

Abstract
This study examined the release of carbamazepine (CBZ) from hydrophobic (Compritol® 888 ATO) and hydrophilic-hydrophobic matrix combination (Compritol® 888 ATO-hydroxpropyl methylcellulose, HPMC). Hydrophobic matrix tablets were prepared by hot fusion technique, while hydrophilic-hydrophobic matrix tablets were prepared by wet granulation technique. The properties of the compressed matrix tablets were determined according to the US Pharmacopoeia. Both matrix formulations displayed a controlled-release profile when compared to the reference formulation (Tegretol® CR 200). The bioavailability of CBZ formulations and Tegretol® CR 200 were evaluated in beagle dogs. Carbamazepine presented a significant higher bioavailability from matrix tablets containing hydrophilic polymer (HPMC) than that obtained from Tegretol® CR200. The average inter-subject plasma concentration variability CV% was the least with tablet containing hydrophilic polymer (HPMC) and was the highest with Tegretol® CR 200 (33.8 and 54.1, respectively). Analysis of variance applied to log 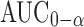 and log C max showed statistical significant differences among the three formulations (P < 0.05). Plotting the fraction of CBZ released in vitro and fraction absorbed showed a statistically significant relationship (R2 = 0.935–0.975) for the three matrix tablets examined.
and log C max showed statistical significant differences among the three formulations (P < 0.05). Plotting the fraction of CBZ released in vitro and fraction absorbed showed a statistically significant relationship (R2 = 0.935–0.975) for the three matrix tablets examined.
Key words: bioavailability study, carbamazepine, controlled release matrix, in vitro/in vivo correlation
INTRODUCTION
Over the past 30 years, significant medical advances have been made in the area of drug delivery with the development of novel dosage forms. Controlled-release formulations have been one of the major focuses in pharmaceutics. Matrix systems appear to be a very attractive approach in controlled-release system (1). Matrix type formulations are prepared from either swellable hydrophilic polymers or non-swellable lipophilic excipients, like waxes and lipids. The use of lipid and wax matrix seems to have a particular advantage due to their chemical inertness against other materials, better characterization of lipidic excipients and formulation versatility and the choice of different drug delivery systems (2). Recently, much attention has been focused on the use of Gelucires as carriers in drug delivery systems. The Gelucire containing only glyceride are used in preparation of controlled-release formulations. In particular, Compritol 888 ATO5 or glyceryl behenate can be used as glyceride base for the preparation of controlled-release dosage forms (3,4). When lipophilic excipients is used as a matrix, the drug substance and the excipients have to be formulated into a solid dispersion; just mixing the ingredients is not enough. In fusion method, no organic solvent or water is required, moreover, in melt methods the drying step is not necessary, a satisfactory retard profile can be achieved with low wax content and the process is therefore applicable to high dose drugs.
In practice for the controlling of drug release from matrix device, polymeric hydrogel is being investigated for controlled-release applications. Hydrogels can be applied for both hydrophilic and hydrophobic drugs. Hydrophilic polymers, in particular cellulose derivatives, have been widely used in the formulation of hydrogel matrices which satisfy the key criteria for the development of controlled-release oral solid dosage forms. Different types of modified cellulose polymers are usually employed, either alone or with other swellable polymers (5) and with hydrophobic polymers (6). A study has been made on the in vitro release from a matrix comprising hydrophobic and hydrophilic (gel-forming) components containing different non-steroidal anti-inflammatory agents (7).
Carbamazepine (CBZ)-a tricyclic iminostilbene derivative is the treatment of choice for simple partial, complex partial and secondarily generalized seizures (8). Carbamazepine shows slow and irregular absorption and unpredictable fluctuations in the plasma levels of both CBZ and its metabolite CBZ 10, 11-epoxide (9) and these, in turn, lead to the occurrence of intermittent side-effects. These can be minimized by increasing the frequency of dosing but at the expense of compliance (10). With immediate-release CBZ doses, failure to adhere to a 3-or 4-time-daily dosing schedule can contribute to variability in serum drug level, which are also affected by many other factors; thus, suggesting the study of modified release formulations as a powerful approach to improve its therapeutic use (11). Barakat and Radwan (12), prepared CBZ-loaded polylactic co-glycolic microspheres which exhibits sustainer release profile over 10 h. Emulsion congealing technique has been successfully used for preparation of CBZ-Precifac®ATO lipospheres (13).
In the present study, various matrix systems were designed and tested for controlled delivery of carbamazepine. The objectives of the study were (1) to evaluate the influence of matrix excipients on the drug release behavior from matrix tablets (2) to investigate both in vitro and in vivo performance of hydrophilic and hydrophobic matrix systems in controlling the release of carbamazepine, and (3) to explore the relationship between in vitro release and in vivo absorption.
EXPERIMENTAL
Materials
Carbamazepine powder, (CBZ; Novartis Pharma, Egypt). Tegretol® CR 200 tablet, (Reference) Batch no. T4171, (Novartis Pharma AG, Basle, Switzerland). Compritol® 888 ATO (USP-NF glyceryl behenate) was a gift from Gattefosse (Saint Priest, France). Diethyl ether (BDH Lab, England). Formic acid 98–100% (Suffolk, England). Phenacetin (BDH Chemicals Ltd, Poole, England). Methanol and acetonitrile hypersolv for HPLC, VWR International Ltd, England.
Preparation of Matrix Tablets
Hydrophobic Matrix Tablets
Composition of hydrophobic matrix is listed in (Table I). Carbamazepine and Compritol® 888 ATO were mixed homogeneously; the blend was then heated (70–80 °C) in a water bath (model GFL Labortechnik GMBH, Germany) with continuous agitation. The molten mass was allowed to cool at room temperature. The congealed solid mass was then sieved, fraction sizes between 710–500 μm were blended with 1% magnesium stearate as a lubricant and 5% Ac-Di-Sol as disintegrant. The granules then compressed into tablets using 9 mm diameter flat-faced punch.
Table I.
Composition of Carbamazepine 200 mg Matrix Tablets
| Ingredients | Matrix F (mg/tablet) | Matrix L (mg/tablet) |
|---|---|---|
| Carbamazepine | 200 | 200 |
| Compritol® 888 ATO | 200 | 6.6 |
| Methocel K15M | – | 52.8 |
| Avicel® | – | 6.6 |
| Ac-Di-Sol | 20 | – |
| Magnesium stearate | 4 | 2.6 |
Hydrophilic Matrix Tablets
Composition of hydrophilic matrix is listed in (Table I). The tablets were prepared by wet granulation technique. Drug, Compritol® 888 ATO, hydroxypropyl methylcellulose and Avicel were mixed thoroughly with a pestle and mortar. A 10% alcoholic solution of PVP was added to this mixture drop wise with continuous mixing. The wet mass was sieved through sieve no 14. The granules were then dried at 60 °C for 6 h using a convection oven. After adding 1% magnesium stearate as a lubricant, the resulting dried and size-distributed granule mixtures (25–35 mesh) were directly compressed into tablets using a tablet machine (Erweka, Germany) equipped with fraction 710–500 μm were lubricated and compressed into tablets using 9-mm flat faced punch.
Tablet Properties
The properties of the compressed tablets, such as drug content, weight variation, hardness, thickness, disintegration time, and friability were determined.
In Vitro Release Studies
The release characteristics of CBZ from tablet formulations or Tegretol® CR were determined according to the USP dissolution II paddle method at a rotation speed of 75 rpm in 900 ml of water containing 1% sodium lauryl sulfate at 37 ± 0.5 °C using dissolution tester (Erweka DT-6, Germany). Five milliliters of samples were taken at appropriate intervals and volumes were replenished with fresh test medium. Collected samples were filtered through 0.45 μm Millipore filters. The concentration of CBZ in the dissolution samples was spectrophotometrically determined at 285 nm using a UV spectrophotometer (Ultrospec 2100 Pro, UV/Visible Spectrophotometer, Cambridge England).
The in vitro release profiles of CBZ formulations and Tegretol® CR were compared using similarity factors, f2, as defined by the following equation (14).
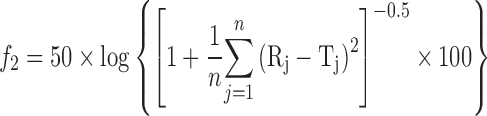 |
1 |
where n is number of time points, and Tj and Rj are percentage release at time point (t) for the reference and test tablet, respectively.
Dissolution efficiency (DE%) was calculated from the area under the dissolution curve at time t (measured using the trapezoidal rule) and expressed as percentage of the area of the rectangle described by 100% dissolution in the same time (15).
The mean dissolution time determined for drug release up to 80% (MDT-80%) was calculated from dissolution data using the following expression (16).
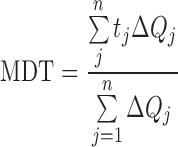 |
2 |
where j is the sample number, n is the number of time increments considered, tˆj is the time at midpoint between tj and tj − 1, and ΔQj the additional amount of drug dissolved in the period of time tj and tj − 1.
Data Analysis
In order to propose the possible release mechanism, the drug release from CBZ formulations was fitted to the following power model (17).
 |
3 |
where 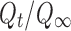 is the fractional drug release percentage at time t. The k is the kinetic constant related to the properties of the drug delivery system and n is the diffusional exponent, which characterizes the drug transport mechanism. A value of n = 0.45, indicates Case I (Fickian) diffusion, 0.45 < n < 0.89 indicates anomalous (non-Fickian) diffusion and n = 0.89 indicates case II transport. This equation can be used to analyze the first 70% of a release curve.
is the fractional drug release percentage at time t. The k is the kinetic constant related to the properties of the drug delivery system and n is the diffusional exponent, which characterizes the drug transport mechanism. A value of n = 0.45, indicates Case I (Fickian) diffusion, 0.45 < n < 0.89 indicates anomalous (non-Fickian) diffusion and n = 0.89 indicates case II transport. This equation can be used to analyze the first 70% of a release curve.
In Vivo Absorption Study
Six male beagle dogs weighing 8–14 kg were used in this study in accordance with a protocol approved by the Institutional Review Board-Use and care of Animals at King Saud University. The study was designed to include a single dose, three-way crossover oral paradigm in which three CBZ products were administered. The animals were housed in polypropylene cages at 25 ± 1 °C and 45–55% relative humidity with a 12-h light/dark cycle. All dogs were fasted overnight prior to the experiment, no food was allowed until a standard meal was served 4 h after dosing. Water was available ad libitum 2 h after dosing and throughout the study period. On each occasion, dogs received orally the following formulations: controlled-release commercial CBZ tablet (Tegretol® CR 200); Formula F; and Formula L. In all cases, a 2-week washout period was allowed between successive treatments. The tested formulations were administered with 100 ml of water. The dog’s leg was shaved and a forefoot vein was cannulated using an 18 gauge cannula. Serial 3-ml blood samples were collected from each dog just prior to dose administration and at each of the following time points: 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 48, and 72 h following oral dosing. Heparinized blood samples were centrifuged immediately for 15 min at 3500 rpm, and the plasma transferred into glass containers to be kept frozen at −20 °C pending analysis. The plasma concentrations of CBZ were determined by the modified high-performance liquid chromatography modified method of Gavini et al. (18).
Chromatographic Conditions
Chromatography was carried out on Shimadzu HPLC equipped with Class VP software for data processing. Samples were analyzed on a reverse-phase Bondapak C18 column attached to a C18 precolumn, detector was set at 220 nm and mobile phase was pumped at a flow rate of 0.8 ml/min. All determinations were made at ambient temperature. The mobile phase consisting of mixture of acetonitrile/methanol/formic acid (0.1%; 10:50:40 v/v, pH 3.34). The mobile phase passed through 0.45 μm Millipore filter and degassed by ultrasonication under vacuum before use. Volume of the injected sample was 50 μl. The amount of CBZ in plasma was expressed as μg ml−1 plasma.
Analytical Procedure of Plasma Samples
An extraction method of assay was performed to calculate CBZ concentration in plasma sample. Briefly, to a series of six 15-ml ground glass stoppard reaction vessels add CBZ in methanol (10 μg ml−1) in amounts of 5, 10, 20, 40 and 80 μl to prepare standard curve in the range 0.25–4 μg ml−1. Add 50 μl phenacetin (as internal standard) in methanol (20 μg ml−1) to these tubes. Add 0.2 ml of blank plasma to each tube in the standard curve series. Add an identical amount of phenacetin to a series of clean 15-ml tubes for the samples. Add 0.2 ml thawed plasma to be analyzed to the sample tubes. Mix the content of the tubes for 30 s with a vortex-type mixer. Add 4 ml diethyl ether to the tubes, mix the tube content vigorously for 30 s. Centrifuge at 3,500 rpm for 15 min, transfer the upper organic layer by Pasteur pipette to a disposable 15 ml culture tube, evaporate the solvent in a gentle current of air at 40 °C. Dissolve the residue in 200 μl of HPLC eluent by vortex-mixing for 20 s, pour into a micro-centrifuge tube, centrifuge at 20,000 rpm at 10 °C for 5 min and inject 50 μl of clear supernatant onto the column.
Validation of Assay Methodology
The assay procedure was validated in term of linearity, precision, accuracy and recovery.
Linearity
A calibration curve was generated from the resulting chromatograms based on the ratio of the peak area of CBZ to IS. A standard curve of five replicates at each data point (0.25, 0.5, 1, 2, and 4 μg ml−1) was constructed and goodness-of-fit was determined by linear regression.
Precision and Reproducibility
Within-and-between-day reproducibility studies were performed on five replicates; five different concentrations of CBZ (in the linear range) were measured three times in a consecutive manner to obtain within-day reproducibility. The between-day analysis of the same concentrations was repeated on 15 separate days. Precision was expressed as coefficient of variation (CV%) calculated from recovered standards assayed in the same day or on different days.
Recovery
The physical recovery of CBZ was determined by comparing the peak-height ratios measured in the extracts of pooled plasma contain known amounts of CBZ with those peak-height ratios measured in unextracted samples supplemented with similar working range of CBZ, keeping the injection volume constant. For purpose of this calculation we added internal standard to the samples just before injection into the chromatograph. Percentage recovery of the drug was calculated from the following equation.
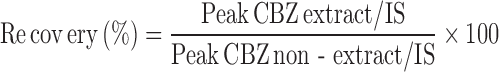 |
4 |
Calculation of CBZ Concentration
Standard curve constructed for peak area ratios are adjusted by linear regression analysis to express peak-area ratio (CBZpeak area/ISpeak area) as a function of CBZ concentration of the standards. The mean best fit linear regression equation was used to estimate the concentrations of CBZ in the unknown plasma samples at different time intervals.
Calculation of CBZ Pharmacokinetic Parameters
Pharmacokinetic parameters were estimated from the individual plasma concentrations versus time profiles. The peak plasma concentration (Cmax), the time to reach the maximum peak (tmax) and the times of CBZ firstly appeared in the plasma (tlag) were obtained as directly measured values. The extent of absorption (AUC0 − t) was calculated using linear trapezoidal rules. Extrapolated AUCs (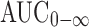 ) were determined by the following equation:
) were determined by the following equation:
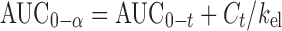 |
5 |
kel was estimated by fitting the logarithm of the concentrations versus time to a straight line over the observed exponential decline.
The Wagner–Nelson model was used to calculate the percentage of CBZ dose absorbed profiles (19).
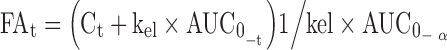 |
6 |
where FAt is the fraction of drug absorbed at time t, Ct is the concentration of drug in the plasma at time t and k is the elimination rate constant. The elimination rate constant, kel, was calculated from the mean plasma concentration-time profile after administration of immediate release tablets. The in vivo absorption values were directly related to in vitro dissolution data to complete the in vitro–in vivo correlations.
Statistical Evaluation of Results
All results were expressed as mean values ± standard deviation (SD). In order to assess the statistical significance between the data, a single factor analysis of variance (ANOVA) was applied with the Tukey multiple comparison procedure, employing Graphpad INSTAT tm, Copyright© 1990–1993, Graphpad Software V 2.04, Ralf Stahlman, Purdue Univ. 931897S. The level of statistical significance was chosen as less than 0.05 (P < 0.05).
RESULTS AND DISCUSSION
Characterization of the Tested Tablets
The physical characterization of the tested CBZ tablets according to the USP 24 compendia requirement with respect to CBZ content, weight, thickness, hardness, friability and disintegration time are given in Table II. For both formulations, drug content ranging from 99.30% to 103.45%, based on the theoretical composition, which evidences content uniformity. It was also verified that the tablets passed the friability test (F < 1%), showing that both formulations lie within the established limits. Dissolution profile of fresh CBZ products was performed using USP 24 dissolution apparatus II, 75 rpm in 1% sodium lauryl sulphate dissolution medium at 37 ± 0.5 °C is given in (Fig. 1). The release kinetics was illustrated in (Table III). Both hydrophobic and hydrophilic matrix tablets tended to exhibit non-Fickian (anomalous) diffusion characteristics (n = 0.654 and 0.637, respectively), while reference tablets showed Fickian release behavior (n = 0.433). The similarity factor (f2) values of hydrophobic and hydrophilic matrix tablets are greater than 50 (61.3 and 56.0, respectively) means an average difference of no more than 10% at the sample time points, ensures equivalence of the test and reference products. The mean dissolution time up to 80% is applied to compare dissolution rates of CBZ products, no significant different was shown with different CBZ products (MDT ranged from 1.9 to 2.1 h).
Table II.
Physical Characterization of Matrix Tablets of Carbamazepine
| Matrix formulation | CBZ content (%) | Weighta (mg) | Thicknessa (mm) | Hardnessa (kg/cm2) | Friabilityb (%) | Disintegrationa time (min) |
|---|---|---|---|---|---|---|
| Formula F | 102.34 ± 1.02 | 425.54 ± 1.35 | 3.0 ± 0.03 | 5.10 ± 0.32 | 0.65 ± 0.02 | 5.0 ± 0.02 |
| Formula L | 98.95 ± 0.98 | 270.3 ± 1.67 | 2.9 ± 0.06 | 6.3 ± 0.19 | 0.34 ± 0.03 | >360 min |
aData are the mean of ten determinations
bData are the mean of two determinations
Fig. 1.

In vitro dissolution profiles of CBZ from three different CBZ products in 1% sodium lauryl sulfate aqueous solution. Each point represents mean values of six tablets
Table III.
Mathematical Modeling and Dissolution Parameters for Carbamazepine Release from Tegretol® CR and Formula F and L Matrix Tablets
| Matrix formulation | Regression coefficient (r) | Kinetic constant k, (h −n) | n | DE (%) | f 2 | MDT (h) |
|---|---|---|---|---|---|---|
| Formula F | 0.9799 | 33.32 | 0.654 | 68.5 | 61.3 | 1.99 |
| Formula L | 0.9929 | 34.90 | 0.637 | 65.4 | 56.0 | 2.10 |
| Tegretol® CR | 0.9989 | 44.46 | 0.433 | 71.3 | – | 1.91 |
Method Validation
A reliable separation of CBZ and the IS using the previously reported chromatographic conditions was demonstrated. Chromatographic performance was good for CBZ with good peak shapes and acceptable retention time for routine activity (3.16 and 4.47 min, respectively for IS and CBZ).
The analytical technique for CBZ in plasma was validated for linearity, accuracy and precision of determination (Table IV).
Table IV.
Validation of HPLC Technique for the Analysis of Carbamaz epine in Dogs Plasma
| Parameters | Values |
|---|---|
| Limit of detection (LOD; μg/ml) | 0.025 |
| Limit of quantitation (LOQ; μg/ml) | 0.25 |
| Linearity | |
| Range (μg/ml) | 0.25–4.0 |
| Regression equation | y = 4.8795 × −0.041 |
| Correlation coefficient (r) | 0.9978–0.9992 |
| Recovery range (%) | 70.45–80.43 |
| Precision | |
| Intra-day R.S.D. (%) | |
| With sample injected 3 times | 2.58 |
| With samples prepared 3 times | 3.71 |
| Inter-day R.S.D. (%) | |
| With sample injected 3 times | 5.74 |
| With samples prepared 3 times | 7.43 |
In Vivo Absorption of CBZ Products in Dogs
The three tablets were administered to dogs, in order to investigate the in vivo absorption profiles. The average CBZ plasma concentration versus time profiles following a single 200 mg CBZ as Tegretol® CR, Formula F, or Formula L are shown in Fig. 2. The highest plasma levels were obtained in dosing of Formula L-tablets. When CBZ tablets Formula L were administered to beagle dogs, drug appeared in plasma almost at the same time in each dog after a ttag of 0.566 ± 0.1 h. The onset of drug absorption from Tegretol® CR tablet was found to be delayed, the mean ttag was 0.682 ± 0.264 ranging from 0.34 to 1 h.
Fig. 2.

Mean plasma concentration-time profiles following oral administration of CBZ tablets to beagle dogs under fasting. Each point represents mean ± SD (n = 6)
The mean pharmacokinetics parameters of CBZ derived from CBZ plasma-time profiles are summarized in (Table V). The mean peak CBZ concentration Cmax after oral administration of Formula L was higher than that of Tegretol® or Formula F tablet. The mean time to reach the peak concentration (tmax) of CBZ was comparable, 6–7 h for the three products. No statistically significant difference (P > 0.05) was observed among the tmax values of samples that were 6.67 ± 2.06, 7.00 ± 1.09 and 7.66 ± 1.50 for the three products Tegretol®, Formula F and Formula L, respectively. There was extremely significant difference (P < 0.001) in the terminal elimination rate constant among the three product. The 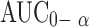 value of 25.27 ± 10.44, 30.09 ± 10.50 and 36.34 ± 10.66 for Tegretol®, Formula F and Formula L respectively were found to be significantly different (P > 0.05). This suggest that Formula L showed the highest rate and extent of drug absorption, whereas, Reference product showed the lowest rate and extent of drug absorption.
value of 25.27 ± 10.44, 30.09 ± 10.50 and 36.34 ± 10.66 for Tegretol®, Formula F and Formula L respectively were found to be significantly different (P > 0.05). This suggest that Formula L showed the highest rate and extent of drug absorption, whereas, Reference product showed the lowest rate and extent of drug absorption.
Table V.
Mean Pharmacokinetic Parameters of Carbamazepine Obtained After Oral Administration of Tegretol® CR, Formula F and L to Beagle Dogs
| Parameters | Products | ||
|---|---|---|---|
| Tegretol® CR | Formula F | Formula L | |
| AUC0–72 h (μg.h.ml−1) | 23.1 ± 9.561 (10.69–31.91) | 26.14 ± 11.705 (15.00–46.16) | 34.25 ± 10.43 (25.36–55.74) |
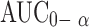 (μg.h.ml−1) (μg.h.ml−1) |
25.27 ± 10.44 (11.59–34.61) | 30.091 ± 10.496 (18.70–48.92) | 36.34 ± 10.67 (26.26–59.08) |
| C max (μg ml−1) | 1.38 ± 0.49 (0.91–2.09) | 1.352 ± 0.549 (0.76–2.01) | 2.18 ± 0.68 (1.34–3.18) |
| t max a (h) | 6.00 (4–10) | 7.00 (6–8) | 8.00 (6–10) |
| MRT (h) | 21.43 ± 4.57 (13.22–26.21) | 34.091 ± 9.749 (23.94–47.35) | 23.76 ± 5.17 (17.25–32.19) |
| t lag (h) | 0.68 ± 0.26 (0.34–1.0) | 0.58 ± 0.31 (0.12–0.91) | 0.57 ± 0.10 (0.41–0.69) |
| F rel(%)b | 119.1 | 143.8 | |
Values in parenthesis are range of data (n = 6)
aMedian data
bRelative bioavailability
The fact that a relationship exist between higher water penetration and faster absorption rate may be an explanation of this result. The gel layer of the Formula L-tablet (HPMC-based matrix) with higher water retention could be disintegrated by GI destructive forces and its in vivo release rate markedly accelerated, while the Formula F-tablet (50% wax-matrix) with low water penetration could absorb water slightly.
The percent coefficient of variation (CV%) for plasma concentration recorded at each sampling time following oral administration of the three CBZ products were calculated. The statistical analysis indicated insignificant differences (P > 0.05) among the three products examined at most sampling time, whereas, significant difference P < 0.05 was indicated at early sampling point, 0.5 and 1 h post dosing. The CV% appeared greater during the absorption phase than during the elimination phase after administration of Formula F. Following administration of Tegretol® CR and Formula L tablet the CV% appeared greater during the elimination phase. The average inter-subject plasma concentration variability CV% was 54.1, 52.9, 33.8 and following administration of Tegretol® CR, Formula F, and Formula L-tablets, respectively. The lower inter-subject variability for Formula L may be due to the presence of hydrophilic polymer HPMC, one of the suitable carriers for enhancement of the water-solubility of drugs as well as for prevention of drugs from re-crystallization in the dissolution medium (20). HPMC also, is known in the literature that it affects the solubility of CBZ polymorph (21). The percent coefficients of variation for selected pharmacokinetic parameters as a function of tested products to manifest the inter-subject variability showed small inter-subject variability for Formula L, with relative standard deviations (CV%) of 35 or less for all pharmacokinetics parameters. Reference and Formula F showed CV% of 63% or 58% or less, respectively, for all parameters. Different pharmacokinetic factors might influence the plasma concentration time profile and thus the in vivo dissolution profile.
As it is recognized from literature that CBZ absorption profile is characterized by irregular plasma levels. The variability of therapeutic efficiency can be attributed to inter-individual sensibility, chronobiologic effect, but also to rates of dissolution which can differ when polymorphs are induced by technologic operation (22). Otsuka et al. (23) showed that hydroxypropyl methylcellulose inhibited the dehydrate formation of CBZ. The presence of hydroxypropyl methylcellulose in sustained release CBZ tablets was also shown to affect the dissolution of the drug due to its inhibiting effect on dihydrate formation (21). The solubility of the anhydrous CBZ is approximately twice that of its dehydrate (24).
Analysis of variance applied to log 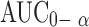 and log Cmax data as recommended by USP 24 are shown in Table VI. There are statistical significant differences between the values of
and log Cmax data as recommended by USP 24 are shown in Table VI. There are statistical significant differences between the values of 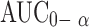 and Cmax calculated for both formulations.
and Cmax calculated for both formulations.
Table VI.
ANOVA of 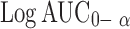 and Log C
max Following Oral Administration of Single Dose of Tegretol® CR, Formula F, and L Tablet (200 mg CBZ) to Six Beagle Dog
and Log C
max Following Oral Administration of Single Dose of Tegretol® CR, Formula F, and L Tablet (200 mg CBZ) to Six Beagle Dog
| Source of variation | Degree of freedom | Sum of squares | Mean square | F |
|---|---|---|---|---|
| Treatments (between columns) | 2 | |||
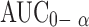
|
0.2182 | 0.1091 | 4.827a | |
| C max | 0.1915 | 0.0958 | 3.892a | |
| Residuals (within columns) | 15 | |||
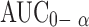
|
0.3389 | 0.0226 | ||
| C max | 0.3686 | 0.0246 | ||
| Total | 17 | |||
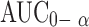
|
0.5570 | |||
| C max | 0.5601 |
aSignificant
In Vitro/In Vivo Relationship
Exploring a relation between the in vivo absorption and in vitro drug release from a controlled-release dosage form is an important part of the dosage form development process (25). Furthermore, it is crucial to develop a reproducible and predictable in vitro dissolution test to be used for optimization of the oral dosage form. A variety of factors affects the in vivo dissolution process, for example physicochemical factor of the drug or physiological factors in the gastrointestinal tract, such as intestinal motility and fluid secretion (26). In this study, the relationship between the in vitro dissolution data and the in vivo pharmacokinetic data was examined by plotting the fraction of drug dissolved (FD) after 0.5, 1, 1.5, 2, 3, and 4 h and the fraction absorbed data (FA) calculated at the same time post dosing (Fig. 3). The linear regression analysis showed that a statistically significant relationship (R2 = 0.935–0.974) existed between the FD and FA for the matrix tablets and was best described by the following equation: y = 1.012 × −0.0231 (Formula L); y = 1.0736 × −0.0428 (Tegretol®); and y = 1.028 × −0.0341 (Formula F). The slope and intercept were close to 1 and 0, respectively, indicating that the in vivo fraction absorbed could be predicted from in vitro dissolution data. Recently it has been proposed that a 1:1 (level A) relationship between in vitro dissolution and in vivo absorption is the most desirable type of correlation for extended-release dosage forms (USP 24) (27).
Fig. 3.

Relationship between the CBZ fraction dissolved in vitro and the CBZ fraction absorbed in vivo at 0.5, 1, 1.5, 2, 3, and 4 h for the three CBZ products
The current food and drug administration (FDA) guidelines for bioequivalence are that two formulations whose rate and extent of absorption differ by −20% to +25% or less are generally considered bioequivalent. This is based on the concept that difference of −20% to +25% would not lead to change in therapy for a patient. According to the literature, CBZ is known as a highly variable drug and, for this reason, many authors (28) have suggested widening the bioequivalence limit from 80–125% to 70–143%.
CONCLUSION
The hydrophobic and hydrophilic matrix formulations developed in this study are a viable oral dosage form of carbamazepine. Both matrix formulations showed higher relative bioavailability of CBZ than the reference Tegretol tablet. The in vitro dissolution test was able to reflect the in vivo absorption with Level A relationship between the three CBZ products.
Acknowledgements
The authors would like to thank the Research Center at College of Pharmacy, King Saud University for providing facilities to carry out the study (Grant No. A T 14-115).
References
- 1.Cardinal J. R. Matrix systems. In: Langer R. S., Wise D. I., editors. Medical Applications of Controlled Releas. Vol I, Classes of Systems. Boca Raton, FL, USA: CRC Press; 1984. pp. 41–67. [Google Scholar]
- 2.Stuchlík M., Žák S. Lipid-based vehicle for oral drug delivery. Biomed Papers. 2001;145:17–26. [PubMed] [Google Scholar]
- 3.Saraiya D., Bolton S. The use of PrecirolÒ to prepare sustained release tablets of theophylline and quinidine gluconate. Drug Dev Ind Pharm. 1990;16:1963–1969. doi: 10.3109/03639049009023634. [DOI] [Google Scholar]
- 4.N. S. Barakat. Formulation and evaluation of some extended-release drugs, Ph.D. dissertation, Alexandria University, College of Pharmacy, Alexandria, Egypt. 1996.
- 5.Katzhendler I., Mäder K., Friedman M. Structure and hydration properties of hydroxypropylmethylcellulose matrices containing naproxen and naproxen sodium. Int J Pharm. 2000;200:161–179. doi: 10.1016/S0378-5173(00)00360-4. [DOI] [PubMed] [Google Scholar]
- 6.Lotfipour F., Nokhodchi A., Saeedi M., Norouzi-Sani S., Sharbafi J., Siahi-Shadbad M. R. The effect of hydrophilic and lipophilic polymers and fillers on the release rate of atenolol from HPMC matrices. IL Farmaco. 2004;59:819–825. doi: 10.1016/j.farmac.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 7.Malamataris S., Ganderton D. Sustained release from matrix system comprising hydrophobic and hydrophilic (gel-forming) parts. Int.J Pharm. 1991;70:69–75. doi: 10.1016/0378-5173(91)90165-K. [DOI] [Google Scholar]
- 8.Bourgeois B. F. D., Wad N. Individual and combined epileptic and neurotoxic activity of carbamazepine and carbamazepine-10, 11-epoxide in mice. J Pharmacol. Exp Ther. 1984;231:411–415. [PubMed] [Google Scholar]
- 9.Perrucca E., Richens A. Clinical pharmacokinetics of antiepileptic drugs. In: Janz D, Frey H. H., editors. Antiepileptic drugs. Handbook of Experimental Pharmacology. Berlin: Springer; 1984. [Google Scholar]
- 10.Cramer J. A., Mattson R. H., Prevey M. C., Scheyer R. D., Ovellette V. L. How often is medication taken as prescribed? A novel assessment technique. J Am Med Ass. 1989;261:3273–3277. doi: 10.1001/jama.261.22.3273. [DOI] [PubMed] [Google Scholar]
- 11.McKee P. J. W., BlackLaw J., Carswell A., Gillham R. A., Brodie M. J. Double dummy comparison between once and twice daily dosing with modified-release carbamazepine in epileptic patients. Br J Clin Pharmacol. 1993;36:257–261. doi: 10.1111/j.1365-2125.1993.tb04226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barakat N. S., Radwan M. A. In vitro performance of carbamazepine loaded to various molecular weights of poly (D,L-lactide-co-glycolide) Drug Delivery. 2006;13:9–18. doi: 10.1080/10717540500308992. [DOI] [PubMed] [Google Scholar]
- 13.Barakat N. S., Yassin A. B. In vitro characterization of carbamazepine-loaded Precifac lipospheres. Drug Delivery. 2006;13:95–104. doi: 10.1080/10717540500313661. [DOI] [PubMed] [Google Scholar]
- 14.Moore J. W., Flanner H. H. Mathematical comparison of curves with an emphasis on in vitro dissolution profiles. Pharmaceut Technol. 1996;20:64–74. [Google Scholar]
- 15.Khan K. A. The concept of dissolution efficiency. J Pharm Pharmacol. 1975;27:48–49. doi: 10.1111/j.2042-7158.1975.tb09378.x. [DOI] [PubMed] [Google Scholar]
- 16.Voegele D., Brockmeier D., Von Hattingberg H. M., Lippold B. C. Die mittlere Auflösezeit-ein parameter zur prüfung von liberationsbedingungen auf vergleichbarkeit. Acta Pharm Techno. 1985;29:167–174. [Google Scholar]
- 17.Ritger P. l., Peppas N. S. A simple equation for disposition of solute release II: Fickian and anomalous release from swellable devices. J Control Rel. 1987;5:37–42. doi: 10.1016/0168-3659(87)90035-6. [DOI] [Google Scholar]
- 18.Gavini E., Hegge A. B., Rassu G., Sanna V., Testa C., Pirisino G., Karlsen J., Giunchedi P. Nasal administration of Carbamazepine using chitosan microspheres: In vitro/in vivo studies. Int J Pharm. 2006;307:9–15. doi: 10.1016/j.ijpharm.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 19.Wagner J., Nelson E. Kinetic analysis of blood levels and urinary excretion in the absorptive phase after single dose of drug. J Pharm. Sci. 1964;53:1392–1404. doi: 10.1002/jps.2600531126. [DOI] [PubMed] [Google Scholar]
- 20.Kushida I., Ichikawa M., Asakawa N. Improvement of dissolution and oral absorption of ER-34122, a poorly water-soluble dual 5-lipoxygenase/cyclooxygenase inhibitor with anti-inflammatory activity by preparing solid dispersion. J Pharm Sci. 2002;91:258–266. doi: 10.1002/jps.10020. [DOI] [PubMed] [Google Scholar]
- 21.Katzhendler I., Azoury R., Friedman M. Crystalline properties of carbamazepine in sustained release hydrophilic matrix tablets based on hydroxypropyl methylcellulose. J Control Rel. 1998;54:69–85. doi: 10.1016/S0168-3659(98)00002-9. [DOI] [PubMed] [Google Scholar]
- 22.Lefebvre C., Guyot-Hermann A. M., Draguet-Brighmans M., Bouché R., Guyot J. C. Polymorphic transitions of carbamazepine during grinding and compression. Drug Dev Ind Pharm. 1986;12:1913–1927. doi: 10.3109/03639048609042617. [DOI] [Google Scholar]
- 23.Otsuka M., Ohfusa T., Matsuda Y. Effect of binders on polymorphic transformation kinetics of carbamazepine in aqueous solution. Colloids and Surfaces B: Biointerfaces. 2000;17:145–152. doi: 10.1016/S0927-7765(99)00111-3. [DOI] [Google Scholar]
- 24.Luhtala S. Effect of sodium lauryl sulphate and polysorbate 80 on crystal growth and aqueous solubility of carbamazepine. Acta Pharm. Nord. 1992;4:85–90. [Google Scholar]
- 25.Qiu Y., Cheskin H. S., Engh K. R., Poska R. P. Once-a-day controlled-release dosage form of divalproex sodium I: Formulation design and in vitro/in vivo investigations. J Pharm Sci. 2003;92:1166–1173. doi: 10.1002/jps.10385. [DOI] [PubMed] [Google Scholar]
- 26.Dressman J. B., Amidon G. L., Reppas C., Shah V. P. Dissolution testing as a prognostic tool for oral drug absorption: Immediate release dosage forms. Pharm Res. 1998;15:11–22. doi: 10.1023/A:1011984216775. [DOI] [PubMed] [Google Scholar]
- 27.United States Pharmacopoeia 24/The National Formulary 21. USP Convention, Rockville, MD, 2000.
- 28.Mayer T., May T. W., Altenmüller D. -M., Sandmann M., Wolf P. Clinical problems with generic antiepileptic drugs: Comparison of sustained-release formulations of Carbamazepine. Clin Drug Invest. 1999;18:17–26. doi: 10.2165/00044011-199918010-00003. [DOI] [Google Scholar]