mg) and the rest in the compressed coat (25 mg) showed a typical zero order release kinetic that could extend drug release >10 h and reasonable time for 75% to be released (t75) (8.92 h). When compared to immediate release Berotec® tablet the MRT was significantly extended from 7.03 ± 0.76 to 10.93 ± 1.25 h (P < 0.001) and HVDt 50%Cmax was also significantly extended from 2.71 ± 0.68 to 6.81 ± 0.67 h with expected prevention of nocturnal asthma.
Keywords: compression coat, extended release, fenoterol hydrobromide, LC/MS/MS, pharmacokinetics, tablets
INTRODUCTION
Asthma is a condition in which the airways are narrowed in response to stimuli that produce inflammation but do not affect the airways in normal lungs. Beta-adrenergic receptor agonists are the best drugs for relieving sudden attacks of asthma by stimulating beta-adrenergic receptors to widen the airways. Non-selective β-adrenergic receptor agonists such as adrenaline and isoprenaline cause side effects such as tachycardia, restlessness, headache and muscle tremors. However, Selective β2-adrenergic receptor agonists like Fenoterol hydrobromide (1-(3, 5-dihydroxy phenyl)-2-[(R)-4 hydroxy-α-methyl phenethyl amino] ethanol hydrobromide) which mainly act on β2-receptors found primarily on cells in the lung have little effect on other organs and cause fewer side effects than non-selective bronchodilators (1). Fenoterol hydrobromide is available in the market either as inhaler for acute asthma or in the form of immediate release oral tablets for chronic asthma. However oral conventional tablets suffers some drawbacks: extensive first pass metabolism (50–60%), high dosing frequency due to short half life time (about 4 h) which reduce patient compliance and therapeutic efficacy besides its inability to prevent nocturnal asthma. Hence a controlled release formulation of fenoterol would maintain its effective plasma concentration for longer than 8 h, subsequently reducing drug administration to twice or even once daily regimen.
From an economical point of view the production of a sustained release tablets by direct compression is of great promise. An innovative excipient that offers good controlled release characteristic together with excellent direct compression properties is required as the essential tool for the development and manufacture of tablets. Compression coated core tablets is one of the approaches for delaying the release of drugs and are simple formulations to manufacture. Kollidon® SR is one of the promising release retarding polymers. It has already been used to delay the release of highly water soluble drugs such as Propranolol HCl and Diphenhydramine HCl (2,3). It shows excellent flowability and can be used as an excipient for direct compression. High molecular weight polyethylene oxides (Polyox®) have been successfully employed in controlled release swellable matrices (4). Polyox® swell and form a gel layer upon contact with water and then erode slowly thus, modulating the drug delivery rate (5). Precirol® ATO5 is a multiple uses excipient for solid dosage forms. Chemically, it is atomized glyceryl palmitostearate. It is synthesized by esterification of glycerol by palmito-stearic acid (C16–C18 fatty acid). The product is then atomized by spray-cooling. It is composed of mono-, di-and tri-glycerides of palmito-stearic acid, and the di-ester fraction being predominant. The estrification of glycerol by long chain fatty acids and the absence of polyethylene glycol esters give them a pronounced hydrophobic character expressed by the low HLB value of 2. Precirol® ATO5 forms an inert matrix, from which the drug is slowly released in the body by erosion/diffusion. It can be incorporated by direct compression technique (Technical Information, Precirol® ATO5, Gattefossè, St. Priest, France). In previous work (6), it was concluded that Precirol® ATO5 can be used as glycerides bases for the preparation of sustained-release dosage form.
The purpose of this study was to formulate extended release matrix and compression coated core tablets of fenoterol using Kollidon® SR, Polyox® WSR 303 and Precirol® ATO5. The effect of changing polymer type and content in the coat and core on the in vitro drug release was studied. Also pharmacokinetics of the prepared extended release compression coated core tablets compared to commercial immediate release tablets was studied in beagle dogs using a new developed LC-MS/MS method.
MATERIALS AND METHODS
Materials
Fenoterol HBr CAS# 1944-12-3, lot # 41k1744 was purchased from Sigma (St. Louis, Mo, USA), Emcompress® (di-basic calcium phosphate di-hydrate USP): obtained from Pen west Pharmaceutical Co. (Patterson, N.J, USA), Kollidon® SR (8 parts polyvinyl acetate + 2 parts polyvinyl pyrrolidon), lot # 08-9010 was a kind gift from BASF Corporation (North mount olive, N.J, USA), Precirol® ATO5 (Atomized Glyceryl Palmito stearate), lot # 26488 was genoursly donated by Gattefossè (St. Priest, France), Polyox® WSR 303-NF grade [Poly (ethylene oxide), Fumed Silica, Calcium as mixed salts] M.wt 7 × 106, lot# 815458 from Dow Chemical Company (Midland, M.I, USA). All other chemicals were of analytical grade.
Differential Scanning Calorimetry (DSC)
In order to investigate possible interaction between fenoterol and the polymeric materials employed such as Kollidon® SR, Polyox® WSR 303 and Precirol® ATO5, DSC analysis was carried out on pure substances and their physical mixtures in equimolar ratios using (Shimadzu DSC-50) instrument equipped with a computerized data station at a rate of 10°C/min between room temperature and 380°C.
Preparation of the Tablets
Preparation of Matrix Tablets
Fenoterol (7.5 mg) together with either kollidon® SR, polyox® 303 or precirol® ATO as release retardants and Emcompress as filler were blended by dry mixing using a laboratory mixer for 10 min after sieving each powder through a 60 mesh sieve. Then, 100 mg of the powder mixture was compressed directly using a single punch tablet machine equipped with concave punch 6.5 mm in diameter. The compression force was selected to be one metric ton (1,000 kg) for 20 s to provide tablets with 5–6 kg hardness. A typical design of all formulations and their codes is shown in Table I.
Table I.
Composition of 100 mg Drug Loaded Matrix Tablets
| Formula | Release Retardant (mg) | Filler (mg) |
|---|---|---|
| Kollidon® SR | Emcompress® | |
| F1 | 40 mg | 52.5 mg |
| F2 | 50 mg | 42.5 mg |
| F3 | 55 mg | 37.5 mg |
| F4 | 60 mg | 32.5 mg |
| F5 | 65 mg | 27.5 mg |
| F6 | 70 mg | 22.5 mg |
| F7 | 75 mg | 17.5 mg |
| Polyox® 303 | Emcompress® | |
| F18 | 40 mg | 52.5 mg |
| F19 | 50 mg | 42.5 mg |
| Precirol® ATO | Emcompress® | |
| F26 | 20 mg | 72.5 mg |
| F27 | 30 mg | 62.5 mg |
| F28 | 40 mg | 52.5 mg |
| F29 | 50 mg | 42.5 mg |
Each formula contains 7.5 mg Fenoterol HBr.
Preparation of Compression Coated Core Tablets
Compression coated core tablets containing 7.5 mg fenoterol were prepared. First, fenoterol, Emcompress® and one of the selected polymers (kollidon® SR, polyox® 303 or precirol® ATO) were mixed in a laboratory mixer after passing each component through sieve 60. Then, 50 mg of this powder mixture were compressed directly to prepare the core tablets using a single press tabletting machine equipped with concave punch 4.5 mm in diameter. The core tablets were then compressed coated using a powder mixture of Emcompress® and the selected polymer at different ratios, by placing 25 mg of this powder mixture in the die, manually centering the previously prepared tablet cores and then loading another 25 mg of the outer layer powder mixture into the die. The contents were then compressed by a single punch tabletting machine fitted with a 6.5 mm diameter concave punch. The crushing strengths of the tablets were kept in the 5–6 kg range and the final tablet weight was 100 ± 5 mg. The compressed coated core tablets were composed of a constant core composition and different coat ratios or the reverse as shown in Table II.
Table II.
Composition of 100 mg Drug Loaded Compression Coated Tablets
| Formula | Core | Coat | ||
|---|---|---|---|---|
| Kollidon® SR | Emcompress® | Kollidon® SR | Emcompress® | |
| F8 | 25 mg | 17.5 mg | 10 mg | 40 mg |
| F9 | 25 mg | 17.5 mg | 20 mg | 30 mg |
| F10 | 25 mg | 17.5 mg | 25 mg | 25 mg |
| F11 | 25 mg | 17.5 mg | 30 mg | 20 mg |
| F12 | 25 mg | 17.5 mg | 40 mg | 10 mg |
| F13 | 25 mg | 17.5 mg | 50 mg | – |
| F14 | 5 mg | 37.5 mg | 25 mg | 25 mg |
| F15 | 10 mg | 32.5 mg | 25 mg | 25 mg |
| F16 | 15 mg | 27.5 mg | 25 mg | 25 mg |
| F17 | 20 mg | 22.5 mg | 25 mg | 25 mg |
| Polyox® 303 | Emcompress® | Polyox® 303 | Emcompress® | |
| F20 | 20 mg | 22.5 mg | 25 mg | 25 mg |
| F21 | 30 mg | 12.5 mg | 25 mg | 25 mg |
| F22 | 40 mg | 2.5 mg | 25 mg | 25 mg |
| F23 | 40 mg | 2.5 mg | 20 mg | 30 mg |
| F24 | 40 mg | 2.5 mg | 30 mg | 20 mg |
| F25 | 40 mg | 2.5 mg | 40 mg | 10 mg |
| Precirol® ATO | Emcompress® | Precirol® ATO | Emcompress® | |
| F30 | – | 42.5 mg | 5 mg | 45 mg |
| F31 | 5 mg | 37.5 mg | 5 mg | 45 mg |
| F32 | – | 42.5 mg | 10 mg | 40 mg |
| F33 | 10 mg | 32.5 mg | 10 mg | 40 mg |
Each formula contains 7.5 mg Fenoterol HBr in the core.
Physical Properties of Tablets
Determination of Tablet Weight Uniformity
Twenty tablets were randomly selected and accurately weighed using an electronic balance (Sartorius GmbH, Gottingen). The results were the mean values of 20 determinations.
Determination of Fenoterol Content Uniformity
Uniformity of fenoterol content was determined in 10 tablets. The contents of fenoterol in each tablet should be comprised between 85 and 115% of average contents.
Determination of Tablet Friability
Friability of tablets was performed according to USP 27.
In Vitro Drug Release Studies
In vitro drug release studies from the prepared matrix tablets and the compression coated core tablets were performed according to the USP paddle method (Apparatus II) thermostated at 37 ± 0.5°C and stirred at 50 rpm. The prepared tablets were placed in 500 ml 0.1N HCl (pH 1.2) for 2 h and Sorenson’s phosphate buffer pH 6.8 for an additional 10 h. At time intervals of 1 h, samples were withdrawn with a syringe filter (pore size 0.45 mm) and replaced with an equal volume of fresh medium. The drug content was spectrophotometrically determined (Shimadzu spectrophotometer, Tokyo, Japan) at 276 nm. Three replications were made for each tablet batch.
In order to compare the drug release profile from the prepared tablets, the time for 75% of the drug to be released (t75) was determined and statistically compared.
In order to determine drug release mechanism from the prepared tablets, the release kinetic data (up to 60% release) were analyzed by the following equation (7)
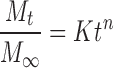 |
where, Mt,  , k and n are the amount of drug released at time t, the total amount of drug in tablet, a constant, and the exponent for the release kinetics, respectively. The value of n for a tablet, n = 0.45 for Fickian (Case I) release, >0.45 but <0.89 for non-Fickian (Anomalous) release and 0.89 for Case II (Zero order) release and >0.89 for super case II type of release.
, k and n are the amount of drug released at time t, the total amount of drug in tablet, a constant, and the exponent for the release kinetics, respectively. The value of n for a tablet, n = 0.45 for Fickian (Case I) release, >0.45 but <0.89 for non-Fickian (Anomalous) release and 0.89 for Case II (Zero order) release and >0.89 for super case II type of release.
In Vivo Study
Animals
Six male beagle dogs weighing 10.5–13.5 kg were used in the present study. The same set of 6 dogs was used in all experiments after washout period of at least 7 days between trials. Food was withdrawn from the dogs the night before each experiment and the animals were not fed until the final blood sample was drawn. The study protocol was reviewed and approved by the ethical committee of Faculty of pharmacy, Cairo University.
One tablet of F16, F23, and F30 as well as the commercially available immediate release tablet (one and half tablet of Berotec®, 5 mg, CID pharmaceutical Co. under license from Boreinger Inglhaeim, Germany) were orally administered to the dogs with 30 ml of water. A heparin-coated disposable syringe was used to collect blood from the femoral vein at each sample collection time. The blood samples were collected at 0.5, 1, 2, 3, 4, 6, 9, 12, 18 and 24 h. The blood was collected in a 2 ml vaccutainer® tube containing di-potassium EDTA and then centrifuged at 3,000 rpm for 12 min. The separated plasma was transferred into tubes, sealed and stored at −20°C until assayed.
LC-MS/MS Assay
For LC-MS/MS analysis, a Shimadzu prominence HPLC system was used, consisting of an HPLC pump (LC-10ADVP), a degasser (DGU-14A), an autosampler (SIL-20A), connected to an API 3200 tandem mass spectrometer (Applied Biosystems, Darmstadt, Germany) equipped with a turbo ion spray [pneumatically assisted electron spray ionization (ESI)] source. The software used for controlling LC and MS was Analyst 1.4.2 (Applied Biosystems). For separation, Symmetry® C18–75 × 4.6 mm, 3.5 μm (Waters, Milford, MA) column was used. The mobile phase consisted of 30% methanol and 70% 5 mM ammonium acetate. The system operated at a flow rate 0.5 ml/min and the volume of injection was 50 μl. The ESI interface was used in positive mode and the turbo ion spray source was heated to 450°C. To obtain predominantly protonated species of the analyte, an ionization voltage of 4,500 V for both fenoterol and salbutamol (the internal standard), an orifice voltage of 35 V and 25 V, collision energy of 40 and 25 for fenoterol and salbutamol were used respectively. The first quadrupole was set to select the protonated ions of fenoterol [M+H]+ (m/z 304.1) and salbutamol [m/z 240.2]. The second quadrupole was then used as a collision chamber using high-purity nitrogen gas as the collision gas at a setting of 7 PSI to produce different product ion and the third quadruple was then used to select the characteristic most intense product ions due to the transition of fenoterol (m/z 304.1) to m/z 106.9 and salbutamol (m/z 240.2) to m/z 148, respectively. PE Sciex Analyst (version 1.4.2) software was used to control the LC-MS/MS and to acquire the output signals from the detector. Also the software used for integration of peak areas and calculation of peak area ratios, calibration curves, and the drug concentration.
Calculations and Calibrations
Stock solutions were prepared containing 1,000 ng/ml and 400 ng/ml of both fenoterol hydrobromide and the internal standard Albuterol (salbutamol) sulfate in water respectively. The prepared solutions were kept in a refrigerator for use within 2 weeks. Calibration curves were constructed by spiking dog plasma to contain 1, 4, 8, 12.5, 25, 50 and 100 ng/ml fenoterol and 80 ng/ml salbutamol. A complete set of calibration samples were analyzed daily together with the unknown samples needed to be identified. Peak area ratios (Fenoterol/Salbutamol) were employed for the calculation of the calibration functions by least square linear regression with 1/× weighing.
Sample Preparation
Aliquots of 800 μl dog plasma samples spiked with 200 μl of salbutamol sulphate solution containing 80 ng salbutamol sulphate as internal standard were placed into 7 ml glass centrifuge tubes. The samples were deproteinized with 2 ml acetonitrile and extracted with 4 ml 2-butanol. After centrifugation, the organic layer was evaporated to dryness using Eppendorf® Vacufuge Concentrator 5301 at 60°C for 2 h. The residue was reconstituted with 250 μl mobile phase and 50 μl of the samples were injected.
Pharmacokinetic Analysis
Plasma concentration-time data of fenoterol was analyzed for each dog by non compartmental pharmacokinetic models using kinetica® software (version 4.4.1). The peak plasma concentrations (Cmax) and the time of its occurrence (Tmax) were directly obtained from the concentration-time data. The area under the plasma concentration-time curve (AUC) from time zero to last measured concentration (AUC0−t) was calculated according to the linear trapezoidal rule. The terminal elimination rate constant (λz) was estimated by linear regression of the terminal portion of the ln(concentration)-time curve, and the elimination half life was calculated.  was the corresponding area extrapolated to infinity by AUC0−t + Ct/λz, where Ct was the last measurable drug concentration.
was the corresponding area extrapolated to infinity by AUC0−t + Ct/λz, where Ct was the last measurable drug concentration.
The sustained release characteristic of a formulation were evaluated by the time span during which the plasma concentrations were at least 50% of the Cmax value (HVDt 50%Cmax which is the width of the plasma concentration profile at 50% of Cmax) (8,9). The HVDt 50% Cmax values were determined from the individual plasma concentration-time profiles. The ratio between the HVDt 50% Cmax values of a test formulation and the immediate release reference formulation (Berotec®) expressed as RD is indicative of sustained release effect: a ratio of 1.5, 2 and >3 indicates, low, intermediate and strong sustained release effect respectively (8).
Statistical Analysis
All statistical analysis was undertaken using ANOVA test followed by Fisher's pairwise least significant difference for multiple comparisons at P ≤ 0.0001 with Stat view statistical software program.
RESULTS AND DISCUSSION
All prepared tablet formulations met the USP 27 requirements for weight variation. Drug uniformity was found to be good where the percentage of drug content was more than 98%. The friability of the prepared tablets was within the compendial limits.
Differential Scanning Calorimetry Study
The DSC thermograms (Fig. 1) showed that, fenoterol HBr exhibited an endothermic peak at 236.27°C while Polyox® WSR 303 and Precirol® ATO5 declared an endothermic peak at 71.26°C and 64.66 respectively. However Kollidon® SR showed no peaks. The physical mixture consisting of fenoterol HBr with each of Kollidon® SR, Polyox® WSR 303 and Precirol® ATO5 at 1:1 ratio showed the aforementioned original peaks with no evidence of any interaction between the drug and the tested polymers.
Fig. 1.

Differential scanning calorimetry thermograms of fenoterol HBr and its physical mixture with each of A. Kollidon® SR, B. Polyox® WSR 303, C. Precirol® ATO5
In Vitro Drug Release
Kollidon® SR
The results of the in-vitro release of fenoterol from different matrix tablets prepared using Kollidon® SR as a release retardant are represented in Fig. 2. It is clear that increasing Kollidon® SR to Emcompress® as a water insoluble excipients ratio decreased the drug release, where increasing Kollidon® SR amounts from 40 mg in F1 to 75 mg in F7 decreased the cumulative percentage of fenoterol released in 12 h from 93.33 to 39.8% (w/w). Also there is a direct relation between Kollidon® SR amount and t75, where F1 showed t75 value of 2.85 h while F7 showed significantly higher t75 value of 22.6 h (Table III). It is obvious from the same table that increasing Kollidon® SR level in the mixture resulted in a decrease in the kinetic release constant (k). The kinetic data of fenoterol release from its matrix tablets F1 to F7 adopting Kollidon® SR (Table III) shows that all formulas have a release exponent value (n) less than 0.45 indicating a Fickian (diffusion) release kinetics. This results support the diffusion mechanisms of Kollidon® SR proposed by both BASF technical information and Shao et al (3).
Fig. 2.

Mean (± s.d.) percent of fenoterol release from Kollidon® SR matrix tablets F1–F7
Table III.
Kinetic Data of Fenoterol Release from Different Formulas
| Formula No. | n | K (hr.−1) | t 75 | r2 |
|---|---|---|---|---|
| 1 | 0.484 | 37.15 | 2.85 | 1 |
| 2 | 0.245 | 33.11 | 8.77 | 0.993 |
| 3 | 0.275 | 27.54 | 13.33 | 0.992 |
| 4 | 0.262 | 22.9 | 20.14 | 0.999 |
| 5 | 0.271 | 20.89 | 20.97 | 0.994 |
| 6 | 0.25 | 21.87 | 21.96 | 0.989 |
| 7 | 0.301 | 17.3 | 22.6 | 0.955 |
| 8 | 0.512 | 21.37 | 8.25 | 0.994 |
| 9 | 0.555 | 14.12 | 13.33 | 0.989 |
| 10 | 0.51 | 13.48 | 18.91 | 0.988 |
| 11 | 0.517 | 11.22 | 22.14 | 0.99 |
| 12 | 0.482 | 10 | 26.05 | 0.989 |
| 13 | 0.39 | 9.9 | 31.47 | 0.979 |
| 14 | 1.2 | 11.74 | 5.08 | 0.984 |
| 15 | 1.09 | 8.66 | 7.62 | 0.991 |
| 16 | 0.91 | 9.88 | 8.92 | 0.998 |
| 17 | 0.929 | 8.89 | 9.96 | 0.998 |
| 18 | 0.736 | 21.37 | 4.57 | 0.995 |
| 19 | 0.788 | 16.21 | 6.72 | 0.996 |
| 20 | 1.69 | 3.46 | 7.54 | 0.993 |
| 21 | 1.08 | 7.21 | 8.04 | 0.996 |
| 22 | 1.61 | 2.57 | 9.49 | 0.996 |
| 23 | 1.04 | 9.12 | 7.62 | 0.999 |
| 24 | 1.36 | 4.54 | 8.67 | 0.996 |
| 25 | 1.13 | 5.34 | 10.51 | 0.994 |
| 26 | 0.609 | 22.9 | 5.52 | 0.988 |
| 27 | 0.668 | 15.13 | 9.22 | 0.988 |
| 28 | 0.634 | 12.88 | 12.63 | 0.983 |
| 29 | 0.661 | 8.26 | 17.79 | 0.942 |
| 30 | 0.84 | 12.88 | 7.72 | 0.992 |
| 31 | 0.878 | 9.79 | 9.52 | 0.998 |
| 32 | 0.884 | 6.95 | 14.31 | 0.992 |
| 33 | 0.937 | 4.95 | 16.72 | 0.991 |
n: Release exponent, K: Kinetic constant, t 75 : Time for 75% fenoterol to be released, r 2 : Correlation coefficient
The same held true regarding compression coated core tablets F8 to F13 which is formed of a fixed core and varied coat compositions. Figure 3 shows that increasing Kollidon® SR amounts in the coat composition, decreased fenoterol release from its tablets where the cumulative percentages of fenoterol release in 12 h decreased from 68.27% in F8 containing 10 mg Kollidon® SR with 40 mg Emcompress® as diluent to 28.59% in F13 containing 50 mg Kollidon® SR with no diluent. It is obvious from the kinetic data listed in Table III that F8 through F13 showed a non-Fickian or anomalous release mechanism based on the release exponent (n) values which were in the range of 0.4 to 0.55.
Fig. 3.

Mean (± s.d.) percent of fenoterol release from Kollidon® SR Compression coated tablets F8–F13
Regarding tablets formed of a fixed coat composition and varied core ratio between Kollidon® SR and Emcompress® (F14, F15, F16, and F17), Fig. 4 shows that increasing Kollidon® SR from 5 mg to 20 mg in the tablet core decreased fenoterol release, where the cumulative fenoterol percent released in 12 h decreased from 100% w/w in F14 to about 78.5% in F17. This also was clear in t75 to a large extent where t75 for F14 (5.08 h) significantly increased to 9.96 h in F17. F14 and F15 showed a release exponent value (n) >1 indicating super case II transport resulting from increased swelling which usually occurs after long time periods. On the other hand F16 and F17 showed a release exponent of 0.91 and 0.929 respectively, indicating a zero order release kinetic or case II transport respectively.
Fig. 4.

Mean (± s.d.) percent of fenoterol release from Kollidon® SR Compression coated tablets F14–F17
It is noteworthy that incorporation of fenoterol in the core tablet and then, coated with a compression coat gave a better release profile than its incorporation in a matrix tablet where it gave a more sustained release effect. This was shown when comparing matrix tablet F1 containing 40 mg Kollidon® SR with compression coated core tablet F16 containing also 40 mg Kollidon® SR, but divided in the core tablet (15 mg) and the rest is in the compressed coat (25 mg). F16 showed a significant higher t75 (8.92 h) compared to 2.85 h in F1 (Table III). Besides, F16 showed a typical zero order release kinetic rather than Fickian drug release in F1. This could be attributed to that, in case of matrix tablets, fenoterol was homogenously dispersed in Kollidon® SR which forms a matrix block upon compression as it is composed of 8 parts of water insoluble polyvinyl acetate and 2 parts of water soluble polyvinyl pyrrolidon (povidon). The povidon component gradually leaches out of the matrix during dissolution thereby creating pores for the drug to diffuse out. The compressed polyvinyl acetate maintains tablet structure during the dissolution process resulting in the desired diffusion and hence sustained-release effect of a water soluble drug as fenoterol HBr (3). However in case of compression coated core tablets, the drug was present in the core surrounded by the matrix block of Kollidon® SR, when the povidon component leaches gradually creating pores for the drug to diffuse, the drug will still have longer path length to diffuse through compared to the matrix tablets hence producing more sustained release effect.
Polyox® WSR 303
The cumulative percentage of fenoterol released from matrix and compression coated tablets containing different amounts of Polyox® WSR 303 is shown in Fig. 5. Matrix tablets F18 and F19 containing 40 and 50 mg Polyox® WSR 303 respectively, showed nearly the same release profile with cumulative percentages of fenoterol released in 12 h of 94.6 and 95.7% respectively. Also t75 of F19 (6.72 h) was not significantly different from F18 (4.57 h). The kinetic treatment data in Table III showed that both F18 and F19 followed a non-Fickian release mechanism of exponent (n) values of 0.73 and 0.78.
Fig. 5.

Mean (± s.d.) percent of fenoterol release from Polyox® 303 matrix tablets (F18 and F19) and compression coated tablets (F20–F25)
For compression coated core tablets, it was found that in tablets having fixed coat composition and varied core composition (F20, F21 and F22), increasing the Polyox® WSR 303 amount in tablet core significantly decreased fenoterol release as shown Fig. 5. The cumulative percentage of fenoterol released in 12 h was 97.5, 92.69 and 86.4% for F20, F21 and F22 respectively. The three formulas showed release exponent (n) values >1 indicating a super case II transport release mechanism resulting from increased swelling which usually occurs after long time period.
The same held true regarding tablet formulas F23 to F25 with a constant core composition and different coat amounts of Polyox® WSR 303 and Emcompress®. Increasing Polyox® WSR 303 amount in the coat composition from 20 mg in F23 to 40 mg in F25 decreased the cumulative amount of fenoterol released in 12 h from 100% w/w to 81.1% respectively (Fig. 5). Also, it significantly increased t75 from 7.62 h to 10.51 h respectively. The release exponent (n) values for F23, F24 and F25 were 1.04, 1.36 and 1.13 respectively indicating super case II transport.
Polyethylene oxides (Polyox®) are hydrophilic, linear, uncross linked molecules. When, matrices containing these types of polymer come into contact with water some forces of attraction (chiefly hydrogen bonding) start acting between polymer and water. Due to the high water affinity of the polymer, these forces are likely to be preferred over polymer–polymer interactions. Therefore the forces holding the polymer segments together is reduced and the polymer chains can swell (10). Water diffuses into the matrix and when its concentration reaches a threshold value, the polymer swells and a gel layer is formed at the beginning to unfold and gradually become solvated. This complex gelatinous layer forms a diffusional barrier that retards further water uptake but can also modulate drug release rate. Water soluble drugs as fenoterol HBr are mainly released by diffusion of dissolved drug molecules across the gel layer formed after polymer swelling, while poorly water soluble drugs are more likely released through the gel erosion. When the release mechanism involves the synchronization of swelling and erosion rates, the matrix may provide zero-order release kinetics (11,12).
Precirol® ATO5
On trying Precirol® ATO5 as a release retardant incorporated as a physical mixture to form fenoterol matrix tablets (F26 to F29), it was found that increasing Precirol® ATO5 amounts from 20 mg in F26 to 50 mg in F29 decreased the cumulative amount of fenoterol released in 12 h from 92.21% to 52.61% respectively (Fig. 6). On the other hand Table III shows that the t75 significantly increased more than 3 folds from F26 (5.52 h) to F29 (17.79 h). The kinetic analysis data listed in Table III showed release exponent values (n) of 0.6, 0.67, 0.63 and 0.66 for F26, F27, F28 and F29 respectively indicating an anomalous diffusion (non-Fickian) release kinetic resulting from coupling between diffusional and macromolecular relaxation mechanisms (13).
Fig. 6.

Mean (± s.d.) percent of fenoterol release from Precirol® matrix tablets (F26–F29) and Compression coated tablets (F30–F33)
Figure 6 shows the cumulative percentage of fenoterol release from compression coated tablets F30 through F33. F30 containing 5 mg Precirol® ATO5 in the coat composition but has polymer free core shows a cumulative amount of fenoterol release in 12 h of 97.03%. However, incorporation of 5 mg Precirol® ATO5 in the core composition as well as the coat (F31), decreased the cumulative amount of fenoterol released in 12 h to 84.1%. The t75 was significantly increased from 7.72 to 9.52 h for F30 and F31 respectively (Table III). Increasing Precirol® ATO5 amount in the coat composition to 10 mg in F32 rather than 5 mg in F30 decreased the cumulative amount of fenoterol release in 12 h to 66.67% compared to 97.03% respectively and significantly increased t75 from 7.72 h in F30 to 14.31 h in F32. This release retardation was further enhanced by incorporation of 10 mg Precirol® ATO5 in the tablet core composition as well as in the coat represented by F33 where the cumulative amount of fenoterol released in 12 h was 57.03% and t75 was significantly increased to 16.72 h. Table III shows that the release exponents (n) of F30, F31 and F32 were 0.84, 0.878 and 0.884 respectively indicating zero order release mechanism while, F33 showed release exponent value 0.937 indicating super case II mechanism.
It is noteworthy that incorporation of precirol® in the tablet coat only gave slower release compared to division of the same amount of polymer in tablet core and coat, where F32 formed of 10 mg Precirol® in the coat gave 66.67% cumulative percent of fenoterol release in 12 h compared to F31 (5 mg in core and 5 mg in coat) which gave 84.1% cumulative percent of fenoterol release after 12 h. Precirol® is a lipid based insoluble wax excipient that control drug release by slow matrix erosion and drug diffusion. Drug release from this insoluble matrix material is mainly dependant upon the rate and extent of water permeation and aqueous solubility of the drug compounds that are embedded in the matrix. Presence of precirol® on the coat only created a waxy hydrophobic barrier against water permeation, hence slowing drug release. However incorporation of part of it in the core tablet with the drug enhanced the rate and extent of water permeation due to the solubility of the drug.
The in vitro drug release studies carried out have shown that by selecting the appropriate pharmaceutical excipients and the manufacturing technique, it would be possible to modify drug release profiles of fenoterol from tablet formulation and achieve sustaining release for up to 8–12 h. This will enable a twice daily regimen in vivo.
One tablet from each of the three employed polymers was chosen for further in vivo study. The parameter of choice was a compromise between t75 being in the range of 7–9 h, the kinetic release exponent being almost zero order and the release profile being 90 ± 15% after 8 h which is the target for a sustained release product as previously reported (14). Hence the chosen Formulas were the compression coated core tablets F16, F23, and F30 prepared using Kollidon® SR, Polyox® WSR 303 and Precirol® respectively.
In Vivo Study
The LC-MS/MS assay has been validated and has a good linearity from 1–100 ng/ml with acceptable within-and between-day reproducibility. The lower limit of fenoterol quantification in plasma was 1 ng/ml.
The mean plasma concentration-time profiles of fenoterol after oral administration of formulated compression coated core tablets and immediate release tablet are shown in Fig. 7. The pharmacokinetic parameters are listed in Table IV. The plasma concentration-time profiles showed that the compression coated core tablets sustained the oral absorption of fenoterol, expressed by the significant lower Cmax, significant delayed Tmax and the significant higher HVDt50%Cmax. The mean Cmax of F16, F23, and F30 were 80.9 ± 1.8, 69.95 ± 3.31 and 61.23 ± 8.06 ng/ml respectively compared to 146.56 ± 21.52 ng/ml for the immediate release tablet. The mean Tmax of F16, F23 and F30 were 5.83 ± 1.83, 6.16 ± 1.6 and 4.33 ± 1.36 h compared to 1.33 ± 0.51 h in case of Berotec® tablet. The HVDt50%Cmax were 6.81 ± 0.67, 6.52 ± 2.11 and 4.92 ± 1.33 for F16, F23 and F30 compared to 2.71 ± 0.68 h in case of Berotec®. Furtermore the MRT was significantly prolonged from 7.03 ± 0.76 h with Berotec® to 10.93 ± 1.25, 12.61 ± 2.16 and 9.49 ± 1.20 with F16, F23 and F30 respectively. The 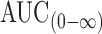 of F16 and F23 was slightly higher than that of Berotec®, however this difference was not statistically significant (P < 0.001).
of F16 and F23 was slightly higher than that of Berotec®, however this difference was not statistically significant (P < 0.001).
Fig. 7.

Mean plasma concentrations—time Profiles of Fenoterol from different formulas
Table IV.
Pharmacokinetic Parameters of Fenoterol after Oral Administration of Different Formulas
| Pharmacokinetic parameter | F16 | F23 | F30 | Berotec® |
|---|---|---|---|---|
| Cmax (ng/mL) | 80.9 ± 1.8 | 69.95 ± 3.31 | 61.23 ± 8.06 | 146.52 ± 21.52 |
| Tmax (h) | 5.83 ± 1.83 | 6.16 ± 1.6 | 4.33 ± 1.36 | 1.33 ± 0.51 |
| AUC 0–24 | ||||
| (ng/ml * h) | 750.669 ± 54.8 | 627.59 ± 92.9 | 468.28 ± 105.01 | 681.66 ± 68.02 |
 (ng/ml * h) (ng/ml * h) |
819.47 ± 40.6 | 718.19 ± 80.03 | 499.28 ± 115.4 | 708.25 ± 59.09 |
| HVDt50%Cmax | ||||
| (h) | 6.81 ± 0.67 | 6.52 ± 2.11 | 4.92 ± 1.33 | 2.71 ± 0.68 |
| RD | 2.51 | 2.41 | 1.82 | 1 |
| K (h−1) | 0.11 ± 0.03 | 0.09 ± 0.03 | 0.13 ± 0.02 | 0.13 ± 0.02 |
| t 1/2 (h) | 6.25 ± 1.73 | 8.68 ± 3.21 | 5.6 ± 0.96 | 5.31 ± 1.05 |
| MRT (h) | 10.93 ± 1.25 | 12.61 ± 2.16 | 9.49 ± 1.20 | 7.03 ± 0.76 |
All formulae between the same brackets are not significantly different from each other but differ significantly from those included in other brackets (P < 0.001)
Cmax (ng/ml): (Commercial) (F16, F23, F30), AUC(0–24) (ng/ml.hr): (Commercial, F16, F23), (F30),  (ng/ml.hr): (Commercial, F16, F23), (F30), t1/2(hr): (Commercial, F16, F23, F30), Tmax (hr): (Commercial), (F16, F23, F30), HVDt50%Cmax (h): (Commercial) (F16, F23, F30), MRT (hr): (Commercial), (F16, F23), (F30)
(ng/ml.hr): (Commercial, F16, F23), (F30), t1/2(hr): (Commercial, F16, F23, F30), Tmax (hr): (Commercial), (F16, F23, F30), HVDt50%Cmax (h): (Commercial) (F16, F23, F30), MRT (hr): (Commercial), (F16, F23), (F30)
F16 formed of Kollidon SR® proved to be superior in sustaining the oral absorption of fenoterol compared to F23 and F30, expressed in its higher RD (2.51) compared to 2.41 and 1.82 for F23 and F30 respectively.
CONCLUSION
The study revealed that Kollidon® SR is a suitable retardant polymer that could be used in the formulation of sustained release compression coated core tablets of fenoterol HBr. The study showed that formula F16 could extend the drug release for 12 h which will enable a twice daily regimen in vivo. When compared to the immediate release tablet Berotec®, the smooth and extended absorption phase coupled with maintenance of plasma concentration for longer duration after administration of compression coated tablets suggests reduced chance of dose-dependent side effects of fenoterol and trying to prevent the nocturnal asthma.
Acknowledgement
The authors would like to thank Genuine Research Center, Cairo, Egypt, for giving them the chance to use LC-MS/MS API 3200.
References
- 1.Goodman and Gillman. The Pharmacological Basis of Therapeutic, 10th Ed., McGraw Hill, London, 2001, p. 229.
- 2.Draganoiu E., Andheira M., Sakr A. Evaluation of the new polyvinylacetate/povidone excipient for matrix sustained release dosage forms. Pharm. Ind. 2001;63:624–629. [Google Scholar]
- 3.Shao Z. J., Farooqi M. I., Diaz S., Krishna A. K., Muhammad N. A. Effects of formulation variables and post-compression curing on drug release from a new sustained-release matrix material: polyvinylacetate-povidone. Pharm. Dev. Technol. 2001;6(2):247–254. doi: 10.1081/PDT-100002201. [DOI] [PubMed] [Google Scholar]
- 4.Kim C. Effects of drug solubility, drug loading, and polymer molecular weight on drug release from Polyox tablets. Drug Dev. Ind. Pharm. 1998;24(7):645–651. doi: 10.3109/03639049809082366. [DOI] [PubMed] [Google Scholar]
- 5.Reynolds T. D., Gehrke S. H., Hussain A. S., Shenouda L. S. Polymer erosion and drug release characterization of hydroxypropyl methylcellulose matrices. J. Pharm. Sci. 1998;87(9):1115–23. doi: 10.1021/js980004q. [DOI] [PubMed] [Google Scholar]
- 6.Saraiya K., Bolton S. Use of Precirol to prepare sustained release tablets of theophylline and quinidine gluconate. Drug Dev. Ind. Pharm. 1990;16(13):1963–1969. doi: 10.3109/03639049009023634. [DOI] [Google Scholar]
- 7.Ritger P. L., Peppas N. A. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J. Contr. Rel. 1987;5:37–42. doi: 10.1016/0168-3659(87)90035-6. [DOI] [PubMed] [Google Scholar]
- 8.Meier J., Nuesch E., Schmidt R. Pharmacokinetic criteria for the evaluation of retard formulations. Eur. J. Clin. Pharmacol. 1974;7(6):429–432. doi: 10.1007/BF00560355. [DOI] [PubMed] [Google Scholar]
- 9.Steinijans V. W. Pharmacokinetic characterization of controlled release formulations. Eur. J. Drug Metab. Pharmacokinet. 1990;15:173–181. doi: 10.1007/BF03190201. [DOI] [PubMed] [Google Scholar]
- 10.Wan L. S., Heng P. W., Wong L. F. Effect of hydroxypropylmethylcellulose on water penetration into a matrix system. Int. J. Pharm. 1991;73:111–116. doi: 10.1016/0378-5173(91)90033-K. [DOI] [Google Scholar]
- 11.Colombo P., Bettini R., Massimo G., Catellani P. L., Santi P., Peppas N. A. Drug diffusion front movement is important in drug release control from swellable matrix tablets. J. Pharm. Sci. 1995;84(8):991–997. doi: 10.1002/jps.2600840816. [DOI] [PubMed] [Google Scholar]
- 12.Maggi L., Segale L., Torre M. L., Ochoa M. E., Conte U. Dissolution behaviour of hydrophilic matrix tablets containing two different polyethylene oxides (PEOs) for the controlled release of a water-soluble drug. Dimensionality study. Biomaterials. 2002;4:1113–1119. doi: 10.1016/S0142-9612(01)00223-X. [DOI] [PubMed] [Google Scholar]
- 13.Efentakis M., Vlachou M. Evaluation of high molecular weight poly(oxyethylene) (Polyox) polymer: studies of flow properties and release rates of furosemide and captopril from controlled-release hard gelatin capsules. Pharm. Dev. Technol. 2000;5(3):339–346. doi: 10.1081/PDT-100100549. [DOI] [PubMed] [Google Scholar]
- 14.Cohen J. L., Hubert B. B., Rhodes C. T. The development of USP dissolution and drug release standards. Pharm. Res. 1990;7:983–987. doi: 10.1023/A:1015922629207. [DOI] [PubMed] [Google Scholar]