

Abstract
The zwitterionic title compound, C13H8F2N4S, is situated on a twofold rotation axis running along the C—S [1.691 (2) Å] single bond. The phenylene ring is twisted out of the tetrazolium plane by 42.18 (7)°. Relatively short distances [3.7572 (9) and 4.0625 (6) Å] between the centroids of the phenylene and tetrazolium rings of neighbouring molecules suggest π–π interactions. The crystal under investigation was a non-merohedral twin, with a 33% twin component.
Related literature
For details of the synthesis, see: Mirkhalaf et al. (1998 ▶); Irving et al. (1971 ▶). For comparison bond distances, see: Allen et al. (1987 ▶). For the indexing of twinned crystals by the CELL_NOW program, see: Bruker (2008 ▶).
Experimental
Crystal data
C13H8F2N4S
M r = 290.29
Monoclinic,

a = 14.500 (3) Å
b = 12.656 (3) Å
c = 6.9066 (14) Å
β = 92.93 (3)°
V = 1265.8 (5) Å3
Z = 4
Mo Kα radiation
μ = 0.27 mm−1
T = 100 K
0.33 × 0.11 × 0.11 mm
Data collection
Bruker APEXII CCD diffractometer
Absorption correction: multi-scan (TWINABS; Bruker, 2008 ▶) T min = 0.915, T max = 0.971
1562 measured reflections
1562 independent reflections
1360 reflections with I > 2σ(I)
Refinement
R[F 2 > 2σ(F 2)] = 0.038
wR(F 2) = 0.101
S = 1.07
1562 reflections
93 parameters
H-atom parameters constrained
Δρmax = 0.32 e Å−3
Δρmin = −0.23 e Å−3
Data collection: APEX2 (Bruker, 2005 ▶); cell refinement: SAINT-Plus (Bruker, 2004 ▶); data reduction: SAINT-Plus; program(s) used to solve structure: SIR97 (Altomare et al., 1999 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: DIAMOND (Brandenburg & Putz, 2005 ▶); software used to prepare material for publication: WinGX (Farrugia, 1999 ▶).
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536809026683/ng2610sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536809026683/ng2610Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C6—H6⋯Si | 0.95 | 2.79 | 3.6828 (19) | 157 |
Symmetry code: (i) 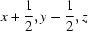 .
.
Acknowledgments
Financial assistance from the National Research Foundation of South Africa and IPCore are gratefully acknowledged.
supplementary crystallographic information
Comment
During the process of synthesizing a series of electronically altered dithizones for the purpose of investigating its effect on the photochromic isomerization reaction of metal dithizonates, several phenyl substituted species were fully oxidized to its dehydrodithizone derivatives. Dehydrodithizones, most probably due to their zwitter-ionic nature, crystallizes much more readily than the parent compound. The yellow meta-fluoro dehydrodithizone crystals, suitable for X-ray crystallography, were isolated from a mixture of polar solvents, i.e. acetone and water.
The title compound crystallizes in the monoclininc space group C2/c (Z = 4) resulting in molecules lying on special positions in the crystal lattice. All bond lengths and angles (see Table 1, Fig. 1) are within range of their expected values (Allen et al., 1987). The phenyl rings adopt a non-parallel arrangement with the dehydrodithizone backbone with dihedral angles of 42.18 (7)° for ring C2—C7, mainly due to their close proximities on the tetrazole moiety. The preferred orientation is supported by the π-π stacking of the phenyl rings of neighbouring molecules (distance between planes = 3.4069 Å, centroid to centroid distance = 3.7572 (9) Å). Similar π-π stacking is also observed between neighbouring tetrazole moieties in a head-to-head fashion (distance between planes = 3.4235 Å, centroid to centroid distance = 4.0625 (6) Å) Additionally, several other close contacts/interactions are noted, among these a rather close contact for C6—H6···S between neighbouring dithizone molecules.
Experimental
Solvents (AR) purchased from Merck and reagents from Sigma-Aldrich were used without further purification. The meta-fluoro derivative of dithizone, (m-FPhNHN)2CS, was prepared from ammonium sulfide and 3-fluoroaniline according to the procedure reported by Mirkhalaf et al., 1998. The synthesis and crystallization of the title compound, meta-fluoro dehydrodithizone, was done according to a procedure reported by Irving et al., 1971. Hereby a solution of (m-FPhNHN)2CS (0.3 g, 0.75 mmol) in dichloromethane (100 ml) was stirred (2 hrs) with a solution of potassium hexacyanoiron (III) (0.72 g) and potassium carbonate (0.70 g) in water (30 ml). After the organic layer was washed with water, the solvent was removed under reduced pressure. From hot acetone and water orange crystals, in 52% yield, were crystallized.
M.p 155 °C (explode). λmax(acetone) 434.9 nm (ε = 1650 dm3 mol-1 cm-1). δH (300 MHz, (CD3)2SO, 7.748 - 7.533 (8 H, m, 2x-C6H4).
Refinement
The aromatic H atoms were placed in geometrically idealized positions (C—H = 0.95 Å) and constrained to ride on their parent atoms with Uiso(H) = 1.2Ueq(C). Initial CheckCIF evaluation indicated possible non-merohedral twinning, and the data was subsequantly treated using CELL_NOW to obtain orientation matrix of the two components. The raw data was then integrated as two components resulting in a HKLF5 format file, which greatly improved refinement parameters and yielded the refined composition of the twinned domains in a 33.1:66.9 ratio.
Figures
Fig. 1.

View of (I) (30% probability displacement ellipsoids). Accented lettering indicate atoms generated by symmetry (-x, y, 1/2 - z).
Fig. 2.

Packing diagram of (I) indicating the π-π interactions
Crystal data
| C13H8F2N4S | F(000) = 592 |
| Mr = 290.29 | Dx = 1.523 Mg m−3 |
| Monoclinic, C2/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -C 2yc | Cell parameters from 1712 reflections |
| a = 14.500 (3) Å | θ = 2.8–28.2° |
| b = 12.656 (3) Å | µ = 0.27 mm−1 |
| c = 6.9066 (14) Å | T = 100 K |
| β = 92.93 (3)° | Needle, red |
| V = 1265.8 (5) Å3 | 0.33 × 0.11 × 0.11 mm |
| Z = 4 |
Data collection
| Bruker X8 APEXII 4K Kappa CCD diffractometer | 1562 measured reflections |
| Radiation source: fine-focus sealed tube | 1562 independent reflections |
| graphite | 1360 reflections with I > 2σ(I) |
| Detector resolution: 8.4 pixels mm-1 | θmax = 28.4°, θmin = 2.1° |
| φ and ω scans | h = −19→19 |
| Absorption correction: multi-scan (TWINABS; Bruker, 2008) | k = 0→16 |
| Tmin = 0.915, Tmax = 0.971 | l = 0→9 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.038 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.101 | H-atom parameters constrained |
| S = 1.07 | w = 1/[σ2(Fo2) + (0.0484P)2 + 0.809P] where P = (Fo2 + 2Fc2)/3 |
| 1562 reflections | (Δ/σ)max < 0.001 |
| 93 parameters | Δρmax = 0.32 e Å−3 |
| 0 restraints | Δρmin = −0.23 e Å−3 |
Special details
| Experimental. The intensity data was collected on a Bruker X8 Apex II 4 K Kappa CCD diffractometer using an exposure time of 180 s/frame. A total of 791 frames were collected with a frame width of 0.5° covering up to θ = 28.36° with 98.9% completeness accomplished. |
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| S | 0 | 0.63767 (5) | 0.25 | 0.01916 (16) | |
| N1 | 0.07681 (9) | 0.44217 (11) | 0.27936 (18) | 0.0170 (3) | |
| N2 | 0.04565 (8) | 0.34445 (11) | 0.26789 (18) | 0.0156 (3) | |
| F | 0.10346 (9) | 0.01182 (11) | 0.57483 (18) | 0.0443 (4) | |
| C1 | 0 | 0.50409 (18) | 0.25 | 0.0158 (4) | |
| C2 | 0.10317 (10) | 0.25414 (13) | 0.3084 (2) | 0.0179 (3) | |
| C3 | 0.07274 (12) | 0.17451 (14) | 0.4266 (2) | 0.0212 (4) | |
| H3 | 0.0134 | 0.1766 | 0.4786 | 0.025* | |
| C4 | 0.13285 (14) | 0.09224 (16) | 0.4647 (3) | 0.0284 (4) | |
| C5 | 0.22040 (14) | 0.08883 (16) | 0.3976 (3) | 0.0321 (5) | |
| H5 | 0.2606 | 0.0316 | 0.4304 | 0.039* | |
| C6 | 0.24883 (12) | 0.17000 (16) | 0.2820 (3) | 0.0310 (4) | |
| H6 | 0.3092 | 0.1687 | 0.2346 | 0.037* | |
| C7 | 0.18989 (11) | 0.25393 (15) | 0.2339 (2) | 0.0243 (4) | |
| H7 | 0.2087 | 0.3094 | 0.1522 | 0.029* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| S | 0.0192 (3) | 0.0169 (3) | 0.0216 (3) | 0 | 0.00337 (19) | 0 |
| N1 | 0.0156 (6) | 0.0179 (7) | 0.0174 (6) | −0.0005 (5) | 0.0011 (4) | −0.0009 (5) |
| N2 | 0.0111 (6) | 0.0197 (7) | 0.0158 (6) | −0.0011 (5) | 0.0000 (4) | −0.0007 (5) |
| F | 0.0556 (8) | 0.0311 (8) | 0.0447 (7) | 0.0079 (6) | −0.0118 (6) | 0.0152 (5) |
| C1 | 0.0159 (10) | 0.0193 (11) | 0.0123 (9) | 0 | 0.0025 (7) | 0 |
| C2 | 0.0142 (7) | 0.0195 (8) | 0.0193 (7) | 0.0017 (6) | −0.0046 (5) | −0.0042 (6) |
| C3 | 0.0209 (8) | 0.0219 (9) | 0.0199 (8) | 0.0016 (7) | −0.0062 (6) | −0.0019 (6) |
| C4 | 0.0329 (11) | 0.0239 (10) | 0.0268 (9) | 0.0044 (8) | −0.0121 (7) | −0.0001 (7) |
| C5 | 0.0283 (10) | 0.0281 (11) | 0.0379 (10) | 0.0139 (8) | −0.0175 (7) | −0.0120 (8) |
| C6 | 0.0165 (8) | 0.0369 (11) | 0.0388 (10) | 0.0066 (7) | −0.0067 (7) | −0.0182 (8) |
| C7 | 0.0165 (8) | 0.0281 (10) | 0.0281 (9) | 0.0001 (6) | −0.0006 (6) | −0.0085 (7) |
Geometric parameters (Å, °)
| S—C1 | 1.691 (2) | C3—C4 | 1.375 (3) |
| N1—N2 | 1.3177 (18) | C3—H3 | 0.95 |
| N1—C1 | 1.3685 (18) | C4—C5 | 1.374 (3) |
| N2—N2i | 1.334 (2) | C5—C6 | 1.377 (3) |
| N2—C2 | 1.434 (2) | C5—H5 | 0.95 |
| F—C4 | 1.352 (2) | C6—C7 | 1.393 (3) |
| C1—N1i | 1.3685 (18) | C6—H6 | 0.95 |
| C2—C7 | 1.383 (2) | C7—H7 | 0.95 |
| C2—C3 | 1.383 (2) | ||
| N2—N1—C1 | 104.75 (13) | F—C4—C5 | 119.28 (18) |
| N1—N2—N2i | 110.19 (8) | F—C4—C3 | 117.58 (18) |
| N1—N2—C2 | 122.82 (12) | C5—C4—C3 | 123.14 (19) |
| N2i—N2—C2 | 126.72 (8) | C4—C5—C6 | 118.76 (18) |
| N1i—C1—N1 | 110.1 (2) | C4—C5—H5 | 120.6 |
| N1i—C1—S | 124.94 (10) | C6—C5—H5 | 120.6 |
| N1—C1—S | 124.94 (10) | C5—C6—C7 | 120.60 (17) |
| C7—C2—C3 | 122.81 (16) | C5—C6—H6 | 119.7 |
| C7—C2—N2 | 117.37 (15) | C7—C6—H6 | 119.7 |
| C3—C2—N2 | 119.74 (14) | C2—C7—C6 | 118.11 (18) |
| C4—C3—C2 | 116.54 (17) | C2—C7—H7 | 120.9 |
| C4—C3—H3 | 121.7 | C6—C7—H7 | 120.9 |
| C2—C3—H3 | 121.7 | ||
| C1—N1—N2—N2i | 0.36 (17) | N2—C2—C3—C4 | −177.74 (14) |
| C1—N1—N2—C2 | −173.99 (11) | C2—C3—C4—F | −177.94 (14) |
| N2—N1—C1—N1i | −0.14 (7) | C2—C3—C4—C5 | 2.5 (3) |
| N2—N1—C1—S | 179.86 (7) | F—C4—C5—C6 | 178.54 (16) |
| N1—N2—C2—C7 | −43.6 (2) | C3—C4—C5—C6 | −1.9 (3) |
| N2i—N2—C2—C7 | 143.01 (18) | C4—C5—C6—C7 | 0.0 (3) |
| N1—N2—C2—C3 | 133.08 (15) | C3—C2—C7—C6 | −0.5 (2) |
| N2i—N2—C2—C3 | −40.3 (2) | N2—C2—C7—C6 | 176.04 (14) |
| C7—C2—C3—C4 | −1.2 (2) | C5—C6—C7—C2 | 1.2 (2) |
Symmetry codes: (i) −x, y, −z+1/2.
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C6—H6···Sii | 0.95 | 2.79 | 3.6828 (19) | 157 |
Symmetry codes: (ii) x+1/2, y−1/2, z.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: NG2610).
References
- Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987). J. Chem. Soc. Perkin Trans. 2, pp. S1–19.
- Altomare, A., Burla, M. C., Camalli, M., Cascarano, G. L., Giacovazzo, C., Guagliardi, A., Moliterni, A. G. G., Polidori, G. & Spagna, R. (1999). J. Appl. Cryst.32, 115–119.
- Brandenburg, K. & Putz, H. (2005). DIAMOND Crystal Impact GbR, Bonn, Germany.
- Bruker (2004). SAINT-Plus Bruker AXS Inc., Madison, Wisconsin, USA.
- Bruker (2005). APEX2 Bruker AXS Inc., Madison, Wisconsin, USA.
- Bruker (2008). TWINABS and CELL_NOW Bruker AXS Inc., Madison, Wisconsin, USA.
- Farrugia, L. J. (1999). J. Appl. Cryst.32, 837–838.
- Irving, H. M. N. H., Kiwan, A. M., Rupainwar, D. C. & Sahota, S. S. (1971). Anal. Chim. Acta, 56, 205–220.
- Mirkhalaf, F., Whittaker, D. & Schiffrin, D. J. (1998). J. Electroanal. Chem.452, 203–213.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536809026683/ng2610sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536809026683/ng2610Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report