

Abstract
Hepatitis C virus (HCV) envelope glycoproteins are highly glycosylated, with generally 4 and 11 N-linked glycans on E1 and E2, respectively. Studies using mutated recombinant HCV envelope glycoproteins incorporated into retroviral pseudoparticles (HCVpp) suggest that some glycans play a role in protein folding, virus entry, and protection against neutralization. The development of a cell culture system producing infectious particles (HCVcc) in hepatoma cells provides an opportunity to characterize the role of these glycans in the context of authentic infectious virions. Here, we used HCVcc in which point mutations were engineered at N-linked glycosylation sites to determine the role of these glycans in the functions of HCV envelope proteins. The mutants were characterized for their effects on virus replication and envelope protein expression as well as on viral particle secretion, infectivity, and sensitivity to neutralizing antibodies. Our results indicate that several glycans play an important role in HCVcc assembly and/or infectivity. Furthermore, our data demonstrate that at least five glycans on E2 (denoted E2N1, E2N2, E2N4, E2N6, and E2N11) strongly reduce the sensitivity of HCVcc to antibody neutralization, with four of them surrounding the CD81 binding site. Altogether, these data indicate that the glycans associated with HCV envelope glycoproteins play roles at different steps of the viral life cycle. They also highlight differences in the effects of glycosylation mutations between the HCVpp and HCVcc systems. Furthermore, these carbohydrates form a “glycan shield” at the surface of the virion, which contributes to the evasion of HCV from the humoral immune response.
Hepatitis C virus (HCV) is a single-stranded positive-sense RNA virus that causes serious liver diseases in humans (31). More than 170 million people worldwide are seropositive for HCV and at risk for developing cirrhosis and hepatocellular carcinoma (50). HCV is a small, enveloped virus that belongs to the Hepacivirus genus in the Flaviviridae family (31). Its genome encodes a single polyprotein precursor of about 3,000-amino-acid residues that is cleaved co- and posttranslationally by cellular and viral proteases to yield at least 10 mature products (31). The two envelope glycoproteins, E1 and E2, are released from the polyprotein by signal peptidase cleavages. These two proteins assemble as noncovalent heterodimers, which are retained mainly in the endoplasmic reticulum (ER) (36), and they are found as large disulfide-linked oligomers on the surfaces of HCV particles (46). HCV glycoproteins are involved in the entry process, and since they are present on the surfaces of viral particles, these proteins are the targets of neutralizing antibodies (4, 21).
E1 and E2 generally contain 4 and 11 N-glycosylation sites, respectively, all of which have been shown to be modified by glycans (19). Despite variability in HCV envelope glycoprotein sequences, the four glycosylation sites of E1 and nine of E2 are highly conserved, suggesting that the glycans associated with these proteins play an essential role in the HCV life cycle (22). Using retroviral particles pseudotyped with genotype 1a (H strain) HCV envelope glycoproteins (HCVpp), recent studies have determined the potential roles played by these glycans in protein folding, HCV entry, and protection against neutralization (14, 19, 22). Indeed, the lack of glycan E1N1, E1N4, E2N8, or E2N10 strongly affects the incorporation of HCV glycoproteins into HCVpp, suggesting that these glycans are important for correct protein folding (19). Furthermore, mutation of glycosylation sites E2N2 or E2N4 alters HCVpp infectivity despite normal incorporation into pseudotyped particles, suggesting a role for the corresponding glycans in viral entry, at least in this model system (19). Finally, glycans at positions E2N1, E2N6, and E2N11 were shown to reduce the sensitivity of HCVpp to antibody neutralization as well as access of the CD81 coreceptor to its binding site on E2, suggesting that glycans also contribute to HCV evasion of the humoral immune response (14, 22).
It has recently been proposed that targeting glycans could be a promising approach to inhibiting viral infection (1). Indeed, HCV, as well as several other viruses with highly glycosylated envelope proteins, can be inhibited by carbohydrate binding agents such as cyanovirin-N and pradimicin A (1, 7, 23). Furthermore, resistance against drugs that target glycans is likely to develop and will probably result in mutations at some glycosylation sites (3, 52). However, since glycans associated with viral envelope proteins play an important role in the viral life cycle, adaptation of viruses to the selective pressure of carbohydrate-binding agents will most likely come at a replicative cost to the virus (2).
Although the role of HCV glycans has been studied using mutant recombinant HCV envelope glycoproteins incorporated into HCVpp, these particles do not recapitulate all the functions of HCV envelope proteins. Cell culture-derived virus (HCVcc) (32, 49, 55) assembles in an ER-derived compartment in association with very low density lipoproteins (17, 26), whereas HCVpp are assembled in a post-Golgi compartment and are not associated with lipoproteins (44). Importantly, this leads to differences between HCVpp and HCVcc in the oligomerization of the envelope glycoproteins (46). It is also important to note that the carbohydrate composition of viral glycoproteins can differ when the same virus is grown in different cell lines (13). Thus, HCVpp that are produced in 293T cells are not the most appropriate model for glycosylation studies, since HCV tropism is restricted to the liver. Furthermore, differences in envelope protein glycosylation have been observed between HCVpp and HCVcc particles (46). Differences in some HCV envelope protein functions were also observed when the HCVpp and HCVcc systems were compared (28, 29, 42, 43). The development of the HCVcc system provides, therefore, the opportunity to characterize the role of E1/E2-associated glycans in the context of authentic infectious virions. Here, we analyzed the role of E1/E2 glycans by introducing point mutations at N-linked glycosylation sites in the context of the HCVcc system. The effects of these mutations on virus replication, particle secretion, infectivity, and sensitivity to neutralizing antibodies were investigated. Our results demonstrate that several glycans play an important role in HCVcc assembly and/or infectivity and reduce access of neutralizing antibodies to their epitopes.
MATERIALS AND METHODS
Cell culture.
293T human embryo kidney cells (HEK293T), Huh-7 human hepatoma cells (35), and Huh-7w7 cells (38) were grown in Dulbecco's modified essential medium (Invitrogen) supplemented with 10% fetal bovine serum.
Antibodies and reagents.
Anti-HCV monoclonal antibodies (MAbs) A4 (anti-E1) (12), 3/11 (anti-E2; kindly provided by J. McKeating, University of Birmingham, United Kingdom) (16), and anti-murine leukemia virus capsid (anti-CA; ATCC CRL1912, clone R187) were produced in vitro by using a MiniPerm apparatus (Heraeus) as recommended by the manufacturer. Anti-NS3 MAb (486D39) (41) was kindly provided by J. F. Delagneau (Bio-Rad, France). Anti-β-actin (C4) MAb was purchased from Santa Cruz Biotechnology. The soluble recombinant form of the CD81 large extracellular loop (CD81-LEL) was produced as a glutathione S-transferase (GST) fusion protein as described previously (24). Purified cyanovirin-N was kindly provided by K. Gustafson (National Institutes of Health, National Cancer Institute, Frederick, MD) (23).
Serum samples.
Sera of four patients chronically infected with HCV were selected for this study. The serum sampling and use for research application was approved by the ethics committee of the University Hospital of Amiens (France). Determination of HCV genotypes was done by sequence analysis of NS5B. Total antibodies were purified using a NAb protein G spin purification kit (Pierce, Rockford, IL).
Site-directed mutagenesis.
In this work, we used a modified version of the plasmid encoding the full-length JFH-1 genome (genotype 2a; GenBank accession number AB237837), kindly provided by T. Wakita (National Institute of Infectious Diseases, Tokyo, Japan) (49). Mutations were introduced into a JFH-1 plasmid containing a Renilla luciferase reporter gene (39) and amino acid changes F172C and P173S, which have been shown to increase viral titers (10), at the C terminus of the core protein. In this construct, the Renilla luciferase gene is fused with the viral open reading frame in a monocistronic configuration. Furthermore, the N-terminal E1 sequence encoding residues 196TSSSYMVTNDC has been modified to reconstitute the A4 epitope (SSGLYHVTNDC) (12) as described previously (20). Mutations of glycosylation sites were constructed by sequential PCR steps as described previously (9), using an Expand High FidelityPLUS PCR system (Roche). Briefly, the AAT and AAC codons encoding the asparagine residue from the Asn-X-Ser/Thr motifs were replaced individually with the CAA or CAG codon encoding a glutamine residue. Mutations were assembled by a second PCR amplification. For the E1N1 to E1N4 mutants, the amplicons were digested with BsiWI and MluI and ligated into a BsiWI/MluI-digested plasmid. For the E2N1 to E2N11 mutants, the amplicons were digested with BsiWI and KpnI and ligated into a BsiWI/KpnI-digested plasmid. E1 and E2 nucleotide sequences were verified for each mutant. For HCV glycoprotein expression analyses, all the mutants were reintroduced in the context of the JFH-1 plasmid devoid of the luciferase reporter gene.
To construct HCVpp mutants, we used a modified version of the pcDNA/E1E2(JFH-1) plasmid. This plasmid encodes HCV envelope glycoproteins from strain JFH-1 (genotype 2a), in which the A4 epitope was reconstituted as described above. Then fragments containing mutation E2N2, E2N4, or E2N7 were excised from mutated full-length JFH-1 plasmids with AscI and BsiWI and ligated into a AscI/BsiWI-digested pcDNA/E1E2(JFH-1) plasmid. E1 and E2 nucleotide sequences were verified for each mutant.
HCVcc replication and infectivity assays.
Plasmids encoding wild-type (WT) and mutated genomes were linearized at the 3′ end of the HCV cDNA with the restriction enzyme XbaI and treated with mung bean nuclease (New England Biolabs). In vitro transcripts were generated using a Megascript kit according to the manufacturer's protocol (Ambion). The in vitro reaction mixture was set up and incubated at 37°C for 4 h, and transcripts were precipitated by the addition of LiCl. Twenty micrograms of RNA was delivered into CD81-deficient Huh-7w7 cells by electroporation as described previously (10). Replication was assessed at 72 and 96 h by measuring Renilla luciferase activities in electroporated cells with a Berthold CentroXS3 LB 960 luminometer as indicated by the manufacturer (Promega). We verified that our luciferase data were in the linear range of the assay. Supernatants containing HCVcc were harvested 96 h after electroporation and filtered through a 0.45-μm-pore-sized membrane for infectivity measurements. HCVcc were incubated for 3 h with Huh-7 cells seeded the day before in 24-well plates. At 72 h postinfection, luciferase assays were performed on infected cells as indicated by the manufacturer (Promega). In additional experiments, we verified that luciferase activity correlates with viral titers (focus-forming units [FFU]/ml), and we obtained a coefficient of determination of 0.9988 (data not shown). In some experiments, we also used plasmids carrying the JFH-1 genome, which carries a large in-frame deletion in the E1/E2 coding region (ΔE1E2) known to alter viral particle release or a nonreplicative genome containing a GND mutation in the NS5B active site (49).
HCVcc intracellular infectivity assay.
Twenty micrograms of RNA was delivered to Huh-7 cells by electroporation as described previously (10). Supernatants containing extracellular HCVcc were harvested 72 h after electroporation, and cell debris was removed by centrifugation for 5 min at 10,000 × g. Cells were washed with phosphate-buffered saline (PBS), harvested by treatment with trypsin, and pelleted at 100 × g for 5 min. Cell pellets were resuspended in complete medium (medium supplemented with 10% fetal bovine serum) and lysed by three freeze-thaw cycles. Cell lysates were clarified by centrifugation at 10,000 × g for 5 min. Supernatants containing extracellular or intracellular virus were collected and used for infection of naïve cells. Three hours postinfection, the inoculum was removed and replaced with complete medium. At 72 h postinfection, cells were assayed for luciferase activity.
HCV core analysis.
HCV core was quantified by a fully automated chemiluminescent microparticle immunoassay according to the manufacturer's instructions (Architect HCVAg; Abbott, Germany) (33, 34).
Production of HCVpp and infection assay.
HCVpp were produced as described previously (5, 36). Supernatants containing the pseudotyped particles were harvested 72 h after transfection and filtered through 0.45-μm-pore-sized membranes. HCVpp were added to Huh-7 cells seeded the day before in 24-well plates and incubated for 3 h at 37°C. The supernatants were then removed, and the cells were incubated in Dulbecco's modified essential medium with 10% fetal bovine serum at 37°C. At 72 h postinfection, luciferase activities were measured as indicated by the manufacturer (Promega).
Neutralization assay.
Twenty micrograms of WT or mutated HCV genomic RNA was delivered into Huh-7 cells by electroporation as described previously (10). Supernatants containing HCVcc were harvested 8 to 9 days after electroporation and filtered through 0.45-μm-pore-sized membranes. Neutralization assays were performed by preincubating HCVcc and antibodies for 2 h at 37°C before they were put in contact with target cells. After 3 h of contact with HCVcc, cells were further incubated for 48 to 72 h with complete medium before luciferase activities were measured as indicated by the manufacturer (Promega). Concentrations resulting in 50% inhibition were measured graphically.
RESULTS
Effect of N-glycosylation site mutations on HCV replication and E1/E2 expression.
To study the role of E1/E2 glycans, the asparagine residues of the Asn-X-Ser/Thr motifs to which glycans are potentially added were mutated to glutamine residues. These mutations are known to prevent glycosylation without affecting the physicochemical properties of the sequon. The mutations were introduced in the context of a modified genotype 2a virus (JFH-1 strain) containing a luciferase reporter gene. Furthermore, to facilitate the detection of E1, the sequence encoding the A4 epitope of the H77 isolate was reconstituted into the JFH-1 genome (20). These mutations did not affect HCV infectivity (data not shown). As previously proposed, mutants were denoted with an N followed by a number corresponding to the relative position of the potential glycosylation site in each glycoprotein (E1N1 to E1N4 for E1 mutants and E2N1 to E2N11 for E2 mutants).
We first assessed the impact of each mutation on RNA replication. As described previously and in order to study replication in a single cycle, we used the CD81-deficient cell line Huh-7w7 for this assay (38, 42). As a control, we used a variant genome carrying a large in-frame deletion in the E1/E2 coding region (ΔE1E2) known to inactivate release of viral particles (49). As shown in Fig. 1 A, luciferase activities in electroporated cells after 72 h and 96 h were similar for the wild-type (WT) and the majority of the mutant viruses, suggesting that the mutations do not affect genomic replication in these viruses. However, a slight decrease in luciferase activity was repeatedly observed for the E1N4 mutant, suggesting that this mutation has a slight effect on RNA replication.
FIG. 1.

Effect of N-glycosylation site mutations on viral-genome replication, E1/E2 expression, and infectious-virion production. (A) Wild-type (WT) and mutated HCV genomes were delivered to CD81-deficient Huh-7w7 cells. Replication was assessed at 72 and 96 h by measuring Renilla luciferase activities in transfected cells. Results are expressed as relative light units (RLU) and are reported as the means ± standard deviations (SDs) of three independent experiments. An assembly-deficient virus (ΔE1E2) and a replication-defective virus (GND) were used as negative controls for assembly and replication, respectively. (B) Forty-eight hours after electroporation of the viral genomes devoid of the luciferase reporter gene, expression of the viral proteins E1, E2, and NS3 was analyzed in cell lysates by Western blotting with specific MAbs (A4 [anti-E1], 3/11 [anti-E2], 486D39 [anti-NS3], and C4 [anti-β-actin]). Mutated E1 and E2 envelope proteins lacking one glycan are indicated as E13g and E210g, respectively. (C) HCVcc produced by Huh-7w7 cells were incubated for 3 h with Huh-7 cells. Luciferase assays were performed on infected cells at 72 h postinfection. Results are expressed as relative light units and are reported as the means ± SDs of three independent experiments. The nonparametric Mann-Whitney test was used to compare the infectivities of the wild-type and mutant HCVcc. Differences were considered statistically significant if P < 0.05 (*).
At 48 h postelectroporation, the expression of mutated envelope proteins was analyzed by Western blotting on cell lysates. This analysis was performed in the context of Huh-7 cells transfected with mutant viruses lacking the luciferase reporter gene. As a control for HCV genome translation, the expression of the NS3 protein was also analyzed. As shown in Fig. 1B, mutated envelope proteins (E13g and E210g) migrated faster than wild-type glycoproteins, indicating that all potential glycosylation sites were modified in the context of the wild-type proteins. The level of detection of E1 and E2 was rather similar for all the E2 mutants. However, in the case of the E1 mutants, the E1 protein was undetectable for the E1N1 mutant (Fig. 1B). Since this mutation did not affect NS3 and E2 expression, the absence of detection of the E1N1 mutant could be due to the effect of the mutation on the recognition of the A4 epitope, which is located close to this glycosylation site.
Effect of N-glycosylation site mutations on infectious-virus production.
To further characterize our glycosylation mutants, infectious-virus production was measured for each mutant and compared to that of the wild-type virus. Supernatants of cells transfected with the ΔE1E2 mutant were used as a negative control to determine the background level of luciferase activity. As shown in Fig. 1C, the production of infectious virus for the E1N1, E2N7, E2N8, and E2N10 mutants was close to that of ΔE1E2. Compared to the wild-type virus, these mutants showed log10 reductions of 1.6, 1.4, 1.8, and 2.2, respectively. The infectivity of the E2N3 and E2N11 mutants was significantly reduced, to approximately 15% of that of the wild-type virus (Fig. 1C). Mutations of glycosylation sites E1N2, E1N4, E2N2, E2N4, and E2N5 led to a 0.2- to 0.3-log reduction of infectivity. In the case of the E1N4 mutant, we cannot exclude that the slight decrease of virus infectivity is due to a slightly lower replication efficiency (compare Fig. 1A and C). Production of infectious virus for the E1N3, E2N1, E2N6, and E2N9 mutants was similar to or slightly increased compared to that of the wild type, with the E2N6 mutant showing the largest increase (0.4 log compared to that of the WT). Although these increases were not statistically significant, this higher level of infectivity could be the result of improved secretion of viral particles or an increased specific infectivity of virions. Together, these results suggest that the E1N1, E2N3, E2N7, E2N8, E2N10, and E2N11 mutations have a negative effect on viral particle assembly and/or secretion or on virus entry.
Effects of N-glycosylation site mutations on HCV assembly and secretion.
It has been shown that significant amounts of infectious virions are present inside infected cells and can be harvested by repetitive freeze-thaw cycles (18). Thus, in order to study the effect of glycosylation site mutations on the secretion of viral particles, we compared the amounts of intracellular and extracellular infectious viruses produced upon transfection of Huh-7 cells with our mutants (Fig. 2 A). It is worth noting that the profile of infectivity of our mutants was the same in Huh-7 and Huh-7w7 cells (compare extracellular infectivity in Fig. 2A and 1C). For each mutant, we calculated the percentage of cell-associated infectivity relative to the total infectivity (intracellular plus extracellular infectivity). As shown in Fig. 2B, the percentages of intracellular infectivity were similar for all the mutants and the WT with about 75% of particles liberated into the culture fluid (ranging between 65% and 85%). These results indicate that glycosylation site mutations had no effect on the efficiency of virion release but rather affected viral particle assembly or specific infectivity.
FIG. 2.

Effect of N-glycosylation site mutations on HCVcc secretion. (A) Huh-7 cells were transfected, and 72 h posttransfection supernatants were collected. In parallel, virus-producing cells were washed and lysed by repetitive freeze-thaw cycles. Extracellular and intracellular infectivities were determined by measuring Renilla luciferase activities in infected cells. Results are expressed as relative light units and are reported as the means ± SDs of three independent experiments. (B) The results of panel A are expressed as percentages of cell-associated infectivity relative to the total infectivity (intracellular plus extracellular infectivity) and are reported as the means ± SDs of three independent experiments.
To distinguish between these two possibilities, we monitored the release of HCV core into the supernatant of electroporated Huh-7w7 cells using a fully automated chemiluminescent microparticle immunoassay as described previously (33, 34). Compared to the wild-type virus, the ΔE1E2 mutant showed 12% core release that may correspond to noninfectious defective particles (Fig. 3). To get a better estimate of the relative infectivities of secreted viral particles, virus infectivities were normalized to the level of core protein secretion (Fig. 3C). Several mutants (E1N3, E2N1, E2N2, E2N5, and E2N6) had a level of core secretion similar to that of the wild type. Since the infectivity of these mutants was also not affected, we concluded that these mutations have no effect on virus infectivity and assembly. For several other mutants (E1N1, E1N4, E2N8, E2N10, and E2N11), core secretion was drastically reduced (Fig. 3). Among these viruses, the E1N1, E2N8, E2N10, and E2N11 mutants also showed a significant decrease in virus infectivity (compare Fig. 1C and 3), indicating that, for these viruses, the decrease in infectivity is likely essentially due to a lower level of particle assembly. In the case of the E1N4 mutant, the low level of core release did not correlate with a decrease in virus infectivity, suggesting that the released particles of this mutant are more infectious than those of the wild type (Fig. 3C). Finally, several mutants (E1N2, E2N3, E2N4, and E2N7) had an intermediate level of secretion compared to that of the wild type. However, among these viruses, only E2N3 and E2N7 had significant reductions in infectivity, suggesting that these two mutants are affected in their entry functions. It is worth noting that in contrast to the other mutants, the E2N9 mutant showed some increase in core secretion, potentially explaining the slight increase in infectivity for this mutant (compare Fig. 1C and 3).
FIG. 3.

Effect of N-glycosylation site mutations on HCVcc assembly. (A and B) Analysis of core release. Mutated HCV genomes were delivered to CD81-deficient Huh-7w7 cells. HCV core release was assessed at 96 h using a fully automated chemiluminescent microparticle immunoassay. Results are expressed as raw data (A) or as percentages of that for the WT (B) and are reported as the means ± SDs of the results of two independent experiments. (C) Infectivity of the glycosylation mutants normalized to the level of core protein secretion. Values obtained for infectious-virion production (Fig. 1C) have been normalized to those obtained for HCV core release (Fig. 3B) to compare the specific infectivities of secreted viral particles.
Effect of the E2N2, E2N4, and E2N7 mutations on genotype 2a HCVpp.
Importantly, we observed some discrepancies when we compared the results obtained with the HCVcc (this work) and HCVpp systems (14, 19). In particular, mutation of glycosylation sites E2N2 and E2N4 had only a slight effect on HCVcc infectivity, whereas the same mutations led to the secretion of noninfectious HCVpp (14, 19). Furthermore, the mutation of the E2N7 glycosylation site strongly reduced HCVcc infectivity, whereas the same mutation resulted in the production of HCVpp as infectious as wild-type particles in spite of a decrease in the incorporation of E1/E2 into retroviral particles. These differences can potentially be due to the model system used to study the glycosylation mutants. However, we cannot exclude that sequence differences between genotypes may be responsible for the different effects observed, since the HCVpp experiments were performed with HCV envelope proteins of genotype 1a. To discriminate between these two possibilities, we produced the E2N2, E2N4, and E2N7 mutants in the context of genotype 2a HCVpp and compared their infectivities to that of HCVpp generated in the presence of wild-type envelope glycoproteins. As shown in Fig. 4 A, the E2N2 and E2N4 mutants were noninfectious, and the infectivity of the E2N7 mutant was reduced to 20% compared to that of the wild type. The levels of expression of E1, E2, and CA were very similar in cell lysates for all of the mutants (Fig. 4B). However, the level of expression of CA was higher in the absence of E1/E2. This difference is likely due to competition for protein expression when cells are transfected with two plasmids instead of one. In HCVpp, the levels of expression of CA were very similar. However, the level of incorporation of E2 into HCVpp was slightly reduced for each mutant. Furthermore, the level of incorporation of E1 was strongly reduced for the E2N2 and E2N4 mutants and slightly reduced for the E2N7 mutant. Altogether, these results suggest that as for genotype 1a (14, 19), mutation of glycosylation sites E2N2 and E2N4 in genotype 2a envelope proteins leads to a defect in the viral entry process of HCVpp even though the lower level of incorporation of E1/E2 into HCVpp can also contribute to reduction of the infectivity of these mutants. The potential increase in the specific infectivity observed in genotype 1a HCVpp containing the E2N7 mutation (14, 19) contrasts with the decrease in infectivity observed for this mutation in the context of genotype 2a (Fig. 4A), suggesting genotype differences for the role of this glycan.
FIG. 4.

Role of glycans E2N2, E2N4, and E2N7 on genotype 2a HCVpp infectivity. (A) Plasmids encoding the E2N2, E2N4, and E2N7 mutants in the context of a genotype 2a (JFH-1) E1/E2 polyprotein were used to generate HCVpp. Infection assays with the luciferase reporter gene were performed by using target Huh-7 cells. Similar inputs of viral particles were used in each experiment, and this was confirmed by comparing the amounts of capsid protein incorporated into HCVpp (see panel B, anti-CA [CA]). Pseudotyped particles produced in the absence of envelope proteins (ΔE1E2) were used as a control. The results are expressed as percentages of wild-type infectivity (WT) and are reported as means ± SDs of three independent experiments. (B) Incorporation of HCV envelope proteins into HCVpp. Particles were pelleted through 20% sucrose cushions and analyzed by Western blotting. HCV envelope glycoproteins and the capsid protein of murine leukemia virus (MLV) were revealed with the following specific MAbs: anti-E1 (A4), anti-E2 (3/11), and anti-CA (R187). Expression of mutant proteins was verified by direct Western blotting of cell lysates.
Glycans E2N1, E2N2, E2N4, E2N6, and E2N11 modulate the neutralizing activity of antibodies.
We and others recently reported that in the HCVpp system, glycans E2N1, E2N6, and E2N11 reduce the accessibility of antibodies to neutralizing epitopes in the CD81 binding region of E2 of genotype 1a (14, 22). We therefore investigated whether HCV glycans can also affect the accessibility of antibodies to neutralizing epitopes in the context of an infectious virus. Due to a defect in their infectivity, the E2N3, E2N7, E2N8, and E2N10 mutants could not be tested in this antibody neutralization assay.
We first tested antibodies purified from the sera of four patients infected with HCV of genotype 1a, 1b, 2, or 4. These antibodies were tested for their ability to neutralize HCVcc expressing wild-type or mutated envelope glycoproteins. As shown in Fig. 5, the results of viral neutralization were very similar and independent of the serum used. These experiments allowed us to classify the mutants into three groups. The first group contains mutants that are neutralized at a level similar to that of the wild-type virus (Fig. 5 and Table 1, the E2N5 and E2N9 mutants). However, in the case of E2N5, neutralization efficiency was slightly reduced, suggesting that this mutation might slightly affect the binding of neutralizing antibodies. A second group was composed of mutants that were highly sensitive to neutralization by antibodies from HCV-seropositive patients (Fig. 5 and Table 1, the E2N2, E2N4, and E2N6 mutants). The removal of the corresponding glycans led to up to a 20-fold reduction in the effective concentration required for half-maximal neutralization (EC50) (Table 1). The third group contains mutants with an intermediate phenotype (Fig. 5 and Table 1, E2N1 and E2N11). Altogether, these data suggest that glycans at positions E2N1, E2N2, E2N4, E2N6, and E2N11 reduce the accessibility of neutralizing antibodies to their epitopes on the E2 glycoprotein.
FIG. 5.

Effect of N-glycosylation site mutations on HCVcc sensitivity to neutralization by antibodies purified from HCV-seropositive sera. Neutralization assays were performed by incubating HCVcc glycosylation mutants or WT HCVcc with various concentrations of antibodies (Ab) purified from the sera of patients infected with HCV genotype 1a (A), 1b (B), 2 (C), or 4 (D). After a 2 h-incubation at 37°C, mixtures of HCVcc and antibodies were put into contact with target cells for 3 h. Luciferase assays were performed on infected cells at 48 to 72 h postinfection. Results are expressed as percentages of infectivity relative to infectivity in the absence of antibodies and are reported as the means ± SDs of three independent experiments.
TABLE 1.
Concentration resulting in 50% inhibition of HCVcc entry
| Virus | 50% neutralization concna (μg/ml) of: |
||||||
|---|---|---|---|---|---|---|---|
| Antibodies from sera positive for indicated HCV genotype |
MAb 3/11 | Lectin CV-Nb | CD81-LEL | ||||
| 1a | 1b | 2 | 4 | ||||
| Wild type | 3.9 (1.0) | >10 (1.0) | 10 (1.0) | >10 (1.0) | >10 (1.0) | 0.3 (1.0) | 4.3 (1.0) |
| HCVcc mutant | |||||||
| E2N1 | 1.4 (2.8) | 1.3 (>7.7) | 1.3 (7.7) | 0.7 (>14.3) | 0.4 (>25.0) | 0.2 (1.5) | 0.6 (7.2) |
| E2N2 | 0.6 (6.5) | 0.7 (>14.3) | 0.7 (14.3) | 0.5 (>20.0) | 0.4 (>25.0) | 0.4 (0.8) | 0.5 (8.6) |
| E2N4 | 0.7 (5.6) | 0.7 (>14.3) | 0.7 (14.3) | 0.6 (>16.7) | 0.6 (>16.7) | 0.3 (1.0) | 0.6 (7.2) |
| E2N5 | >10 (<0.4) | >10 (ND) | >10 (<1.0) | >10 (ND) | >10 (ND) | 0.5 (0.6) | >10 (<0.4) |
| E2N6 | 0.6 (6.5) | 0.6 (>16.7) | 0.7 (14.3) | 0.5 (>20.0) | 2.8 (>3.6) | 0.4 (0.8) | 0.5 (8.6) |
| E2N9 | 4.3 (0.9) | 2.9 (>3.4) | 8.1 (1.2) | 2.2 (>4.5) | >10 (ND) | 0.5 (0.6) | >10 (<0.4) |
| E2N11 | 1.5 (2.6) | 1.0 (>10.0) | 1.6 (6.3) | 0.8 (>12.5) | 2.4 (>4.2) | 0.5 (0.6) | 2.5 (1.7) |
Values in parentheses are differences (n-fold) from the 50% neutralization concentration of the wild-type. ND, not determined.
CV-N, cyanovirin-N.
We previously reported that the neutralizing activity of MAbs directed against epitopes located in the CD81 binding region is increased in the absence of E2N1, E2N6, or E2N11 glycans in the context of the HCVpp system. We therefore analyzed the sensitivity of HCVcc E2 glycosylation mutants to neutralization by one of these MAbs. We chose the neutralizing MAb 3/11, whose epitope is well characterized (25). As shown in Fig. 6 A and Table 1, the E2N1, E2N2, E2N4, E2N6, and E2N11 mutants were also more sensitive than the WT to neutralization by MAb 3/11, suggesting that the corresponding glycans mask a single region at the surface of the E2 envelope glycoprotein. The greater sensitivity of the E2N1, E2N2, and E2N4 mutants compared to that of E2N6 and E2N11 is likely due to the location of E2N1, E2N2, and E2N4 glycans, which may be closer to the 3/11 epitope (see Discussion). It must be noted that WT and E2 glycosylation mutants showed similar sensitivity to inhibition by cyanovirin-N, an inhibitor of HCV entry (Fig. 6B and Table 1) (23), confirming that the effect of glycosylation site mutations on the sensitivity to polyclonal and monoclonal neutralizing antibodies was not due to differences in the assembly and/or infectivity of HCVcc particles. To exclude a potential influence of antigen concentration on mutant neutralization compared to that of the wild-type virus, we performed neutralization assays with diluted WT or E2N6 virus. However, we did not observe any difference in the sensitivity to MAb 3/11 between diluted viruses and nondiluted viruses (data not shown), indicating that large differences in antigen concentrations are not responsible for the increased sensitivity to neutralizing antibodies. Together, these results indicate that E2N1, E2N2, E2N4, E2N6, and E2N11 glycans contribute to the masking of neutralizing epitopes on E2.
FIG. 6.

Effect of N-glycosylation site mutations on HCVcc sensitivity to inhibition by MAb 3/11 or cyanovirin-N. Inhibition assays were performed by incubating HCVcc glycosylation mutants or wild-type HCVcc (WT) with various concentrations of MAb 3/11 (A) or cyanovirin-N (CV-N) (B). After a 2 h-incubation at 37°C, mixtures were put into contact with target cells for 3 h. Luciferase assays were performed on infected cells at 48 to 72 h postinfection. Results are expressed as percentages of infectivity relative to infectivity in the absence of inhibitory protein and are reported as the means ± SDs of three independent experiments.
Glycans E2N1, E2N2, E2N4, and E2N6 modulate CD81 binding to E2.
In addition to an effect on neutralization, we previously reported that in the HCVpp system, glycans E2N1, E2N6, and E2N11 reduce the accessibility of a soluble form of CD81 to its binding region on E2 (22). We therefore investigated whether HCV glycans affect the accessibility of CD81 to its binding region in the context of an infectious virus. To this end, we analyzed the sensitivity of E2 glycosylation mutants to inhibition by a soluble form of the CD81 large extracellular loop (CD81-LEL). As shown in Fig. 7 and Table 1, our results indicate that glycans E2N1, E2N2, E2N4, and E2N6 are more sensitive to inhibition with CD81-LEL, suggesting that these four glycans also modulate CD81 binding to E2. Surprisingly, we did not observe any effect of E2N11 glycosylation site mutation on sensitivity to CD81-LEL inhibition, whereas glycan E2N11 modulated both neutralizing antibodies and CD81 binding to E2 in the HCVpp model. Altogether, our data indicate that in the context of HCVcc, glycans at positions E2N1, E2N2, E2N4, and E2N6 modulate both CD81 and neutralizing antibody binding to E2.
FIG. 7.

Effect of N-glycosylation site mutations on HCVcc sensitivity to inhibition by CD81-LEL. Inhibition assays were performed by incubating HCVcc glycosylation mutants or wild-type HCVcc (WT) with various concentrations of CD81-LEL. After a 2 h-incubation at 37°C, mixtures were put into contact with target cells for 3 h. Luciferase assays were performed on infected cells at 48 to 72 h postinfection. Results are expressed as percentages of infectivity relative to infectivity in the absence of inhibitory protein and are reported as the means ± SDs of three independent experiments.
DISCUSSION
HCV envelope glycoproteins play a pivotal role in the HCV life cycle. These proteins participate in the assembly of infectious particles and play a major role in viral entry, since they enable interaction with specific cell surface receptors and induce fusion between the viral envelope and the membrane of early endosomes in the host cell. HCV envelope proteins are highly glycosylated and generally contain 4 and 11 N-linked glycans on E1 and E2, respectively, that can play a major role in protein folding, in viral entry, and in modulation of the immune response. Site-directed mutagenesis studies allowed us to study for the first time the role of N-linked glycans in the context of authentic HCV virions. Our results highlight differences in the effects of glycosylation mutations between the HCVpp and HCVcc systems. They also indicate that several glycans play an important role in HCVcc assembly and/or infectivity. Furthermore, neutralization experiments demonstrated that at least five glycans on E2 (denoted E2N1, E2N2, E2N4, E2N6, and E2N11) strongly reduce the sensitivity of HCVcc to antibody neutralization. Together, our data indicate that the glycans associated with HCV envelope glycoproteins play roles at different steps of the viral life cycle.
Several mutations affect HCV assembly (E1N1, E2N3, E2N8, E2N10, and E2N11). In the case of the E1N1, E2N8, and E2N10 mutants, a strong impairment of assembly was observed. Indeed, the secretion level of these mutants was in the same range as that of the negative-control virus with the E1/E2 region deleted. Furthermore, the supernatants of the cells transfected with the genomic RNA of these mutants were noninfectious, a finding that is in agreement with the effect of the mutations on particle assembly. Interestingly, the same mutations were shown to decrease E1/E2 heterodimerization in the context of recombinant proteins, and they also affected the incorporation of envelope proteins at the surfaces of HCVpp (19). Altogether, these results indicate that glycans E1N1, E2N8, and E2N10 are important for the folding and heterodimerization of HCV envelope glycoproteins and thus for viral particle assembly. Another mutant (E1N4), which also showed some decrease in E1/E2 heterodimerization and affected the incorporation of envelope proteins at the surfaces of HCVpp (19), also showed a decrease in HCVcc secretion. However, in this case, the secreted particles seemed more infectious than those of the wild type (Fig. 3C). Mutations of the E2N3 and E2N11 glycosylation sites had an intermediate effect on virus assembly. Indeed, these mutations reduced HCVcc infectivity and somewhat decreased the release of HCVcc.
Glycans associated with viral envelope proteins can modulate the entry functions of these proteins by modifying their receptor affinities or by affecting their fusion activities (45, 51). It is worth noting that the removal of a glycan at positions E2N1 and E2N6 increases HCVcc infectivity, even though the increase was not statistically significant (Fig. 1C). Importantly, these mutants were also more sensitive to CD81-LEL inhibition (Fig. 7). Furthermore, Owsianka et al. identified conserved residues involved in CD81 interaction close to these glycosylation sites (37). Finally, it has also been shown that a soluble form of E2 lacking a glycan at position E2N1 or E2N6 exhibited increased binding to CD81 (14). Altogether, these results suggest that the higher level of fitness of the E2N1 and E2N6 mutants may be due to a better interaction of E2 with CD81. Interestingly, we and others recently reported the emergence of adaptive mutants presenting a shifting site at position E2N1 or lacking a glycan at position E2N6 after serially passaging JFH-1 or J6/JFH-1 in cell culture (8, 10, 42). However, in the human host, these glycans must be important in limiting recognition of the CD81 binding site by neutralizing antibodies and in avoiding too-rapid elimination of HCV by the immune system (see below). This may explain the high level of conservation of these glycosylation sites in HCV genomic sequences (22).
The mutation of the E2N7 glycosylation site strongly reduced HCVcc infectivity (5% compared to that of the WT), with only a slight decrease in viral particle secretion, indicating a defect in virus entry. Interestingly, the same mutation also reduced the infectivity of genotype 2a HCVpp but resulted in the production of genotype 1a HCVpp as infectious as wild-type particles, suggesting a genotype-specific role for the E2N7 glycan. This is not surprising, since this glycosylation site is among the less conserved and is even absent in sequences from genotypes 3 and 6 (22).
For some mutants, the entry functions of HCV envelope glycoproteins were differently affected by glycan loss in the HCVcc system (this work) and in the HCVpp system (14, 19) (Table 2). In addition to differences observed for the E2N3 mutant (see above), mutation of the glycosylation sites E2N2 and E2N4 had only a slight effect on HCVcc infectivity, whereas the same mutations led to the secretion of noninfectious HCVpp particles in genotypes 1a and 2a. These disparities are likely due to differences in the assembly process between HCVpp and HCVcc. Indeed, HCVcc assemble in an ER-derived compartment, whereas HCVpp are assembled in a post-Golgi compartment (44). This implies that the glycans associated with HCV envelope proteins are not exposed in the same way to the Golgi glycosidases and glycosyltransferases in these two viral systems, and they are therefore not necessarily similarly processed, as recently observed (46). Furthermore, the differences in the assembly processes can also lead to differences in the presentation of the proteins at the surface of the particle as well as differences in protein-protein interactions between the envelope glycoproteins. Indeed, at the surface of HCVpp, functional E1 and E2 glycoproteins have been shown to form noncovalent heterodimers (36), whereas the majority of HCV envelope glycoproteins associated with HCVcc particles are high-molecular-weight complexes stabilized by disulfide bonds (46). Finally, we cannot exclude that the presence of very-low-density lipoproteins associated with HCVcc may affect the properties of the envelope proteins (17, 26). Differences in the entry functions of HCV envelope glycoproteins between HCVpp and HCVcc have indeed already been reported (28, 29, 42, 43).
TABLE 2.
Summary of the properties of glycosylation mutants
| Virus | HCVcc infectivitya | HCVpp infectivitya,b | Core releasec | Sensitivity to neutralizationd |
|---|---|---|---|---|
| Wild type | +++ | +++ | ++ | + |
| Mutant | ||||
| E1N1 | +/− | ++ | − | ND (+) |
| E1N2 | ++ | + | + | ND (+) |
| E1N3 | +++ | ++ | ++ | ND (+) |
| E1N4 | ++ | + | +/− | ND (+) |
| E2N1 | +++ | ++ | ++ | ++ |
| E2N2 | ++ | − (−) | ++ | ++e |
| E2N3 | + | +++ | + | ND (+) |
| E2N4 | ++ | − (−) | + | ++e |
| E2N5 | ++ | ++ | ++ | + |
| E2N6 | +++ | ++ | ++ | ++ |
| E2N7 | +/− | +++ (+) | + | ND (−) |
| E2N8 | − | − | +/− | ND |
| E2N9 | +++ | +++ | ++ | + |
| E2N10 | − | − | − | ND |
| E2N11 | + | + | +/− | ++ |
Percentage of infectivity: +++, >90%; ++, between 30% and 90%; +, between 10% and 30%; +/−, between 2% and 10%; −, <2%.
Infectivity of HCVpp of genotype 1a as previously reported (19). Values in parentheses are results obtained for HCVpp of genotype 2a.
Percentage of core release: ++, >75%; +, between 30% and 75%; +/−, between 12% and 30%; −, <12%.
Sensitivity to antibody neutralization: +, similar to that of the wild type; ++, more than 5-fold increase in sensitivity to neutralization with most antibodies tested; −, decrease in sensitivity to neutralization. Results in parentheses were obtained with HCVpp of genotype 1a only (22). ND, not determined.
Results obtained with the HCVcc system only.
Our data demonstrate that at least five glycans on E2 (denoted E2N1, E2N2, E2N4, E2N6, and E2N11) reduce the sensitivity of HCVcc to antibody neutralization, indicating that these glycans limit the recognition of neutralizing epitopes on the surface of E2. Indeed, removal of these glycans led to a greater sensitivity to neutralization by antibodies from HCV-seropositive patients as well as by MAb 3/11. Importantly, for some of these mutants, the reduction in the EC50 was more than 20-fold (Table 1). These data are in agreement with those obtained for the HCVpp system, at least for the E2N1, E2N6, and E2N11 mutants (14, 22). The sensitivity of the E2N2 and E2N4 mutants to antibody-mediated neutralization could not be tested in the HCVpp system because these mutants were not infectious in the pseudoparticle system. Interestingly, four of these five glycans (denoted E2N1, E2N2, E2N4, and E2N6) also modulate inhibition by the soluble form of CD81, in accordance with previous observations (14, 22), suggesting that the CD81 binding site is an important neutralizing-antibody target protected by glycans. A putative structural model of HCV glycoprotein E2 has recently been proposed. In this model, based on class II fusion proteins, E2 consists of three separate domains and a stem region connected to the transmembrane domain (30). Importantly, glycosylation sites E2N1, E2N2, and E2N6 are located in domain I (DI), which contains determinants for CD81 (Fig. 8). It is expected that the orientation of E2 on the surface of the viral particle would expose the CD81 binding site to the target cells. Glycosylation site E2N4, which is in domain II (DII), is also located in close proximity to the CD81 binding site. Together, these four glycosylation sites (E2N1, E2N2, E2N4, and E2N6) surround the CD81 binding site (Fig. 8), providing structural evidence for the role of these glycans in the modulation of the humoral immune response.
FIG. 8.
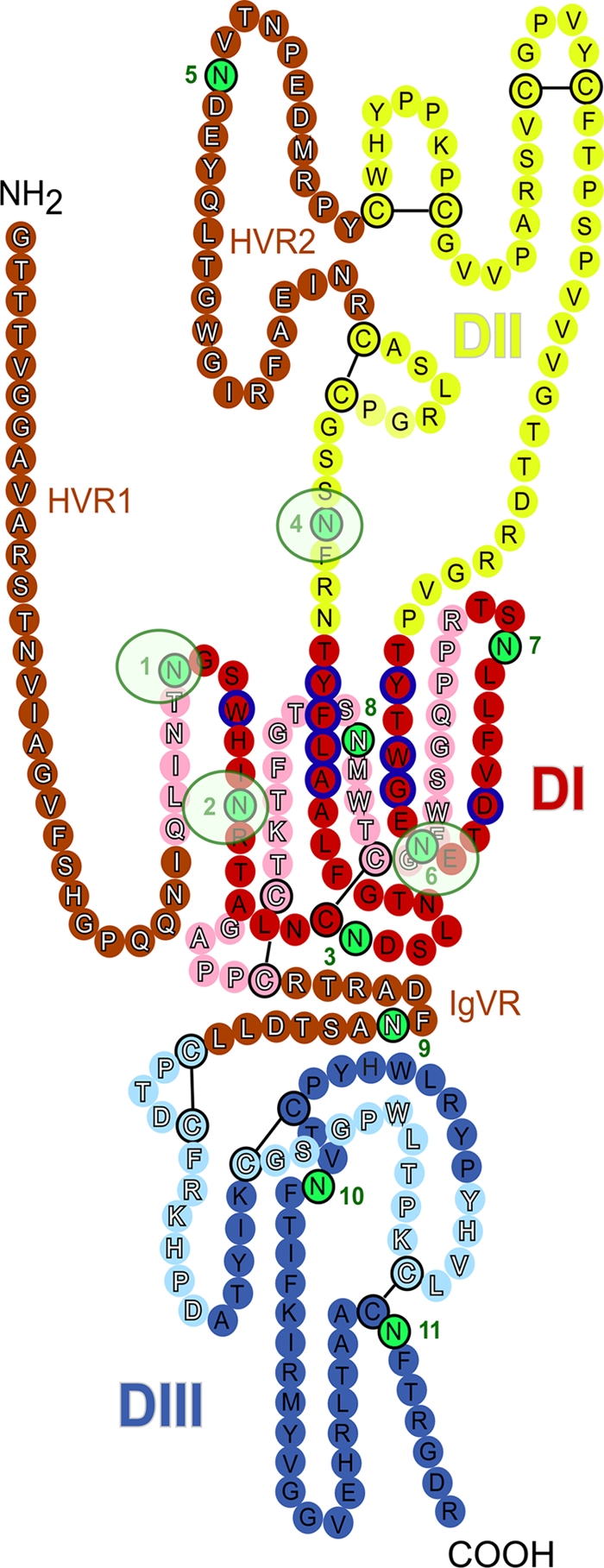
Localization of N-linked glycans on the model of HCV glycoprotein E2. The linear sequence of the JFH-1 E2 ectodomain without the stem region is represented as a chain of beads (colored circles) labeled with the corresponding amino acid and threaded onto a class II fold, which is an adapted version of the model recently published by Krey et al. (30). The three putative domains are presented in red (DI), yellow (DII), and blue (DIII), and the variable regions (HVR1, HVR2, and IgVR) are indicated in brown. Circles in pale and bright colors represent residues in the background and foreground of the domains, respectively, and are labeled in white and black fonts. Disulfide bonds are indicated by black bars. Glycosylation sites are shown by green circles numbered sequentially. DI domain residues that participate in CD81 binding are outlined in blue. It has been suggested that additional residues in the DIII domain can also affect CD81 binding (40). However, mutation of these residues might affect E2 folding, suggesting an indirect role for these residues (27). Glycans affecting CD81 binding are highlighted by light green circles.
The E2N11 glycan affects virus neutralization without affecting CD81 binding. The lack of an effect of the E2N11 mutation on inhibition by soluble CD81 is in agreement with the position of this glycosylation site in domain III (DIII), far from the CD81 binding region (Fig. 8) (30). However, the effect of this mutation on virus neutralization by MAb 3/11, which binds to the CD81 binding region of DI (25), suggests that the E2N11 glycan can also partially contribute to reduction of the accessibility of the CD81 binding region to antibodies. The size difference between MAb 3/11 (150 kDa) and the GST-CD81-LEL (36 kDa) can potentially explain why the mutation did not affect inhibition by CD81, whereas it led to better neutralization by MAb 3/11. Furthermore, to affect neutralizing antibodies targeting epitopes in DI, the E2N11 glycan must be located closer to DI than is suggested by the two-dimensional representation in the E2 model. One possibility is that in the oligomeric form of the envelope proteins, DIII from one molecule is close to DI from an adjacent E2 protein on the viral particle. This would be the case if E2 forms antiparallel dimers, as is the case with flaviviruses. Alternatively, we cannot exclude that the E2N11 mutation affects the DI-DIII interaction, which indirectly leads to better accessibility of the CD81 binding region to neutralizing antibodies. Interestingly, mutation of residues in DIII can affect CD81 binding (40); however, these mutations might affect E2 folding, suggesting an indirect role for these residues (27).
Besides modulating accessibility to the CD81 binding site, the glycans associated with HCV envelope proteins reduce the accessibility of other regions of the protein moiety. Indeed, one third of the molecular weight of the E1/E2 heterodimer corresponds to glycans. Interestingly, in the case of HIV, it has been proposed that the glycoprotein gp120 presents an immunologically “silent face” that consists of heavily glycosylated regions of gp120 that may appear as self to the immune system (53). Taking into account the size of one glycan, one can also suppose that the presence of a large concentration of glycans on the surface of an HCV particle could also limit the immunogenicity of the envelope proteins. This suggests that, as observed for gp120, HCV envelope glycoproteins likely contain immunologically silent regions, with the associated glycans forming a “glycan shield” at the surface of the virion. Together with other mechanisms (6, 11, 15, 47, 48, 54), this may explain, at least in part, how HCV evades the humoral immune response.
Acknowledgments
We thank Sophana Ung for technical assistance. We also thank T. Wakita, J. McKeating, J. F. Delagneau, K. Gustafson, C. M. Rice, B. Bartosch, F. L. Cosset, T. Pietschamnn, and R. Bartenschlager for providing us with reagents.
This work was supported by the Agence Nationale de Recherche sur le Sida et les Hépatites Virales (ANRS) and by a Marie Curie Research Training Network grant (MRTN-CT-2006-035599). C.-I.P was supported by postdoctoral program grant POSDRU/89/1.5/S/60746 from the European Social Fund. J.D. is an international scholar of the Howard Hughes Medical Institute.
Footnotes
Published ahead of print on 15 September 2010.
REFERENCES
- 1.Balzarini, J. 2007. Targeting the glycans of glycoproteins: a novel paradigm for antiviral therapy. Nat. Rev. Microbiol. 5:583-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balzarini, J. 2005. Targeting the glycans of gp120: a novel approach aimed at the Achilles heel of HIV. Lancet Infect. Dis. 5:726-731. [DOI] [PubMed] [Google Scholar]
- 3.Balzarini, J., K. Van Laethem, S. Hatse, M. Froeyen, W. Peumans, E. Van Damme, and D. Schols. 2005. Carbohydrate-binding agents cause deletions of highly conserved glycosylation sites in HIV GP120: a new therapeutic concept to hit the Achilles heel of HIV. J. Biol. Chem. 280:41005-41014. [DOI] [PubMed] [Google Scholar]
- 4.Bartosch, B., J. Bukh, J. C. Meunier, C. Granier, R. E. Engle, W. C. Blackwelder, S. U. Emerson, F. L. Cosset, and R. H. Purcell. 2003. In vitro assay for neutralizing antibody to hepatitis C virus: evidence for broadly conserved neutralization epitopes. Proc. Natl. Acad. Sci. U. S. A. 100:14199-14204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bartosch, B., J. Dubuisson, and F. L. Cosset. 2003. Infectious hepatitis C pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 197:633-642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bartosch, B., G. Verney, M. Dreux, P. Donot, Y. Morice, F. Penin, J. M. Pawlotsky, D. Lavillette, and F. L. Cosset. 2005. An interplay between the hypervariable region 1 of the hepatitis C virus E2 glycoprotein, the scavenger receptor BI, and high-density lipoprotein promotes both enhancement of infection and protection against neutralizing antibodies. J. Virol. 79:8217-8229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bertaux, C., D. Daelemans, L. Meertens, E. G. Cormier, J. F. Reinus, W. J. Peumans, E. J. Van Damme, Y. Igarashi, T. Oki, D. Schols, T. Dragic, and J. Balzarini. 2007. Entry of hepatitis C virus and human immunodeficiency virus is selectively inhibited by carbohydrate-binding agents but not by polyanions. Virology 366:40-50. [DOI] [PubMed] [Google Scholar]
- 8.Bungyoku, Y., I. Shoji, T. Makine, T. Adachi, K. Hayashida, M. Nagano-Fujii, Y. Ide, L. Deng, and H. Hotta. 2009. Efficient production of infectious hepatitis C virus with adaptive mutations in cultured hepatoma cells. J. Gen. Virol. 90:1681-1691. [DOI] [PubMed] [Google Scholar]
- 9.Cormack, B. 1 May 2001, posting date. Unit 8.5, Directed mutagenesis using the polymerase chain reaction, p. 8.5.1-8.5.10. In F. M. Ausubel, R. Brent, R. E. Kingston, A. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.), Current protocols in molecular biology. John Wiley and Sons, Hoboken, NJ. doi: 10.1002/0471142727.mb0805s37. [DOI] [PubMed]
- 10.Delgrange, D., A. Pillez, S. Castelain, L. Cocquerel, Y. Rouillé, J. Dubuisson, T. Wakita, G. Duverlie, and C. Wychowski. 2007. Robust production of infectious viral particles in Huh-7 cells by introducing mutations in HCV structural proteins. J. Gen. Virol. 88:2495-2503. [DOI] [PubMed] [Google Scholar]
- 11.Dreux, M., T. Pietschmann, C. Granier, C. Voisset, S. Ricard-Blum, P. E. Mangeot, Z. Keck, S. Foung, N. Vu-Dac, J. Dubuisson, R. Bartenschlager, D. Lavillette, and F. L. Cosset. 2006. High density lipoprotein inhibits hepatitis C virus-neutralizing antibodies by stimulating cell entry via activation of the scavenger receptor BI. J. Biol. Chem. 281:18285-18295. [DOI] [PubMed] [Google Scholar]
- 12.Dubuisson, J., H. H. Hsu, R. C. Cheung, H. B. Greenberg, D. G. Russell, and C. M. Rice. 1994. Formation and intracellular localization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia and Sindbis viruses. J. Virol. 68:6147-6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Etchison, J. R., and J. J. Holland. 1974. Carbohydrate composition of the membrane glycoprotein of vesicular stomatitis virus grown in four mammalian cell lines. Proc. Natl. Acad. Sci. U. S. A. 71:4011-4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Falkowska, E., F. Kajumo, E. Garcia, J. Reinus, and T. Dragic. 2007. Hepatitis C virus envelope glycoprotein E2 glycans modulate entry, CD81 binding, and neutralization. J. Virol. 81:8072-8079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farci, P., A. Shimoda, D. Wong, T. Cabezon, D. De Gioannis, A. Strazzera, Y. Shimizu, M. Shapiro, H. J. Alter, and R. H. Purcell. 1996. Prevention of hepatitis C virus infection in chimpanzees by hyperimmune serum against the hypervariable region 1 of the envelope 2 protein. Proc. Natl. Acad. Sci. U. S. A. 93:15394-15399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flint, M., C. Maidens, L. D. Loomis-Price, C. Shotton, J. Dubuisson, P. Monk, A. Higginbottom, S. Levy, and J. A. McKeating. 1999. Characterization of hepatitis C virus E2 glycoprotein interaction with a putative cellular receptor, CD81. J. Virol. 73:6235-6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gastaminza, P., G. Cheng, S. Wieland, J. Zhong, W. Liao, and F. V. Chisari. 2008. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J. Virol. 82:2120-2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gastaminza, P., S. B. Kapadia, and F. V. Chisari. 2006. Differential biophysical properties of infectious intracellular and secreted hepatitis C virus particles. J. Virol. 80:11074-11081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goffard, A., N. Callens, B. Bartosch, C. Wychowski, F. L. Cosset, C. Montpellier-Pala, and J. Dubuisson. 2005. Role of N-linked glycans in the functions of hepatitis C virus envelope glycoproteins. J. Virol. 79:8400-8409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goueslain, L., K. Alsaleh, P. Horellou, P. Roingeard, V. Descamps, G. Duverlie, Y. Ciczora, C. Wychowski, J. Dubuisson, and Y. Rouille. 2010. Identification of GBF1 as a cellular factor required for hepatitis C virus RNA replication. J. Virol. 84:773-787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Helle, F., and J. Dubuisson. 2008. Hepatitis C virus entry into host cells. Cell. Mol. Life Sci. 65:100-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Helle, F., A. Goffard, V. Morel, G. Duverlie, J. McKeating, Z. Y. Keck, S. Foung, F. Penin, J. Dubuisson, and C. Voisset. 2007. The neutralizing activity of anti-hepatitis C virus antibodies is modulated by specific glycans on the E2 envelope protein. J. Virol. 81:8101-8111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Helle, F., C. Wychowski, N. Vu-Dac, K. R. Gustafson, C. Voisset, and J. Dubuisson. 2006. Cyanovirin-N inhibits hepatitis C virus entry by binding to envelope protein glycans. J. Biol. Chem. 281:25177-25183. [DOI] [PubMed] [Google Scholar]
- 24.Higginbottom, A., E. R. Quinn, C. C. Kuo, M. Flint, L. H. Wilson, E. Bianchi, A. Nicosia, P. N. Monk, J. A. McKeating, and S. Levy. 2000. Identification of amino acid residues in CD81 critical for interaction with hepatitis C virus envelope glycoprotein E2. J. Virol. 74:3642-3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hsu, M., J. Zhang, M. Flint, C. Logvinoff, C. Cheng-Mayer, C. M. Rice, and J. A. McKeating. 2003. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. U. S. A. 100:7271-7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang, H., F. Sun, D. M. Owen, W. Li, Y. Chen, M. Gale, Jr., and J. Ye. 2007. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc. Natl. Acad. Sci. U. S. A. 104:5848-5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iacob, R. E., Z. Keck, O. Olson, S. K. Foung, and K. B. Tomer. 2008. Structural elucidation of critical residues involved in binding of human monoclonal antibodies to hepatitis C virus E2 envelope glycoprotein. Biochim. Biophys. Acta 1784:530-542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johansson, D. X., C. Voisset, A. W. Tarr, M. Aung, J. K. Ball, J. Dubuisson, and M. A. Persson. 2007. Human combinatorial libraries yield rare antibodies that broadly neutralize hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 104:16269-16274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kapadia, S. B., H. Barth, T. Baumert, J. A. McKeating, and F. V. Chisari. 2007. Initiation of hepatitis C virus infection is dependent on cholesterol and cooperativity between CD81 and scavenger receptor B type I. J. Virol. 81:374-383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krey, T., J. d'Alayer, C. M. Kikuti, A. Saulnier, L. Damier-Piolle, I. Petitpas, D. X. Johansson, R. G. Tawar, B. Baron, B. Robert, P. England, M. A. Persson, A. Martin, and F. A. Rey. 2010. The disulfide bonds in glycoprotein E2 of hepatitis C virus reveal the tertiary organization of the molecule. PLoS Pathog. 6:e1000762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lemon, S. M., C. Walker, M. J. Alter, and M. Yi. 2007. Hepatitis C virus, p. 1253-1304. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 32.Lindenbach, B. D., M. J. Evans, A. J. Syder, B. Wolk, T. L. Tellinghuisen, C. C. Liu, T. Maruyama, R. O. Hynes, D. R. Burton, J. A. McKeating, and C. M. Rice. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623-626. [DOI] [PubMed] [Google Scholar]
- 33.Mederacke, I., H. Wedemeyer, S. Ciesek, E. Steinmann, R. Raupach, K. Wursthorn, M. P. Manns, and H. L. Tillmann. 2009. Performance and clinical utility of a novel fully automated quantitative HCV-core antigen assay. J. Clin. Virol. 46:210-215. [DOI] [PubMed] [Google Scholar]
- 34.Morota, K., R. Fujinami, H. Kinukawa, T. Machida, K. Ohno, H. Saegusa, and K. Takeda. 2009. A new sensitive and automated chemiluminescent microparticle immunoassay for quantitative determination of hepatitis C virus core antigen. J. Virol. Methods 157:8-14. [DOI] [PubMed] [Google Scholar]
- 35.Nakabayashi, H., K. Taketa, K. Miyano, T. Yamane, and J. Sato. 1982. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res. 42:3858-3863. [PubMed] [Google Scholar]
- 36.Op De Beeck, A., C. Voisset, B. Bartosch, Y. Ciczora, L. Cocquerel, Z. Keck, S. Foung, F. L. Cosset, and J. Dubuisson. 2004. Characterization of functional hepatitis C virus envelope glycoproteins. J. Virol. 78:2994-3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Owsianka, A. M., J. M. Timms, A. W. Tarr, R. J. Brown, T. P. Hickling, A. Szwejk, K. Bienkowska-Szewczyk, B. J. Thomson, A. H. Patel, and J. K. Ball. 2006. Identification of conserved residues in the E2 envelope glycoprotein of the hepatitis C virus that are critical for CD81 binding. J. Virol. 80:8695-8704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rocha-Perugini, V., M. Lavie, D. Delgrange, J. Canton, A. Pillez, J. Potel, C. Lecoeur, E. Rubinstein, J. Dubuisson, C. Wychowski, and L. Cocquerel. 2009. The association of CD81 with tetraspanin-enriched microdomains is not essential for hepatitis C virus entry. BMC Microbiol. 9:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rocha-Perugini, V., C. Montpellier, D. Delgrange, C. Wychowski, F. Helle, A. Pillez, H. Drobecq, F. Le Naour, S. Charrin, S. Levy, E. Rubinstein, J. Dubuisson, and L. Cocquerel. 2008. The CD81 partner EWI-2wint inhibits hepatitis C virus entry. PLoS One 3:e1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rothwangl, K. B., and L. Rong. 2009. Analysis of a conserved RGE/RGD motif in HCV E2 in mediating entry. Virol. J. 6:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rouillé, Y., F. Helle, D. Delgrange, P. Roingeard, C. Voisset, E. Blanchard, S. Belouzard, J. McKeating, A. H. Patel, G. Maertens, T. Wakita, C. Wychowski, and J. Dubuisson. 2006. Subcellular localization of hepatitis C virus structural proteins in a cell culture system that efficiently replicates the virus. J. Virol. 80:2832-2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Russell, R. S., J. C. Meunier, S. Takikawa, K. Faulk, R. E. Engle, J. Bukh, R. H. Purcell, and S. U. Emerson. 2008. Advantages of a single-cycle production assay to study cell culture-adaptive mutations of hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 105:4370-4375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sainz, B., Jr., N. Barretto, and S. L. Uprichard. 2009. Hepatitis C virus infection in phenotypically distinct Huh7 cell lines. PLoS One 4:e6561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sandrin, V., P. Boulanger, F. Penin, C. Granier, F. L. Cosset, and B. Bartosch. 2005. Assembly of functional hepatitis C virus glycoproteins on infectious pseudoparticles occurs intracellularly and requires concomitant incorporation of E1 and E2 glycoproteins. J. Gen. Virol. 86:3189-3199. [DOI] [PubMed] [Google Scholar]
- 45.Sterjovski, J., M. J. Churchill, A. Ellett, L. R. Gray, M. J. Roche, R. L. Dunfee, D. F. Purcell, N. Saksena, B. Wang, S. Sonza, S. L. Wesselingh, I. Karlsson, E. M. Fenyo, D. Gabuzda, A. L. Cunningham, and P. R. Gorry. 2007. Asn 362 in gp120 contributes to enhanced fusogenicity by CCR5-restricted HIV-1 envelope glycoprotein variants from patients with AIDS. Retrovirology 4:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vieyres, G., X. Thomas, V. Descamps, G. Duverlie, A. H. Patel, and J. Dubuisson. 2010. Characterization of the envelope glycoproteins associated with infectious hepatitis C virus. J. Virol. 84:10159-10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Voisset, C., A. Op De Beeck, P. Horellou, M. Dreux, T. Gustot, G. Duverlie, F. L. Cosset, N. Vu-Dac, and J. Dubuisson. 2006. High-density lipoproteins reduce the neutralizing effect of hepatitis C virus (HCV)-infected patient antibodies by promoting HCV entry. J. Gen. Virol. 87:2577-2581. [DOI] [PubMed] [Google Scholar]
- 48.von Hahn, T., J. C. Yoon, H. Alter, C. M. Rice, B. Rehermann, P. Balfe, and J. A. McKeating. 2007. Hepatitis C virus continuously escapes from neutralizing antibody and T-cell responses during chronic infection in vivo. Gastroenterology 132:667-678. [DOI] [PubMed] [Google Scholar]
- 49.Wakita, T., T. Pietschmann, T. Kato, T. Date, M. Miyamoto, Z. Zhao, K. Murthy, A. Habermann, H. G. Krausslich, M. Mizokami, R. Bartenschlager, and T. J. Liang. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791-796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wasley, A., and M. J. Alter. 2000. Epidemiology of hepatitis C: geographic differences and temporal trends. Semin. Liver Dis. 20:1-16. [DOI] [PubMed] [Google Scholar]
- 51.Willett, B. J., E. L. McMonagle, N. Logan, A. Samman, and M. J. Hosie. 2008. A single site for N-linked glycosylation in the envelope glycoprotein of feline immunodeficiency virus modulates the virus-receptor interaction. Retrovirology 5:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Witvrouw, M., V. Fikkert, A. Hantson, C. Pannecouque, B. R. O'Keefe, J. McMahon, L. Stamatatos, E. de Clercq, and A. Bolmstedt. 2005. Resistance of human immunodeficiency virus type 1 to the high-mannose binding agents cyanovirin N and concanavalin A. J. Virol. 79:7777-7784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wyatt, R., P. D. Kwong, E. Desjardins, R. W. Sweet, J. Robinson, W. A. Hendrickson, and J. G. Sodroski. 1998. The antigenic structure of the HIV gp120 envelope glycoprotein. Nature 393:705-711. [DOI] [PubMed] [Google Scholar]
- 54.Zhang, P., L. Zhong, E. B. Struble, H. Watanabe, A. Kachko, K. Mihalik, M. L. Virata-Theimer, H. J. Alter, S. Feinstone, and M. Major. 2009. Depletion of interfering antibodies in chronic hepatitis C patients and vaccinated chimpanzees reveals broad cross-genotype neutralizing activity. Proc. Natl. Acad. Sci. U. S. A. 106:7537-7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhong, J., P. Gastaminza, G. Cheng, S. Kapadia, T. Kato, D. R. Burton, S. F. Wieland, S. L. Uprichard, T. Wakita, and F. V. Chisari. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 102:9294-9299. [DOI] [PMC free article] [PubMed] [Google Scholar]