

Abstract
A proposed mechanism for irreversible ischemic liver damage has been peroxidation of membrane phospholipids by free radicals. However, the hepatocyte is laden with enzymes which are antioxidants and, therefore, ought to be relatively resistant to oxidative injury. To test the hypothesis that free radical damage from ischemia and reperfusion of the liver is a non parenchymal cell process, we studied an in vivo model of ischemia. A point of transition from reversible to irreversible ischemia was defined at ≥60 min of total ischemia by serial measurements of ATP at control, end of ischemia, and end of reperfusion periods (n = 6 each). Nonparenchymal cells were separated out of 10 livers in each ischemic group using a Percoll gradient. Second derivative spectroscopy did not detect conjugated dienes in any hepatocellular fraction, total cellular, mitochondrial, or microsomal, but did in the nonparenchymal cell fractions of livers from the 60- and 90-min ischemia groups. This in vivo study shows that irreversible ischemia in the rat liver is associated with free radical lipid peroxidation, but that the non parenchymal cells rather than hepatocytes are the focus of this injury.
INTRODUCTION
A variety of mechanisms are responsible for the damage to the liver with ischemia and reperfusion. One process which is of great interest is the initiation of lipid peroxidation by oxygen-derived free radicals and, in particular, the superoxide anion [1, 2]. Free radicals are generated upon reperfusion after prolonged ischemia and are hypothesized to initiate a cascade of oxidative reactions which results in irreversible damage to cellular and organellar membranes [3, 4]. The exact mechanism by which this damage to membranes occurs is uncertain and depends on the particular model which is studied and the experimental methodology used to ascertain the processes involved [13].
Much evidence supports the hypothesis that toxic injury to the liver involves the production of free radicals with subsequent membrane damage by lipid peroxidation [5–7]. However, most of the support for this mechanism of irreversible cellular damage comes from models of hepatic injury involving alcohol [8], iron overload [9], paraquat and diquat [10], acetaminophen [11], and carbon tetrachloride [40]. In models of ischemic damage, the evidence is mixed; certain organ systems (e.g., brain mitochondria) support the role of lipid peroxidation [12], while many others do not (e.g., in situ canine heart and whole rat liver) [14, 15].
Recently, much attention in the field of injury of the liver due to ischemia/reperfusion has been focused on the role of the microvasculature and, in particular, its susceptibility to damage relative to the hepatic parenchyma [16–20]. Xanthine oxidase has a controversial role in the generation of free radicals with reperfusion, but is an enzyme which is associated with membrane damage induced by free radicals [18, 35]. Since, xanthine oxidase has been localized to the cytosol of capillary endothelial cells and, in particular, those of the liver sinusoids [21–23], we posed the hypothesis that the hepatic sinusoidal endothelium is most sensitive to free radical damage. Therefore, if any hepatic tissue should be the site of lipid peroxidation after ischemia/reperfusion in vivo, the vascular endothelium rather than the hepatocytes is most susceptible. In this report, we individually assessed the role of parenchymal and non parenchymal hepatic tissues in the process of membrane damage induced by lipid peroxidation and attempted to clarify some of the controversies that are involved in this process.
MATERIALS AND METHODS
Animal surgical procedure
An in vivo model of hepatic ischemia/reperfusion in the rat was studied in which the branches of the portal vein and hepatic artery to the left lateral and median lobes were occluded for 30, 60, and 90 min using microvascular clips. One hour of reperfusion was also studied. Male, inbred rats of the Lewis strain (250–300 g, Charles River Breeding Lab, Wilmington, MA) were used, and anesthesia consisted of inhalational Metofane.
Standardization of severity of ischemia
An initial group (n = 6/group) was studied to standardize the severity of the ischemic injury. This was of critical importance to exactly determine whether the periods of ischemia used in this model were completely reversible, partially reversible, or irreversible.
Tissue assays: ATP
Tissue samples were collected by freeze-clamping with liquid nitrogen-cooled tongs from ischemic and control lobes at 30-min intervals throughout ischemia and reperfusion. ATP was assayed using the method of Lamprecht and Trautschold [38] and reported as micromoles/gram tissue.
Tissue assays: Superoxide anion
A separate group (n = 10/group) was studied for the concurrent production of superoxide anion and for the generation of the products of lipid peroxidation in each ischemic group. Tissue was collected at the end of ischemia and after 30 min of reperfusion for superoxide levels. The samples were homogenized in 0.25 M sucrose and centrifuged at 3500g and 100,000g to generate cytosolic fractions. Superoxide levels were assayed according to the method of Green and Hill [39] and expressed as nanomoles of cytochrome c reduced/milliliter. This was performed in order to confirm that reperfusion was associated with the generation of oxygen derived free radicals.
Assay for conjugated dienes
Products of lipid peroxidation were assayed in samples collected at the end of ischemia and after 1, 2, 6, and 12 hr of reperfusion by the determination of the presence of conjugated dienes. Tissue was collected and prepared for analysis by two distinct methods to allow the individual evaluation of parenchymal and nonparenchymal cell populations. First, tissue was homogenized in 0.25 M sucrose and sequentially centrifuged at 3500g, 25,000g, and 100,000g to generate whole cell, mitochondrial, microsomal, and cytosolic fractions. Second, separate samples were digested in 0.2% protease, 0.5% collagenase, and 0.2% deoxyribonuclease, and the nonparenchymal cells were separated from the hepatocytes on a Percoll gradient. Samples were stored at −70°C.
For the determination of conjugated dienes, lipid extracts of the samples were prepared in 2:1 chloroform: methanol [24]. Aliquots of the lipid fraction were evaporated to dryness under a vacuum at room temperature and dissolved in hexane. Conjugated dienes were detected in the hexane solutions by the simultaneous calculation of the second derivative of the absorbance spectrum between 220 and 300 nm using a Hitachi 557 recording spectrophotometer. This method has greatly enhanced sensitivity when compared to the standard uv method and is appropriate for in vivo studies (Fig. 1).
FIG. 1.
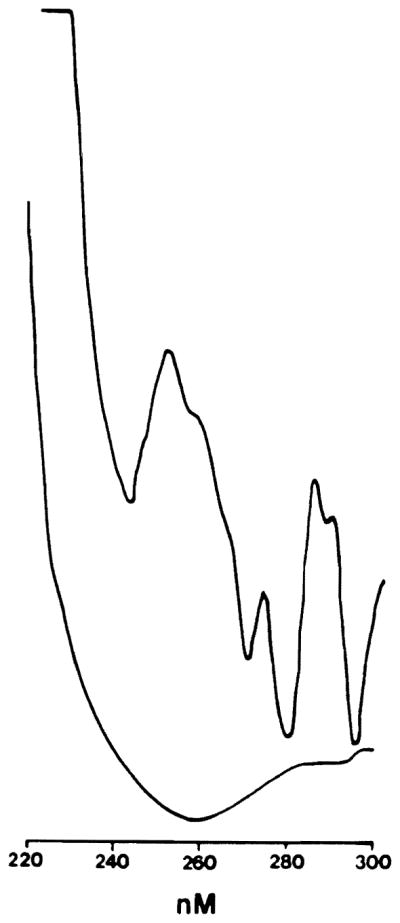
Tracing from Hitachi spectrophotometer. The bottom line is the standard print out of the absorbance for 220–300 nM. The top line is the second derivative of the absorbance at each wavelength and shows how a “shoulder” in the standard printout is transformed into a distinct peak at 242 nM (cis, trans diene).
Materials used
Collagenase (C0130), protease (P5147), sucrose (S5), and deoxyribonuclease (P5025) were purchased from Sigma. Chloroform, hexane, and methanol were all HPLC grade (Fischer Scientific). Percoll was purchased from Pharmacia (Uppsala, Sweden).
RESULTS
Changes in ATP with ischemia and reperfusion
The control level of ATP (n = 12) was 2.98 ± 0.08 μm/g and decreased to 1.71 ± 0.07, 0.63 ± 0.07, and 0.30 ± 0.09 (n = 3 each) at the end of 30, 60, and 90 min of ischemia (Fig. 2). During reperfusion, the livers subjected to 30 min of ischemia recovered to ATP levels of 2.88 ± 0.06 μm/ml, while the rats subjected to 60 and 90 min of ischemia partially recovered to 1.57 ± 0.06 and 0.75 ± 0.08, respectively. Therefore, easily reversible, severe, and completely irreversible periods of normothermic ischemia were represented in this model.
FIG. 2.
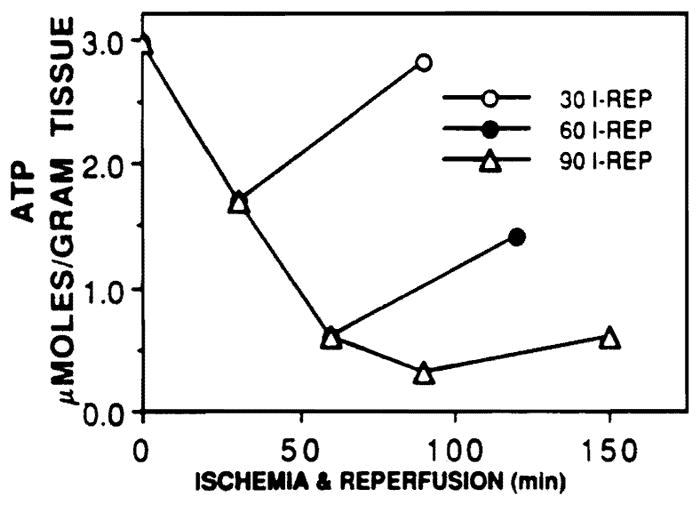
Amount of ATP present in whole liver extracts at the end of ischemia and reperfusion for 30, 60, and 90 min of ischemia. The amount of ATP remained decreased as the length of ischemia increased, but the ability of the liver to regenerate ATP also diminished greatly after 60 and 90 min of ischemia.
Generation of superoxide anion with ischemia and reperfusion
Superoxide levels (Fig. 3) increased from 0.13 nanomoles cytochrome c reduced/ml before ischemia to a mean level of 0.76 at the end of ischemia and 2.53 during reperfusion. Samples taken from control lobes of the experimental rats showed no increase in the level of superoxide at any of the time points studied.
FIG. 3.
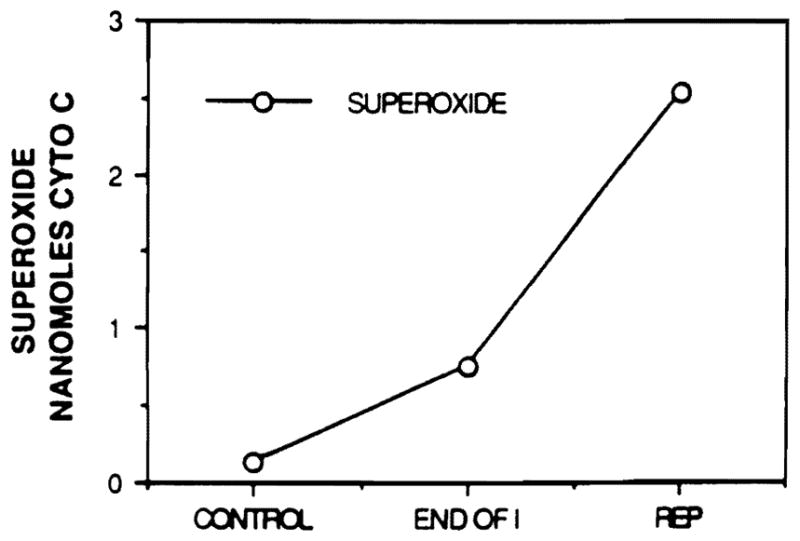
The generation of superoxide anion with ischemia and reperfusion. Superoxide anion was generated in small amounts during ischemia, but increased greatly with reperfusion. The amount of anion produced did not vary with the length of ischemia.
Detection of conjugated dienes: Hepatocellular fractions
In the samples that were sequentially centrifuged (n = 10), there was no evidence of the formation of conjugated dienes in hepatic total cellular, mitochondrial, microsomal, or cytosolic fractions (Table 1). This held true for 30, 60, and 90 min of ischemia and up to 12 hr of reperfusion. This was also true in the hepatocyte pellets of the digested samples. Hepatocellular fractions from control and ischemic rats which were left overnight exposed to room air showed significant production of conjugated dienes.
TABLE 1.
Percentage of Cellular Fractions Containing Conjugated Dienes
| Ischemia (%) | Reperfusion (%) | |
|---|---|---|
| Hepatocellular (n = 10 each) | ||
| Total cellular | 0 | 0 |
| Mitochondrial | 0 | 0 |
| Microsomal | 0 | 0 |
| Cytosolic | 0 | 0 |
| Endothelial/Kupffer cell (n = 10 each) | ||
| 30-min ischemia | 0 | 0 |
| 60-min ischemia | 0 | 20 |
| 90-min ischemia | 0 | 80 |
Note. Percentage of all fractions in each experimental group in which conjugated dienes could be found. All livers which contained conjugated dienes during the 12 hr of reperfusion retained them throughout the remainder of reperfusion.
Detection of conjugated dienes: Nonparenchymal cell fractions
When the nonparenchymal cell layer was studied, 20% of the samples in the 60-min group and 80% in the 90-min group contained conjugated dienes during reperfusion (Table 1). None of the livers subjected to 30 min of ischemia, control samples, or livers at the end of ischemia had produced conjugated dienes. All livers which contained conjugated dienes at any point during reperfusion, retained them throughout the 12 hr of reperfusion that were studied.
DISCUSSION
Injury to the liver after a period of ischemia is a complex process which involves not only the deprivation of oxygen and nutrients, but also the accumulation of by-products of metabolism during the cessation of blood flow. In addition, reperfusion appears to be a phase of additional tissue injury [25]. Therefore, the study of any model of ischemic injury requires appraisal of the events which occur during both ischemia and reperfusion and should include ischemic periods of varying severity so as to properly categorize the importance of the damaging events. The mechanism of ischemic injury appears to be the gradual loss of normal cellular homeostasis and, in particular, membrane-associated processes which regulate ion concentrations within and between subcellular compartments [26–31].
The generation of highly reactive oxidative molecules is mostly reserved for reperfusion which occurs when oxygen is reintroduced and hypoxic cell populations become reactivated or hyperactivated. Peroxidation of membrane phospholipids by oxygen-derived free radicals is a well-accepted mechanism of liver damage from toxic substances [5–7], and it has been logically assumed that the generation of free radicals during ischemia/reperfusion would cause cellular damage via similar mechanisms. However, recent studies have challenged this premise by failing to document the presence of free radicals or the products of lipid peroxidation in livers damaged by ischemia, even during reperfusion [13–15]. Each of these studies analyzed hepatocellular tissue fractions or whole liver tissue which was 70–92.5% hepatocytes by weight (32).
Another aspect of the controversy is that although some studies have found lipid peroxidation to be important in hepatic damage (e.g., endotoxin) [6], the assays which were used to detect the products of lipid peroxidation were performed in an in vivo model. Unfortunately, these assays are not always appropriate and are not specific for the products of lipid peroxidation. Therefore, we have not only individually studied the different cell populations of the liver, but have also applied an assay which is sensitive, specific, and appropriate for an in vivo model.
Our hypothesis is based on the premise that the non-parenchymal cells of the liver are of critical importance in ischemia/reperfusion injury of the liver and that the liver has the ability to subsequently recover normal function [33–35]. This hypothesis is based upon two assumptions. The first assumption is that the hepatocyte is particularly well suited to resist oxidative damage under physiologic conditions since it is laden with antioxidants, especially glutathione and catalase. Much of the work reported on toxic damage of the liver is done under pharmacologic and not physiologic conditions [5–11, 40]. The second assumption is that the nonparenchymal cells of the liver, primarily endothelial and Kupffer cells, are an active population containing the enzyme systems necessary for, but not the cellular defenses against, oxidative damage [19, 22, 23, 36]. Therefore, the primary focus of liver damage may be the vascular endothelium via lipid peroxidation with secondary damage to the hepatocytes [35].
We applied second derivative spectroscopy to the detection of conjugated dienes. This technique is highly specific and sensitive for the detection of products of lipid peroxidation [24]. Additionally, we undertook a careful examination of both hepatocyte and nonparenchymal cell fractions in an in vivo model of liver ischemia which studied both reversible and irreversible damage. In this model, superoxide radicals are generated with ischemia/reperfusion, but especially during reperfusion. Additionally, the concentration of superoxide anion did not vary with the duration of ischemia. Our findings show that lipid peroxidation does occur in livers which have been severely damaged and not in those which have been reversibly damaged. Of greater importance is the localization of this process to the nonparenchymal cell portion of the liver. Under no circumstances did lipid peroxidation occur in nonischemic livers (any fraction) or in hepatocyte fractions, even in livers which had been irreversibly damaged. The nonparenchymal cell fractions prepared by this method ultimately have less than 10% contamination from cells other than endothelial and Kupffer cells when cultured. The contribution of other nonparenchymal cells to the production of conjugated dienes is minimal. Therefore, lipid peroxidation does occur in ischemic/reperfusion damage to the liver, but it is most probably an endothelial/Kupffer cell process.
The findings of this study explain some of the previous discrepancies as to whether lipid peroxidation is involved in liver damage from ischemia/reperfusion. Careful separation and examination of the fractions which contain nonparenchymal cells avoided disposition of the Kupffer and endothelial cells and avoided the concealment of lipid peroxidation in nonparenchymal cells by the examination of only whole tissues.
In summary, this study has shown that superoxide anion is generated during damage to the liver by ischemia and reperfusion, but that the amount produced did not vary with the duration of ischemia. Therefore, the ability of the liver to withstand free radicals seems to diminish as the ischemia lengthens. Second, in this in vivo model, lipid peroxidation was associated with reperfusion after prolonged periods of ischemia which again attests to a diminution of the antioxidant defenses of the liver. Last, lipid peroxidation is an event of the nonparenchymal cell portion of the liver and not of the hepatocyte. These findings further stress the relative susceptibility and importance of the microvasculature of the liver in ischemia/reperfusion.
Footnotes
Supported by Research Grants from the Veterans Administration and Project Grant No. DK 29961 from the National Institutes of Health, Bethesda, MD.
References
- 1.Tien M, Svingen BA, Aust SD. Superoxide dependent lipid peroxidation. Fed Proc. 1981;40:179. [PubMed] [Google Scholar]
- 2.Fridovich I. The biology of oxygen radicals. Science. 1978;201:875. doi: 10.1126/science.210504. [DOI] [PubMed] [Google Scholar]
- 3.McCord JM. Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med. 1985;312:159. doi: 10.1056/NEJM198501173120305. [DOI] [PubMed] [Google Scholar]
- 4.McCord JM. Oxygen-derived radicals: A link between reperfusion injury and inflammation. Fed Proc. 1987;46:2402. [PubMed] [Google Scholar]
- 5.Trenti T, Botti B, Desai MA, Predieri G, Masinin A. Structural and functional properties of rat liver mitochondria in experimental iron overload. I Iron accumulation and lipoperoxidation. IRCS Med Sci. 1986;14:837. [Google Scholar]
- 6.Sugino K, Dohi K, Yamada K, Kawasaki T. The role of lipid peroxidation in endotoxin-induced hepatic damage and the protective effect of antioxidants. Surgery. 1987;101:746. [PubMed] [Google Scholar]
- 7.Corongui FP, Poli G, Dianzani MU, Cheeseman KH, Slater TF. Lipid peroxidation and molecular damage to poly-unsaturated fatty acids in rat liver. Recognition of two classes of hydroperoxides formed under conditions in vivo. Chem Biol Int. 1986;59:147. doi: 10.1016/s0009-2797(86)80062-x. [DOI] [PubMed] [Google Scholar]
- 8.Klein SM, Cohen G, Liever CS, Cedarbaum AI. Increased microsomal oxidation of hydroxyl radical scavenging agents and ethanol after chronic consumption of ethanol. Arch Biochem Biophys. 1983;223:425. doi: 10.1016/0003-9861(83)90606-9. [DOI] [PubMed] [Google Scholar]
- 9.Bacon BR, Tavill AS, Brittenham GM, et al. Hepatic lipid peroxidation in vivo in rats with chronic iron overload. J Clin Invest. 1983;71:429. doi: 10.1172/JCI110787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burk RF, Lawrence RA, Lane JM. Liver necrosis and lipid peroxidation in the rat as a result of paraquat and diquat administration. J Clin Invest. 1980;65:1024. doi: 10.1172/JCI109754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wendel A, Feuerstein S. Drug-induced lipid peroxidation in mice. Biochem Pharmacol. 1981;30:251. doi: 10.1016/0006-2952(81)90576-1. [DOI] [PubMed] [Google Scholar]
- 12.Majewski MD, Strosznajder J, Lazarewicz J. Effect of ischemic anoxia and barbiturate anesthesia on free radical oxidation of mitochondrial phospholipids. Brain Res. 1978;158:423. doi: 10.1016/0006-8993(78)90685-6. [DOI] [PubMed] [Google Scholar]
- 13.Jaeschke H, Smith CV, Mitchell JR. Reactive oxygen species during ischemia-reperfusion injury in isolated perfused rat liver. Fed Proc. 1987;46:410. doi: 10.1172/JCI113441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitchell JR, Smith CV, Hughes H, Lena M, Jaeschke H, Michael L, Entman ML. No evidence for reactive oxygen damage in ischemia-reflow injury. Clin Res. 1987;35:642A. [PubMed] [Google Scholar]
- 15.Silver EH, Szabo S. Role of lipid peroxidation in tissue injury after hepatic ischemia. Exp Mol Pathol. 1983;38:69. doi: 10.1016/0014-4800(83)90099-0. [DOI] [PubMed] [Google Scholar]
- 16.Ward PA, Johnson KJ, Till GO. Oxygen radicals and microvascular injury of lungs and kidney. Acta Physiol Scand. 1986;548:79. [PubMed] [Google Scholar]
- 17.Freeman BA, Young SL, Crapo JD. Liposome-mediated augmentation of superoxide dismutase in endothelial cells prevents oxygen injury. J Biol Chem. 1983;258(12):534. [PubMed] [Google Scholar]
- 18.Parks DA, Granger DN. Ischemia-induced vascular changes: Role of xanthine oxidase and hydroxyl radicals. Amer J Physiol. 1983;245:G285. doi: 10.1152/ajpgi.1983.245.2.G285. [DOI] [PubMed] [Google Scholar]
- 19.Ratych RE, Chuknyiska RS, Bulkley GB. The primary localization of free radical generation after anoxia/reoxygenation in isolated endothelial cells. Surgery. 1987;102:122. [PubMed] [Google Scholar]
- 20.McKeown CMB, Edwsrds V, Phillips MJ, Harvey PRC, Petrunka CN, Strasberg SM. Sinusoidal lining cell damage: The critical injury in cold preservation of liver allografts in the rat. Transplantation. 1988;46:178. [PubMed] [Google Scholar]
- 21.Bruder G, Heid H, Jarasch E, Keenan TW, Mather IH. Characteristics of membrane-bound and soluble forms of xanthine oxidase from milk and endothelial cells of capillaries. Biochim Biophys. 1982;701:357. doi: 10.1016/0167-4838(82)90239-4. [DOI] [PubMed] [Google Scholar]
- 22.Jarasch E, Bruder G, Heid HW. Significance of xanthine oxidase in capillary endothelial cells. Acta Physiol Scand. 1986;548:39. [PubMed] [Google Scholar]
- 23.Parks DA, Granger DN. Xanthine oxidase: Biochemistry, distribution and physiology. Acta Physiol Scand. 1985;548:87. [PubMed] [Google Scholar]
- 24.Corongui FP, Milia A. An improved and simple method for determining diene conjugation in autoxidized polyunsaturated fatty acids. Chem Biol lnterations. 1983;44:289. doi: 10.1016/0009-2797(83)90056-x. [DOI] [PubMed] [Google Scholar]
- 25.Jones DP, Kennedy FG, Anderson BS, Aw TY, Wilson E. When is a mammalian cell hypotoxic? Insights from studies of cells versus mitochondria. Mol Physiol. 1985;8:473. [Google Scholar]
- 26.Chaudry IH, Clemens MG, Baue AE. Alterations in cell function with ischemia and shock and their correction. Arch Surg. 1981;116:1309. doi: 10.1001/archsurg.1981.01380220053009. [DOI] [PubMed] [Google Scholar]
- 27.Chien KR, Abrams J, Serroni A, Martin JT, Farber JL. Accelerated phospholipid degradation and associated membrane dysfunction in irreversible, ischemic liver cell injury. J Biol Chem. 1978;253:4809. [PubMed] [Google Scholar]
- 28.Chien KR, Abrams J, Pfau RG, Farber JL. Prevention by chlorpromazine of ischemic liver cell death. Amer J Pathol. 1977;88:539. [PMC free article] [PubMed] [Google Scholar]
- 29.Chien KR, Farber JL. Microsomal membrane dysfunction in ischemic rat liver cells. Arch Biochem Biophys. 1977;180:191. doi: 10.1016/0003-9861(77)90025-x. [DOI] [PubMed] [Google Scholar]
- 30.Cheung JY, Leaf A, Bonventre JV. Mitochondrial function and intracellular calcium in anoxic cardiac myocytes. Amer J Physiol. 1986;250:C18. doi: 10.1152/ajpcell.1986.250.1.C18. [DOI] [PubMed] [Google Scholar]
- 31.Cheung JY, Bonventre JV, Malis CD, Leaf A. Calcium and ischemic injury. N Engl J Med. 1986;314:1670. doi: 10.1056/NEJM198606263142604. [DOI] [PubMed] [Google Scholar]
- 32.Kuiper J, Zijlstra FJ, Kamps JAAM, van Berkel TJC. Identification of prostaglandin D2 as the major eicosanoid from liver endothelial and Kupffer cells. Biochim Biophys Acta. 1988;959:143. doi: 10.1016/0005-2760(88)90025-2. [DOI] [PubMed] [Google Scholar]
- 33.Otto G, Wolff H, Uerlings I, Gellert K. Preservation damage in liver transplantation. Transplantation. 1986;42:122. doi: 10.1097/00007890-198608000-00003. [DOI] [PubMed] [Google Scholar]
- 34.Parks DA, Bulkley GA, Granger DN. Role of oxygen free radicals in shock, ischemia, and organ preservation. Surgery. 1983;94:428. [PubMed] [Google Scholar]
- 35.Rao P, Walsh T, Makowka L, Rubin R, Snyder J, Starzl TE. Submitted for publication. Purine nucleoside phosphorylase (PNP)–A new marker for ischemia/reperfusion oxidative liver injury. [Google Scholar]
- 36.Zweier JL, Kuppusamy P, Lutty GA. Measurement of endothelial cell free radical generation: Evidence for a central mechanism of free radical injury in postischemic tissues. Proc Natl Acad Sci USA. 1988;85:4046. doi: 10.1073/pnas.85.11.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Metzger J, Dore SP, Lautenberg BH. Oxidant stress during reperfusion of ischemic liver: No evidence for a role of xanthine oxidase. Hepatology. 1988;8:580. doi: 10.1002/hep.1840080324. [DOI] [PubMed] [Google Scholar]
- 38.Lamprecht W, Trsutschold I. Determination with hexokinase and glucose-6-phosphate dehydrogenase. In: Bergmeyer HU, et al., editors. Methods of Enzymatic Analysis. 2. Vol. 4. New York: Verlag. Chemie; 1974. p. 2101. [Google Scholar]
- 39.Green MJ, Hill HAD. In: Methods in Enzymology. Packer L, editor. Vol. 105. New York: Academic Press; 1984. p. 3. [Google Scholar]
- 40.Recknagel RO, Ghoshal AK. Quantitative estimation of peroxidative rat liver microsomal and mitochondrial lipids after carbon tetrachloride poisoning. Exp Mol Pathol. 1966;5:413. doi: 10.1016/0014-4800(66)90023-2. [DOI] [PubMed] [Google Scholar]