

Abstract
The promyelocytic leukemia (PML) protein is expressed in the diffuse nuclear fraction of the nucleoplasm and in matrix-associated structures, known as nuclear bodies (NBs). PML NB formation requires the covalent modification of PML to SUMO. The noncovalent interactions of SUMO with PML based on the identification of a SUMO-interacting motif within PML seem to be required for further recruitment within PML NBs of SUMOylated proteins. RNA viruses whose replication takes place in the cytoplasm and is inhibited by PML have developed various strategies to counteract the antiviral defense mediated by PML NBs. We show here that primary fibroblasts derived from PML knockout mice are more sensitive to infection with encephalomyocarditis virus (EMCV), suggesting that the absence of PML results in an increase in EMCV replication. Also, we found that EMCV induces a decrease in PML protein levels both in interferon-treated cells and in PMLIII-expressing cells. Reduction of PML was carried out by the EMCV 3C protease. Indeed, at early times postinfection, EMCV induced PML transfer from the nucleoplasm to the nuclear matrix and PML conjugation to SUMO-1, SUMO-2, and SUMO-3, leading to an increase in PML body size where the viral protease 3C and the proteasome component were found colocalizing with PML within the NBs. This process was followed by PML degradation occurring in a proteasome- and SUMO-dependent manner and did not involve the SUMO-interacting motif of PML. Together, these findings reveal a new mechanism evolved by EMCV to antagonize the PML pathway in the interferon-induced antiviral defense.
The PML (promyelocytic leukemia) gene was originally identified through its fusion with the RARα gene in the t(15;17) translocation found in acute promyelocytic leukemia (APL) (14). PML, also known as TRIM19, is expressed in the diffuse nuclear fraction of the nucleoplasm and in matrix-associated structures, known as nuclear bodies (NBs) (17, 30). PML functions as the organizer of PML NBs, which are dynamic structures harboring numerous transiently and permanently localized proteins (35). The RBCC/TRIM motif, which contains a C3HC4 (RING finger) zinc-binding domain, two cysteine/histidine-rich motifs (the B boxes B1 and B2), an a helical coiled-coil region (RBCC), is embedded within the PML protein and is required for PML NB formation (28). Due to alternative splicing, many PML isoforms are synthesized, and they are classified into seven groups, designated PML I to PML VII (reviewed in reference 28). They share the N-terminal region (exons 1 to 3), which encodes the RBCC motif, whereas they differ in their C-terminal regions. Posttranslational modification of PML by SUMO (small ubiquitin-like modifier), a ubiquitin-like protein of 11 kDa, is another requirement for PML NB formation. SUMO is covalently coupled to PML through its lysines 65, 160, and 490 via a process called SUMOylation (29). The noncovalent interaction of SUMO with PML through a SUMO-interacting motif (SIM; also named SBD for SUMO binding domain) (46) has been suggested to be important for further recruitment within PML NBs of SUMOylated proteins.
Interferons (IFNs), a large family of secreted proteins, regulate antiviral, antitumor, and immunological responses through IFN-stimulated gene expression (8, 13). The PML gene is directly induced upon IFN treatment, resulting in an increase in the size and number of PML NBs (10, 49). Several pathways have been implicated in resistance to viral infection in IFN-treated cells (45), one of which implicates PML and PML NBs (21, 43).
Previously, we have shown, that PMLIII expression in U373MG and CHO cells confers resistance to human foamy virus (HFV) in a p53-independent way by complexing the HFV transactivator, Tas, preventing its direct binding to viral DNA (44), and to poliovirus in a p53-dependent way by inducing PML-dependent p53 activation leading to apoptosis in infected cells (38).
EMCV and mengovirus are serologically related members of the encephalomyocarditis virus species of the Cardiovirus genus of the Picornaviridae family.
The genome of cardiovirus is a plus-stranded RNA in which translation is initiated from an internal ribosome entry site located in the 5′ untranslated region. This results in the synthesis of a single, large polyprotein (16). Picornaviral proteins and their precursors take their names (L, P1, P2, and P3) from their sequential locations within the polyprotein. The cardioviruses encode a nonstructural protein, called the leader (L) protein. The four P1 peptides are the capsid proteins, 1A, 1B, 1C, and 1D (4). The middle portion P2 contains peptides 2A, 2B, and 2C. The P3 peptides, 3A, 3B (also called VPg), 3C protease (3Cpro), and 3D polymerase (3Dpol), are closely associated with genome replication. The 3Dpol is a central element of viral RNA replication complexes (51). 3B is the peptide covalently linked to the 5′ end of the genome (37). 3Cpro is the central enzyme in the viral cleavage cascade. EMCV effectively inhibits cellular antiviral defense and reprograms cellular metabolism for massive virus replication by several mechanisms: shutting down cellular transcription (3Cpro and its precursors) and translation (2A and L proteins) (33), altering nuclear-cytoplasmic traffic (L protein) (34), and blocking IFN-α/β gene transcription (L protein) (25). Whereas EMCV replication occurs in the cytoplasm, it has been reported that in early steps of infection, 2A, 3B, 3Cpro, and 3Dpol are found in bright, punctate spots in the nucleus (2), and L is localized only in the cytoplasm.
Here, we report that EMCV targeted PML by altering its localization and expression both in cells treated with IFN and in cells stably expressing PML. Early postinfection, EMCV induced PML conjugation to SUMO-1, SUMO-2, and SUMO-3 and its transfer to the nuclear matrix, resulting in an increase in PML NB size. Also we found that PML colocalized with 3Cpro and the proteasome component within the NBs and that the expression of the 3Cpro alone in the absence of the other viral proteins decreased PML expression. This process leads to PML degradation in a proteasome- and SUMO-dependent manner. The PML SIM was not required for EMCV-induced PML SUMOylation or degradation. In addition, EMCV-induced PML degradation required the RING domain, the C-terminal region of PMLIII, and its localization within the NBs. Our results revealed a new strategy used by EMCV to counteract the PML antiviral pathway through mechanisms implicating SUMOylation and nuclear degradation.
MATERIALS AND METHODS
Materials.
Recombinant human IFN-α2 was from Schering (NJ),and IFN-γ was from Roussel-Uclaf (Romainville, France). The following antibodies were used: mouse monoclonal anti-PML (clone PGM3), rabbit polyclonal anti-PML (clone H-238), and rabbit polyclonal anti-SUMO-1 (FL101) (all from Santa Cruz Biotechnology). Rabbit anti-SUMO-2/SUMO-3 ([SUMO-2/3] Sentrin-2) was obtained from Invitrogen, rabbit polyclonal antibody against proteasome 20S was from Biolmol International, and horseradish peroxidase (HRP)-conjugated mouse monoclonal anti-β-actin (clone AC-15) was from Sigma. Rabbit antiviral protein antibodies were raised against formaldehyde-inactivated mengovirus capsid as previously described (19). Mouse monoclonal and rabbit polyclonal anti-3C antibodies were used as described previously (1).
Cell cultures and cell treatments.
U373MG, L929, HeLa, and Chinese hamster ovary (CHO) cells, as well as mouse embryonic fibroblasts (MEFs) from wild-type (WT)or knockout PML (PML−/−) mice (52), immortalized by the simian virus 40 (SV40) T antigen, were grown at 37°C in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum. CHO cells (transfected with the empty vector or stably expressing PMLIII [CHO-PMLIII] or PMLIII mutants) were kept in medium supplemented with 0.5 mg/ml hygromycin. U373MG cells transfected with empty vector or stably expressing PMLIII (U373MG-PMLIII) were kept in medium supplemented with 0.5 mg/ml of G418 (11). The proteasome inhibitor, epoxomicin, was used at a concentration of 1 μM.
Virus stocks.
L929 cells (2 × 107) seeded in flask cultures were infected with EMCV at a multiplicity of infection (MOI) of 1 by absorption in 1 ml of medium without serum. After 1 h, virus was removed and replaced by 10 ml of medium containing 2% fetal calf serum (FCS), and cells were incubated at 37°C for 24 h. Cultures were then frozen and thawed three times and centrifuged to remove cellular debris. Supernatants were serially diluted, and virus titers were measured by a plaque assay on L929 cells and expressed as the number of PFU/ml of supernatant. Mengovirus stocks were produced and titrated similarly on HeLa cells. These virus stocks (2 × 108 to 5 × 108 PFU/ml) were used in medium containing 2% FCS at the MOI and times indicated in the figure legends. Recombinant vaccinia viruses (VV) expressing mengovirus precursor polypeptide P1, P2, or P3 (VV-P1, VV-P2, or VV-P3, respectively), as well as the mature 3AB, 3Cpro, or 3D protein (VV-3AB, VV-3C, or VV-3D, respectively), were produced by standard procedures as previously described (19). Expression was confirmed by immunoprecipitation of [35S]methionine-labeled cytoplasmic extracts prepared from CV1 cells infected with the different recombinant vaccinia viruses, using rabbit antiserum raised against mengovirus capsid (for P1-expressing vaccinia virus) or 3D polypeptide (for 3D- and P3-expressing vaccinia viruses) (data not shown). Expression of 3Cpro polypeptide by VV-3C recombinant vaccinia virus was confirmed by Western blot analysis (this study).
Interferon bioassay.
Infection of cells, harvesting, acid treatment of supernatants to inactivate virus, neutralization, and priming of fresh L929 monolayers were performed. L929 cells were challenged with vesicular stomatitis virus (VSV). The amount of IFN required to produce 50% inhibition of the cytopathic effect was determined by comparing the protective effects with those observed with the NIH standard mouse IFN-β (G602-9902-511).
Northern blot analysis.
Total RNA was extracted with a Bioprobe Systems RNA Extraction Kit Standard. After samples were blotted on nitrocellulose membranes (Schleicher and Schuell), Northern blot analysis was performed with random priming (Boehringer Mannheim) using a radiolabeled EcoRI PML fragment (14) or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as probes.
Cell infection, DNA, and siRNA transfection.
Cells were infected with EMCV at the MOI indicated in the figure legends. Cells were seeded in six-well plates and transfected with 2 μg of plasmid DNA or small interfering RNA (siRNA) using Lipofectamine 2000 or Lipofectamine RNAiMax transfection reagent, respectively (Invitrogen), according to the manufacturer's recommendations. Cells were transfected with His6-tagged SUMO-1, SUMO-2, or SUMO-3. The nomenclature of SUMO-2 and SUMO-3, which differ in only three amino acids, is not fully consistent. In accordance with the NCBI protein database, we refer to the entry P61956 as SUMO-2 and to P55854 as SUMO-3. The following siRNAs were used: against SUMO-1, GGACAGGAUAGCAGUGAGA-dTdT; against SUMO-2, AGGGAUGAAUCUGUAACUUAA-dTdT, and against SUMO-3 GAGGCAUACACCACUUAGU-dTdT. Cells were incubated for 48 h at 37°C, infected, and then prepared for Western blot analysis.
Immunofluorescence staining and confocal microscopy.
Infected or transfected cells were fixed in 4% paraformaldehyde for 15 min at 4°C and permeabilized for 5 min with 0.1% Triton X-100 in phosphate-buffered saline (PBS). Cells were stained using anti-PML, rabbit polyclonal antiviral proteins, or anti-3C antibodies. Cells were washed twice and incubated for 1 h with the appropriate Alexa Fluor 488- or 594-conjugated secondary antibody (1/500 dilution; Molecular Probes, Inc.). Coverslips were mounted using Vectashield (Vector Laboratories). For confocal analysis, images were sequentially collected by confocal laser scanning on a Leica TCS-NT/SP instrument equipped with an air-cooled argon-krypton mixed-gas laser and an Apochromat 63× (1.32 numerical aperture) oil immersion objective. Images were processed using Adobe Photoshop (Adobe Systems, Mountain View, CA).
Cellular fractionation and nuclear matrix isolation.
For in situ fractionation, cells grown on polylysine-coated coverslips were washed three times with ice-cold PBS. After cells were washed, they were sequentially extracted as follows: with 0.5% Triton X-100 in TMS buffer (50 mM Tris-HCl, pH 7.4, 5 mM MgSO4, 250 mM sucrose) for 5 min at room temperature ([RT] fraction 1); 50 U/ml RNase-free DNase RQ1 in TMS at 37°C for 30 min supplemented with ammonium sulfate to a final concentration of 0.25 mM (fraction 2); 2 M NaCl in TM buffer (10 mM Tris-HCl, pH 7.4, 0.02 mM MgSO4) twice for 15 min at RT(fraction 3); and 50 μg/ml RNase A in TM buffer for 15 min at RT (fraction 4). The final monolayer of extracted cells contained the nuclear matrix (fraction 5). The nuclear matrix preparations were fixed with methanol at −20°C for 3 min and then processed for indirect immunofluorescence microscopy as described above.
Cell fractionation and Western blot analysis.
Cells were washed and resuspended in PBS, lysed in hot Laemmli sample buffer, and boiled for 5 min. For cell fractionation, cells were dissociated and washed twice in PBS by centrifugation at 1,200 × g for 5 min. The cytoplasmic and nucleoplasm fractions were extracted by incubating the pellet in 50 μl of radioimmunoprecipitation assay (RIPA) buffer for 20 min on ice, followed by centrifugation at 15,000 × g for 15 min to separate the RIPA-soluble fraction from the pellet. This RIPA-insoluble fraction was suspended in 50 μl of PBS. Fifty microliters of 2× Laemmli buffer was added to each fraction, and the samples were boiled for 5 min before Western blot analysis. Protein extracts were analyzed on a 10% SDS-PAGE gel and transferred onto a nitrocellulose membrane. The proteins were blocked on the membranes with 10% skim milk in Tris-buffered saline (TBS) for 2 h and incubated overnight at 4°C with rabbit polyclonal anti-PML, monoclonal anti-3C, antiviral proteins, anti-SUMO-1, anti-SUMO-2/3, or anti-actin antibodies. The blots were then washed extensively in PBS-Tween and incubated for 1 h with the appropriate peroxidase-coupled secondary antibodies (Amersham). All of the blots were revealed by chemiluminescence (ECL; Amersham). If necessary the membranes were reprobed; they were incubated in 0.1 M glycine, pH 2.9, for 30 min, washed twice in PBS-Tween, and then blocked and incubated as described above. To estimate the apparent molecular mass of the polypeptides, the proteins of interest were compared to prestained molecular weight standards (Bio-Rad Laboratories, Richmond, CA).
Purification of His6-tagged SUMO PML conjugates.
One day after transfection, cells were separated in two flasks for each condition. One of the two flasks was infected with EMCV at an MOI of 5 for 2 h. Cells were then lysed in 4 ml of 6 M guanidinium-HCl, 0.1 M Na2HPO4/NaH2PO4, and 0.01 M Tris-HCl, pH 8.0, plus 5 mM imidazole and 10 mM β-mercaptoethanol per 75-cm3 flask. After sonication to reduce viscosity, the lysates were mixed with 50 μl of Ni2+-nitrilotriacetic acid (NTA)-agarose beads (Qiagen) prewashed with lysis buffer and incubated for 2 h at room temperature. The beads were successively washed with the following: 6 M guanidinium-HCl, 0.1 M Na2HPO4/NaH2PO4, 0.01 M Tris-HCl, pH 8.0, 10 mM β-mercaptoethanol; 8 M urea, 0.1 M Na2HPO4/NaH2PO4, 0.01 M Tris-HCl, pH 8.0, 10 mM β-mercaptoethanol, 6 M guanidinium-HCl, 0.1 M Na2HPO4/NaH2PO4, 0.01 M Tris-HCl, pH 6.3, 10 mM β-mercaptoethanol (buffer A); buffer A plus 0.2% Triton X-100; and buffer A plus 0.1% Triton X-100. After the last wash with buffer A the beads were eluted with 200 mM imidazole. The eluates were subjected to SDS-PAGE (10%), and the proteins were transferred to a nitrocellulose membrane. Western blotting was performed with anti-PML antibodies.
Construction of expression vectors and cell lines.
PMLIII cDNA was used to generate PMLIII with a mutation of three lysines to arginines (3KR) and PMLIII SIM as previously described (40). The mengovirus 3Cpro and its precursor 3CD were amplified using primers corresponding to unique restriction sites, start or stop codons, and the first and last 20 nucleotides of the genes (GenBank accession number DQ288856). The genes were inserted under cytomegalovirus (CMV) promoter into pcDNA3 vector (Invitrogen) using BamHI and NotI sites. The expression of 3Cpro and 3CD was verified with in vitro translation and Western blot assays.
Stable expression of PMLIII and PMLIII mutants.
CHO cells stably expressing PMLIII and the PMLIII mutants PMLIII Stop504 (a stop at codon 504), PMLIII Stop381, and PMLIII C57/60 (in which cysteines at positions 57 and 60 were mutated to serines) were obtained through lipofection (Gibco/BRL) with pSG5 constructs cotransfected with dithiobis(succinimidylproprionate) (DSP)-derived constructs and subsequent hygromycin selection as previously described (11).
Stable expression of PMLIII and PML mutants (PMLIII 3KR and PMLIII SIM) was obtained by a lentivirus-based strategy as described previously (40). Virus stock production and infection were as described by Zennou et al. (53) to transduce and express PMLIII variants (WT, 3KR, and SIM) in the U373MG cell line. Briefly, virus particles were produced after transient cotransfection of U373MG cells with the p8.91 encapsidation plasmid, the pHCMV-G vector encoding the vesicular stomatitis virus envelope, and the pTRIP vector encoding PMLIII and its mutants, using a standard calcium phosphate method.
RESULTS
EMCV infection induced early, large PML NB formation and a late decrease in PML protein level.
We have shown previously that PMLIII expression confers resistance to vesicular stomatitis virus (VSV), influenza virus, and HFV but not to EMCV (11). Two cell lines, CHO and U373MG, stably expressing PMLIII were used to investigate the fate of PML during EMCV infection. CHO cells overexpressing PMLIII were infected with EMCV for 2 h and 12 h at a multiplicity of infection (MOI) of 1 and then subjected to immunofluorescence staining using antibodies against PML or viral proteins. At 2 h postinfection, we noticed an increase in PML NB size while at 12 h postinfection these bodies became undetectable (Fig. 1A). To determine the effect of EMCV on PML protein expression, CHO-PMLIII or U373MG-PMLIII cells were infected with EMCV for 12 h at increasing MOIs, and total protein extracts were analyzed by Western blotting (Fig. 1B and data not shown). In the uninfected cells two forms of PML were detected, a higher and fainter form likely representing the conjugated PML-SUMO form and a lower form representing the unmodified form. A decrease in the level of expression of both forms was observed following infection as a function of the increase in the MOI (Fig. 1B). This decrease was associated with the appearance of cleavage products in extracts from infected cells (Fig. 1B).
FIG. 1.

EMCV infection led to a decrease in the PML level in PMLIII-expressing cells or in IFN-treated cells. (A) Alteration of PML NB in EMCV-infected cells. CHO-PMLIII cells were left uninfected or were infected for 2 and 12 h with EMCV at an MOI of 1. Double-immunofluorescence staining was performed using monoclonal anti-PML antibody visualized by Alexa Fluor 594 and rabbit antiviral protein antibodies followed by Alexa Fluor 488 labeling. (B) CHO-PMLIII cells were infected for 12 h at an MOI of 0.1, 0.2, 1, 5, or 10. (C) CHO-PMLIII or U373MG-PMLIII cells were infected at an MOI of 5 for 2, 4, or 6 h. The different total cell extracts from uninfected and infected cells were analyzed by Western blotting and revealed by rabbit anti-PML antibody. The same blots were reprobed with anti-actin antibody. Molecular size markers are indicated on the left. (D) EMCV infection led to PML decrease in IFN-treated U373MG cells. U373MG cells, treated with 1,000 units/ml of IFN-α for 24 h, were not infected or were infected with EMCV at an MOI of 5 for 12 or 16 h. The different total cell extracts were analyzed by Western blotting and revealed by rabbit anti-PML antibody. The same blot was reprobed with anti-Sp100, anti-PKR, or anti-actin antibodies. Molecular size markers are indicated on the right. The unmodified form of PML is indicated by an arrowhead, and the cleavage product is indicated by an arrow.
It was of interest to determine the time course of PML degradation over the time of EMCV infection. To address this question, CHO-PMLIII and U373MG-PMLIII cells were infected with EMCV at an MOI of 5 for short periods of time. In both cell lines, EMCV infection led to PML decrease as early as 4 h postinfection (Fig. 1C). To assess whether the endogenous PML expression was also altered, U373MG cells treated with IFN for 24 h either were left uninfected or were infected with EMCV for 12 or 16 h. A decrease in IFN-induced PML and the presence of cleavage products were also observed in infected cells. In these (Fig. 1B and D) and the following experiments, the cleavage products were detected only during long periods of infection.
From the same blot, we analyzed the fate of two other IFN-induced proteins, which were the protein kinase PKR and the NB-associated protein Sp100. During EMCV infection, the expression of PKR was not affected, whereas a small decrease in the different isoforms of Sp100 was observed with a loss of the higher, fainter form. This result is in agreement with a finding indicating that downregulation of PML results in an apparent reduction of Sp100 (20, 36).
EMCV infection induced degradation of PML in a proteasome-dependent manner.
To test if EMCV infection interfered with PML mRNA synthesis, cells stably expressing PMLIII or transfected with the empty vector were not infected or were infected with EMCV at an MOI of 5. After 4 or 8 h of infection, total RNA was isolated and analyzed by Northern blotting. The PML mRNA level was not altered during EMCV infection (Fig. 2A). These results suggest that the observed decrease in PML protein level is a consequence of its degradation.
FIG. 2.

(A) EMCV infection did not alter PML mRNA levels. CHO cells transfected with the empty vector (CHO-pSG5) or overexpressing PMLIII were not infected or were infected with EMCV at an MOI of 5. Total RNA extracted as described in Materials and Methods at 4 and 8 h postinfection for cells overexpressing PMLIII and at 8 h postinfection for control samples (20 μg of RNA by lane) was analyzed for PML and GAPDH. (B) The proteasome inhibitor did not alter viral protein expression and abrogated EMCV-induced PMLIII degradation. Total cell extracts were prepared from CHO-PMLIII cells noninfected or infected for 4 h with EMCV at an MOI of 5 in the absence or presence of epoxomicin. Twenty micrograms of protein extract of each sample was analyzed by Western blotting using antiviral protein antibodies (left panel; virus antigens are indicated), anti-PML, or anti-Actin antibodies (right panel); the unmodified form of PML is indicated by an arrowhead. (C) Confocal microscopy analysis of PML and 20S in EMCV-infected cells. U373MG-PMLIII cells were not infected or were infected with EMCV at an MOI of 5 for 2 h. Double-immunofluorescence staining was performed using monoclonal anti-PML antibody visualized by Alexa Fluor 594 and rabbit anti-20S antibody followed by Alexa Fluor 488 labeling.
To determine if the proteasome pathway is implicated in the decrease of PML expression during EMCV infection, CHO-PMLIII cells were infected with EMCV in the absence or the presence of the proteasome inhibitor epoxomicin. First, we analyzed the effect of epoxomicin on virus multiplication and viral protein expression. Cells prepared in duplicate were infected with EMCV at an MOI of 5 for 4 h in the presence or the absence of epoxomicin. One series was harvested and lysed by freezing and thawing, and the virus titers were determined by plaque assay in L929 cells without proteasome inhibitor; the second series was washed in PBS, and cell extracts were analyzed by Western blotting for viral protein expression. In both the absence and presence of epoxomicin, the titer yield values were 5 × 104 PFU/ml, and the profiles of viral protein expression were similar under both conditions (Fig. 2B, left panel), indicating that under our conditions the proteasome inhibitor did not affect viral protein expression or virus yields.
Interestingly, epoxomicin addition reduced EMCV- or mengovirus-induced PMLIII degradation, suggesting that this process occurred in a proteasome-dependent way (Fig. 2B, right panel, and data not shown). Since EMCV interferes with host cell translation (23), we cannot exclude the possibility that PML is not translated as efficiently during EMCV infection, which could further reduce the levels of PML in infected cells. However, the fact that epoxomicin strongly decreased EMCV-induced PML degradation demonstrates that the reduced level of PML for the most part is due to proteasome-mediated degradation (Fig. 2B, right panel).
To further investigate this observation, immunofluorescence labeling using anti-PML and anti-proteasome 20S antibodies was performed on U373MG-PMLIII cells not infected or infected with EMCV. In uninfected U373MG-PMLIII cells, the proteasome was found in the cytoplasm and in the nucleus. In the infected cells (2 h postinfection), PML NBs increased in size, and the proteasome presented punctate labeling colocalizing with PML in PML NBs (Fig. 2C).
3C protease expression led to PML degradation.
In our study, we demonstrated that both mengovirus and EMCV promoted PML degradation through a proteasome-dependent way (Fig. 2B and data not shown), supporting the use of recombinant vaccinia viruses expressing mengovirus precursor and mature polypeptides to track down the viral proteins responsible for virus-induced PML degradation. For this, we have constructed recombinant vaccinia viruses (VV) expressing mengovirus precursor protein P1, P2, or P3 and used them to infect CHO-PMLIII cells. Cell extracts analyzed by immunoblotting with anti-PML antibody revealed a decrease in PML protein level only in the cells infected with VV-P3 or mengovirus (Fig. 3A). This decrease was not observed when the cells were infected with VV, VV-P1, or VV-P2 (Fig. 3A). The P1 precursor is composed only of the viral structural proteins and requires 3Cpro for processing. VV-P1-infected cells behaved the same as mock-infected cells. The P2 precursor, which also did not alter PML expression, consists of self-cleaving 2A protein (autocatalytic cleavage between 2A/2B) and 2BC precursor; the 3Cpro is responsible for 2BC processing.
FIG. 3.
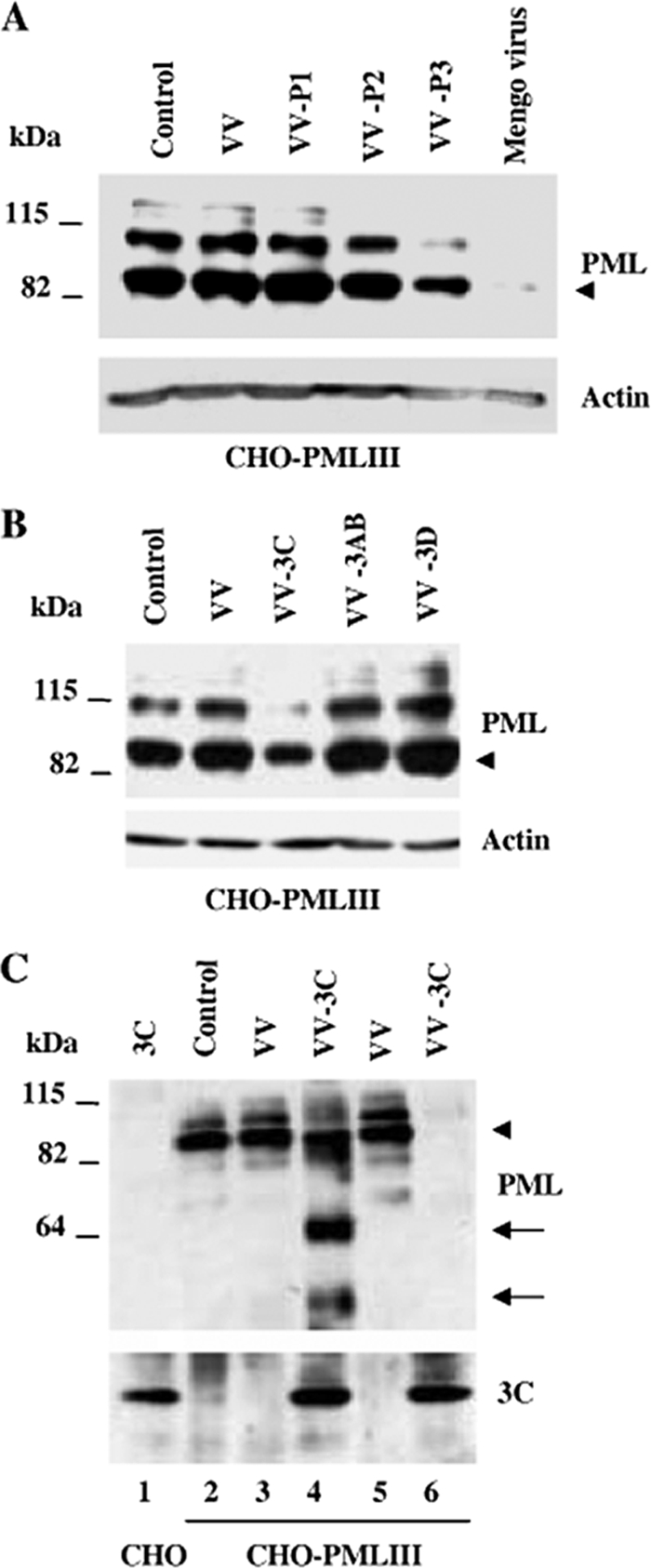
3Cpro expression induced PML cleavage and degradation. (A) CHO-PMLIII cells were infected with VV, VV-P1, VV-P2, or VV-P3 at an MOI of 1 for 8 h. (B) CHO-PMLIII cells were infected with VV, VV-3AB, VV-3C, and VV-3D at an MOI of 1 for 8 h. (C) CHO-PMLIII cells were infected with VV or VV-3C at an MOI of 1 for 12 h (lanes 2, 3, and 4) or 16 h (lanes 5 and 6); extracts from CHO cells transfected with pcDNA 3C vector were used as a control for 3Cpro expression (lane 1). Total cell extracts were analyzed by Western blotting using anti-PML, anti-actin, or anti-3C antibodies. The unmodified form of PML is indicated by an arrowhead, and the cleavage products are indicated by arrows.
Infection of CHO-PMLIII cells at a high MOI of VV, VV-P1, or VV-P2 did not alter PML expression (data not shown), demonstrating that the effect was carried out by only the viral polyprotein P3.
P3 consists of self-cleaving 3Cpro and released proteins 3AB, 3C, and 3D (39); therefore, we set out to investigate the role of these products in PML degradation. We have constructed recombinant VV expressing mature 3AB, 3Cpro, or 3D and used them to infect CHO-PMLIII cells.
CHO-PMLIII cells were infected at an MOI of 1 with VV, VV-3C, VV-3AB, or VV-3D. Remarkably, a decrease in PML protein level was observed only with VV-3C infection, suggesting that the 3Cpro is the viral protein responsible for PML degradation (Fig. 3B). To confirm this result, CHO-PMLIII cells were infected with VV or VV-3C at an MOI of 1 for a longer period of time, i.e., 12 h (Fig. 3C, lanes 3 and 4) or 16 h (lanes 5 and 6). CHO cells transiently transfected with pcDNA3-3C were used as a control for 3Cpro expression (lane 1). Protein extracts were subjected to immunoblotting using anti-PML or anti-3C antibodies (Fig. 3C). At both times of infection, infection with the control VV did not alter the PML level. In contrast, at 12 h postinfection, 3Cpro expression led to a loss of the higher form of PML, likely representing the conjugated PML-SUMO form. Interestingly, this loss was associated with the appearance of lower-molecular-weight products, probably corresponding to cleavage products (Fig. 3C, lane 4). At 16 h postinfection, PML and the SUMO-conjugated form of PML were totally undetectable (Fig. 3C, lane 6). 3Cpro was expressed during the infection, as shown by reprobing the blot with anti-3C antibody (Fig. 3C, lanes 4 and 6). Taken together, these results strongly suggest that 3Cpro expression in mengovirus- or EMCV-infected cells induced PMLIII cleavage and degradation.
EMCV infection shifted PML from the nucleoplasm to the nuclear matrix in PML-transfected cells and IFN-treated cells.
PML is localized in the nucleoplasm fraction and in the matrix-bound NBs (38, 54). To biochemically detect whether EMCV induced the transfer of PML from the nucleoplasm to the nuclear matrix, the RIPA-soluble fraction (the cytoplasm and most of the nucleoplasm) and RIPA-insoluble fractions (the nuclear matrix and some chromatin components) from cells overexpressing PMLIII, either not infected or infected for 2 h with EMCV at MOIs of 1 and 5, were analyzed by immunoblotting using anti-PML antibody. As previously reported (38) in uninfected cells, most of the PML was found in the RIPA-soluble fraction (see Fig. 5A). Interestingly, EMCV infection shifted PML from the nucleoplasm to the nuclear matrix and promoted the appearance of higher-molecular-weight products, which correspond to the conjugated forms of PML together with the emergence of lower-molecular-weight cleavage products (Fig. 4A). In addition, the higher PML conjugated forms and the PML cleavage products were detected only in the nuclear matrix and were more abundant at a higher MOI of 5. Reprobing the blot with anti-3C antibody showed that 3Cpro was properly expressed and mainly found in the nuclear matrix fraction (Fig. 4A).
FIG. 4.

(A) EMCV infection shifted PML toward the nuclear matrix. RIPA-soluble (R) and -insoluble (P) fractions from CHO-PMLIII cells, not infected or infected with EMCV at MOIs of 1 and 5 for 2 h, were analyzed by Western blotting with anti-PML and anti-3C antibodies. The unmodified form of PML is indicated by an arrowhead, and the cleavage product is indicated by an arrow. (B) PML localization in IFN-treated cells. Total extracts from U373MG cells untreated or treated with IFN-α or IFN-γ for 24 h were analyzed by Western blotting using anti-PML antibody (left panel). EMCV infection induced the transfer of PML from the nucleoplasm to the nuclear matrix in IFN-treated cells (right panel). U373MG cells were untreated or treated with IFN-α or IFN-γ for 24 h and then not infected or infected with EMCV at an MOI of 5 for 2 h. RIPA-soluble (R) and -insoluble (P) fractions from the different samples were analyzed by Western blotting with anti-PML antibody.
We have shown previously that different PML isoforms were increased in response to IFN (10). To determine the localization of PML in IFN-treated cells, we analyzed by Western blotting with anti-PML antibody the RIPA-soluble and -insoluble fractions. As expected, PML isoforms were increased in response to IFN-α or IFN-γ compared to levels in IFN-untreated cells. Moreover, both types of IFN increased PML expression in the RIPA-soluble but not in the RIPA-insoluble fraction (Fig. 4B, left panel). Interestingly, at 2 h postinfection EMCV induced a transfer of PML from the nucleoplasm to the nuclear matrix in the untreated and the IFN-α- or IFN-γ-treated cells (Fig. 4B, right panel). Taken together, these results show, first, that EMCV induced the transfer of PML from the nucleoplasm to the nuclear matrix both in cells expressing PML and in IFN-treated cells and, second, that this process preceded PML degradation in infected cells.
EMCV infection promoted early localization of 3Cpro and PML into PML NBs.
It has been reported that early after EMCV infection of different cell lines, 3Cpro formed bright, punctate spots in the nuclei (2). We asked whether 3Cpro expressed alone in transfected cells could be found in the nucleus (Fig. 5A). To demonstrate this, HeLa cells were transfected with pcDNA3-3C, pcDNA3-3CD, or pcDNA3-vector; cells were fixed 18 h later, stained with 4′,6′-diamidino-2-phenylindole (DAPI) and with monoclonal antibodies (MAbs) to 3Cpro, and subjected to confocal microscopy (Fig. 5A). The 3Cpro signal was brightly seen in cell nuclei as dispersed foci for both 3Cpro- and 3CD-expressing cells but not in cells transfected with pcDNA3-vector. This confirms that 3Cpro or 3CD is able to localize in the host nucleus without additional viral protein support.
FIG. 5.

3Cpro localization in transfected cells and in infected cells stably expressing PMLIII. (A) HeLa cells were transfected with pcDNA3, pcDNA3-3Cpro, or pcDNA3-3CD. Cells were fixed 18 h later, and immunofluorescence was performed with anti-3C antibody. DAPI staining is shown. (B) 3Cpro colocalized with PML within the NBs. Double-immunofluorescence staining was performed on nuclear matrix isolated from CHO-PMLIII cells infected with EMCV at an MOI of 5 for 2 h using monoclonal anti-PML antibody visualized by Alexa Fluor 594 and rabbit anti-3C antibody followed by Alexa Fluor 488.
To determine whether PML and 3Cpro colocalized in EMCV-infected cells, cells stably expressing PMLIII were left uninfected or were infected for 2 h with EMCV, and immunofluorescence staining, using anti-PML and anti-3C antibodies, was performed on isolated nuclear matrix. Interestingly, PML and 3Cpro were found colocalizing within PML NBs (Fig. 5B), thus reinforcing the observations described above.
Nuclear localization, RING domain, and the C-terminal domain of PML were required for its EMCV-mediated degradation.
To determine the domains implicated in EMCV-induced PML degradation, we analyzed the fate of PML and its mutants using CHO cells stably expressing a cytoplasmic PML mutant (PMLIII Stop381), a C-terminal mutant (PMLIII Stop504), and a RING finger PML mutant (PMLIII C57/60) which is found only in the nucleoplasm due to a loss of SUMO binding properties (48). The schematic representation of the domain structure of the PMLIII isoform is presented in Fig. 6A. Immunoblotting using anti-PML antibody showed that while PMLIII WT was sensitive to EMCV-induced degradation, PMLIII Stop381, PMLIII Stop504, and PMLIII C57/60, were resistant (Fig. 6B). These results demonstrate that this degradation process required the nuclear localization, the RING finger, and the C-terminal region of PML. Interestingly, compared to the uninfected cells, PMLIII Stop504 was shifted from the nucleoplasm toward the nuclear matrix in the extracts from CHO-PMLIII Stop504 cells infected with EMCV (Fig. 6C, top panel). The high-molecular-weight products may represent the PML SUMO-conjugated forms, as suggested by results using antibodies against SUMO-1 and SUMO-2/3 (Fig. 6C, lower panels). The same blot reprobed with anti-3C antibody showed that 3Cpro was expressed mainly in the matrix fraction but was unable to alter PMLIII Stop504 expression (Fig. 6C). Thus, the resistance of PMLIII Stop504 to EMCV-induced degradation was not due to its inability to be targeted to the nuclear matrix.
FIG. 6.

EMCV-induced PML degradation required its RING domain and its C-terminal region. (A) Schematic representation of the domain structure of the PMLIII isoform (residues 1 to 641). PML domains include the RING finger (R), the B1 and B2 boxes, the coiled-coil (CC) motif, the nuclear localization domain (NLS), three SUMOylation sites (K65, K160, and K490), and the SUMO-interacting motif (SIM). The localization of PMLIII mutants is also shown. (B) PMLIII (residues 1 to 641), PMLIII Stop504, and PMLIII C57/60 stably expressed in CHO cells infected with EMCV at an MOI of 5 for 8 h. The different cell extracts were analyzed by Western blotting and revealed by rabbit anti-PML and anti-actin antibodies. (C) Resistance of PMLIII Stop504 to EMCV-induced degradation did not reflect its inability to be targeted to the nuclear matrix. RIPA-soluble and -insoluble fractions from CHO cells stably expressing PMLIII Stop504, left uninfected or infected with EMCV at an MOI of 5 for 2 h, were analyzed by Western blotting with anti-PML, anti-3C, anti-SUMO-1, and anti-SUMO-2/3 antibodies.
EMCV-induced PML degradation required its covalent but not its noncovalent binding to SUMO.
In addition to the covalent SUMO modification of PML on lysines 65, 160, and 490 (29), a noncovalent interaction of PML with SUMO through a domain named SIM was more recently reported (46). To evaluate the role of the covalent and noncovalent bindings of SUMO to PML in EMCV-induced PML SUMOylation and degradation, U373MG cells expressing PMLIII, PMLIII 3KR (with SUMO target lysines 65, 160, and 490 mutated into arginines), and PMLIII SIM (mutated in its SIM core sequence, VVVI hydrophobic amino acids) were infected with EMCV for 2 h. As expected, no SUMO-modified forms were detected with PMLIII 3KR in either infected or uninfected cells (Fig. 7A). SUMO-modified PML was detected in control cells expressing PMLIII and PMLIII SIM (Fig. 7A). Interestingly, EMCV induced at 2 h postinfection a higher conjugation of PML to SUMO in cells expressing PMLIII and PMLIII SIM (Fig. 7A). At 8 h postinfection (MOI of 5), EMCV induced the degradation of PMLIII and PMLIII SIM but failed to trigger the degradation of PMLIII 3KR (Fig. 7B), demonstrating that this process necessitated the covalent conjugation of PML to SUMO. These results also show that the SIM of PML was not required for either EMCV-induced PML SUMOylation or degradation.
FIG. 7.

(A) EMCV infection increased PMLIII and PMLIII SIM SUMOylation. U373MG cells stably expressing PMLIII, PMLIII SIM, or PMLIII 3KR were infected with EMCV at an MOI of 5 for 2 h. (B) EMCV-induced PML degradation necessitated its covalent but not its noncovalent binding to SUMO. U373MG cells stably expressing PMLIII, PMLIII SIM, or PMLIII 3KR were infected with EMCV at an MOI of 5 for 8 h. The different cell extracts were analyzed by Western blotting and revealed by rabbit anti-PML or anti-actin antibodies. The unmodified form of PML is indicated by an arrowhead.
EMCV-induced PML degradation required PML SUMOylation.
To investigate the role of SUMO paralogs in EMCV-induced PML SUMOylation, PMLIII was coexpressed in HEK293 cells with the empty vector or pcDNA3-His6-tagged SUMO-1, SUMO-2, or SUMO-3. One day postinfection, cells were not infected or were infected with EMCV at an MOI of 5. At 2 h postinfection, the cell extracts were purified on Ni2+-NTA-agarose beads and subjected to immunoblotting with anti-PML antibody. Interestingly, PML was found conjugated to SUMO-1, SUMO-2, and SUMO-3, with an increase in PML modifications in the presence of EMCV infection (Fig. 8A, lower panel, lanes 4, 6, and 8). The modifications by SUMO-2 and SUMO-3 were more apparent due to the capacity of SUMO-2/3 to form polymeric chains on PML (50).
FIG. 8.

(A) EMCV infection increased the conjugation of PML to SUMO. HEK293 cells were cotransfected with PMLIII-expressing vector and pcDNA3, His6-tagged SUMO-1, -SUMO-2, or SUMO-3, and cell extracts from uninfected or infected cells for 2 h at an MOI of 5 were purified on Ni2+-NTA-agarose beads. The inputs and the purified extracts were analyzed by Western blotting using anti-PML antibody. (B and C) Role of SUMO paralogs in EMCV-induced PML degradation. U373MG-PMLIII cells transfected with control siRNA scramble or siRNA specific for SUMO-1 or SUMO-2/3 were infected with EMCV at an MOI of 5 for 8 h. Cells extracts were analyzed by Western blotting with anti-SUMO-1 and anti-SUMO-2/3 (B) or anti-PML and anti-actin antibodies (C). The unmodified form of PML is indicated by an arrowhead.
To investigate the respective contributions of SUMO-1 and SUMO-2/3 to the EMCV-induced PML degradation, we used specific small interfering RNA (siRNA) to knock down the expression of each SUMO paralog in U373MG-PMLIII cells. The efficacy of siRNA SUMO-1 and siRNA SUMO-2/3 is shown in Fig. 8B. As expected, EMCV induced PML degradation in U373MG-PMIII cells transfected with a scrambled siRNA. In contrast, knockdown of SUMO-1 or SUMO-2/3 resulted in a decrease in EMCV-induced PML degradation (Fig. 8C), suggesting that SUMO-1 and SUMO-2/3 contributed to EMCV-induced PML proteolysis.
EMCV replication increased in the absence of PML.
PML knockout mice exhibited enhanced lymphocytic choriomeningitis virus (LCMV) and VSV production, and their derived PML−/− MEFs are more sensitive to rabies virus growth (6, 7). To determine if these cells are more susceptible to EMCV infection, WT MEFs and PML−/− MEFs were infected with EMCV at different MOIs, and the cell extracts were subjected to immunoblotting using antiviral protein antibodies. In the absence of PML, more viral proteins were produced than in PML WT MEFs (Fig. 1). In the infected PML−/− MEFs, viral proteins were detected at an MOI of 0.05, and their expression levels were enhanced as a function of the MOI. In contrast, in the infected WT MEFs, viral proteins were very slightly detected at an MOI of 0.1, and their expression levels increased at an MOI of 0.5, but their levels remained lower than those in PML−/− MEFs at any tested low MOI (Fig. 9). The viral production, determined by plaque assays, indicated higher viral titers of 2 × 107 (MOI of 0.1) and 4 × 108 PFU/ml (MOI of 0.5) in PML−/− MEFs than the 1 × 106 and 2 × 107 PFU/ml in WT MEFs. This result shows a 20-fold decrease in viral production in the presence of PML. EMCV infection of the PML WT MEFs might have led to IFN induction and secretion, which in turn would inhibit viral replication. The presence of IFN in the culture medium was assayed by testing the ability of cell-free medium from EMCV-infected WT and PML−/− MEFs to induce antiviral activity in L929 cells. The supernatants from both cell lines have an IFN titer of 4 IU/ml, clearly demonstrating that resistance in PML WT MEFs was not due to higher IFN production during EMCV infection. In addition, this result is in agreement with a previous report showing that upon infection with EMCV, little if any IFN-α/β is produced due to L protein inhibition of IRF-3 activation (26).
FIG. 9.

Absence of PML expression in PML−/− MEFs resulted in increased expression of viral proteins. WT MEFs and PML−/− MEFs were infected with EMCV for 8 h at different MOIs. Protein extracts were analyzed by Western blotting and revealed by antiviral protein antibodies.
DISCUSSION
In the nucleus of IFN-treated cells or PML-expressing cells, most of the PML is found in the diffuse nuclear fraction of the nucleoplasm, and a small fraction is in the nuclear matrix-associated NBs (38, 41, 54; also the present paper). The transfer of PML to the nuclear matrix is dependent on PML posttranslational modification. Indeed, cell treatment with agents that induce PML SUMOylation, such as arsenic trioxide or poliovirus, results in PML degradation (32) or the PML NB-associated p53 (38), respectively.
We report here that early during the EMCV infection, PML was transferred from the nucleoplasm to the nuclear matrix, both in IFN-treated and PMLIII-expressing cells. Later during the infection, EMCV induced a decrease in PML expression. Furthermore, we showed that among the different viral proteins tested (P1, P2, and P3 precursors as well as P3-encoded mature polypeptides), the expression of 3Cpro alone was sufficient to induce PML degradation. Consistently, 3Cpro translocates into the nucleus of infected (1, 3) or transfected (this paper) cells, is localized in the nuclear fraction (47), is mainly associated to the nuclear matrix, and colocalizes with PML within the NBs early during viral infection. This suggests that a cognate interaction between 3Cpro and PML could be responsible for 3Cpro-mediated PML degradation. We cannot exclude the possibility that other viral proteins which are targeted to the nucleus during EMCV infection, such as 2A (1), or which interfere with nucleocytoplasmic trafficking, such as L (42), could synergize with 3Cpro and amplify PML degradation in EMCV-infected cells. This interesting question will obviously deserve future studies.
A possible molecular mechanism for 3Cpro-induced PML degradation relies on direct PML cleavage by the 3Cpro. EMCV or mengovirus 3C proteases were reported to process the viral polyprotein at the scissile pairs Q/G, Q/S, or even E/S in some cases (24, 39). The PMLIII Stop504 mutant is not cleaved and degraded following expression of 3Cpro, which suggests that potential 3Cpro cleavage sites in PML would be located downstream of codon 504. The dipeptide Q/G is not present within the C-terminal domain of PMLIII, but dipeptides Q/S and E/S are found at positions 576/577 and 618/619, respectively. Although beyond the scope of this study, it would be interesting to determine whether PML is a direct substrate for 3Cpro cleavage at the 576/577 and/or 618/619 dipeptides.
It has been reported recently that EMCV induces retinoic acid-inducible gene (RIG-1) degradation that is mediated by 3Cpro (5, 18), suggesting that PML and RIG-1 degradation during EMCV infection may constitute mechanisms for attenuating the innate response to viral infection.
We next showed that EMCV, early postinfection, led to an increase of PMLIII modification by SUMO-1, SUMO-2, and SUMO-3 and the recruitment of the proteasome core components resulting in PML proteasome-dependent degradation. PML degradation did not require its SIM domain but necessitated its SUMOylation, as shown by the resistance of SUMOylation-deficient mutant PMLIII 3KR in this process. It also required the nuclear localization of PML, its RING finger, and the C-terminal region of PMLIII. In addition, downregulation of SUMO paralogs decreased EMCV-induced PML degradation, further demonstrating the requirement of the covalent modification by SUMO. The fate of other PML isoforms during EMCV infection and the implication of 3Cpro in mediating PML SUMOylation are under investigation.
The pathways involved in regulating SUMOylation remain largely unknown. PML SUMOylation is an important posttranslational modification that controls PML localization, stability, and interaction with other proteins (27). Recent findings indicate that SUMO can act as a signal for the recruitment of E3 ubiquitin ligases, which leads to the ubiquitylation and degradation of the modified protein (22).
It has been reported that VSV infection induces IFN regulatory factor 3 (IRF-3) and IRF-7 SUMOylation, resulting in the restriction of the extent and duration of virus-stimulated IFN transcription and thereby contributing to downregulation of IFN production postactivation (31).
Taken together, these results could suggest that virus-induced protein SUMOylation leads to a counteraction of antiviral defense mechanisms.
Several pathways have been implicated in resistance to viral infection in IFN-treated cells, one of which implicates PML protein and PML NBs (21, 43). The antiviral effect of PML has been observed in vivo since PML deficiency renders mice more susceptible to the arenavirus LCMV and the rhabdovirus VSV infections (7). These in vivo observations corroborate previous findings showing that fibroblasts derived from PML knockout mice exhibit enhanced LCMV growth (15) and are more sensitive to infection with rabies virus, another member of the rhabdovirus family (6). We additionally demonstrated in this report that PML−/− MEFs derived from knockout mice and immortalized by SV40 T antigen (p53 is inactive) are more sensitive to EMCV infection, suggesting that the absence of endogenous PML resulted in an increase in EMCV replication. Studies are under way to determine which PML isoform(s) confers resistance to EMCV in a p53-independent way.
Some RNA viruses whose replication takes place in the cytoplasm and is inhibited by PML have developed different ways to counteract PML NBs (12, 21). LCMV infection results in alteration of PML NBs mediated by a small nonstructural protein named Z. In infected and transfected cells, the RING finger protein Z associates with PML NBs and delocalizes PML from the NBs to the cytoplasm, where both proteins interact with the elongation factor eIF-4E, reducing its affinity for the 5′ mRNA cap structure and inhibiting cellular translation (9). The rabies viral protein P also directly binds via its C-terminal region to the RING finger of PML and causes the redistribution of PML into the cytoplasm where the proteins colocalize (6). An increase in PML NB size is observed in infected cells or cells expressing P3, an N-terminal truncated version of the phosphoprotein P, which is believed to mediate this effect during infection (6).
In conclusion, starting at 2 h postinfection, EMCV induced the transfer of PML to the nuclear matrix and its modification to SUMO-1, SUMO-2, and SUMO-3. This results in an increase in PML body size, where 3Cpro and the proteasome component colocalized with PML within the NBs. This process was followed by PML degradation in a proteasome- and SUMO-dependent way. Our work reveals a new mechanism by which EMCV counteracts the antiviral activity of PML by promoting its degradation in a SUMO-dependent process.
Acknowledgments
This work was supported by the Centre National de la Recherche Scientifique and a grant from the Association pour la Recherche sur le Cancer. B.E.M. was supported by a fellowship from the Fondation de la Recherche Médicale.
We thank M. Pampin and B. Pavie for their advice in this study.
We declare that we have no competing interests.
Footnotes
Published ahead of print on 8 September 2010.
REFERENCES
- 1.Aminev, A. G., S. P. Amineva, and A. C. Palmenberg. 2003. Encephalomyocarditis viral protein 2A localizes to nucleoli and inhibits cap-dependent mRNA translation. Virus Res. 95:45-57. [DOI] [PubMed] [Google Scholar]
- 2.Aminev, A. G., S. P. Amineva, and A. C. Palmenberg. 2003. Encephalomyocarditis virus (EMCV) proteins 2A and 3BCD localize to nuclei and inhibit cellular mRNA transcription but not rRNA transcription. Virus Res. 95:59-73. [DOI] [PubMed] [Google Scholar]
- 3.Amineva, S. P., A. G. Aminev, A. C. Palmenberg, and J. E. Gern. 2004. Rhinovirus 3C protease precursors 3CD and 3CD′ localize to the nuclei of infected cells. J. Gen. Virol. 85:2969-2979. [DOI] [PubMed] [Google Scholar]
- 4.Arnold, E., M. Luo, G. Vriend, M. G. Rossmann, A. C. Palmenberg, G. D. Parks, M. J. Nicklin, and E. Wimmer. 1987. Implications of the picornavirus capsid structure for polyprotein processing. Proc. Natl. Acad. Sci. U. S. A. 84:21-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barral, P. M., D. Sarkar, P. B. Fisher, and V. R. Racaniello. 2009. RIG-I is cleaved during picornavirus infection. Virology 391:171-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blondel, D., T. Regad, N. Poisson, B. Pavie, F. Harper, P. P. Pandolfi, H. De The, and M. K. Chelbi-Alix. 2002. Rabies virus P and small P products interact directly with PML and reorganize PML nuclear bodies. Oncogene 21:7957-7970. [DOI] [PubMed] [Google Scholar]
- 7.Bonilla, W. V., D. D. Pinschewer, P. klenerman, V. Rousson, M. Gaboli, P. P. Pandolfi, R. M. Zinkernagel, M. S. Salvato, and H. Hengartner. 2002. Effects of promyelocytic leukemia protein on virus-host balance. J. Virol. 76:3810-3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borden, E. C., G. C. Sen, G. Uze, R. H. Silverman, R. M. Ransohoff, G. R. Foster, and G. R. Stark. 2007. Interferons at age 50: past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 6:975-990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borden, K. L., E. J. Campbelldwyer, G. W. Carlile, M. Djavani, and M. S. Salvato. 1998. Two RING finger proteins, the oncoprotein PML and the arenavirus Z protein, colocalize with the nuclear fraction of the ribosomal P proteins. J. Virol. 72:3819-3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chelbi-Alix, M. K., L. Pelicano, F. Quignon, M. H. M. Koken, L. Venturini, M. Stadler, J. Pavlovic, L. Degos, and H. de Thé. 1995. Induction of the PML protein by interferons in normal and APL cells. Leukemia 9:2027-2033. [PubMed] [Google Scholar]
- 11.Chelbi-Alix, M. K., F. Quignon, L. Pelicano, M. H. M. Koken, and H. de The. 1998. Resistance to virus infection conferred by the interferon-induced promyelocytic leukemia protein. J. Virol. 72:1043-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chelbi-Alix, M. K., A. Vidy, J. El Bougrini, and D. Blondel. 2006. Rabies viral mechanisms to escape the IFN system: the viral protein P interferes with IRF-3, Stat1, and PML nuclear bodies. J. Interferon Cytokine Res. 26:271-280. [DOI] [PubMed] [Google Scholar]
- 13.Chelbi-Alix, M. K., and J. Wietzerbin. 2007. Interferon, a growing cytokine family: 50 years of interferon research. Biochimie 89:713-718. [DOI] [PubMed] [Google Scholar]
- 14.de Thé, H., C. Lavau, A. Marchio, C. Chomienne, L. Degos, and A. Dejean. 1991. The PML-RAR alpha fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell 66:675-684. [DOI] [PubMed] [Google Scholar]
- 15.Djavani, M., J. Rodas, I. S. Lukashevich, D. Horejsh, P. P. Pandolfi, K. L. Borden, and M. S. Salvato. 2001. Role of the promyelocytic leukemia protein PML in the interferon sensitivity of lymphocytic choriomeningitis virus. J. Virol. 75:6204-6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duke, G. M., M. A. Hoffman, and A. C. Palmenberg. 1992. Sequence and structural elements that contribute to efficient encephalomyocarditis virus RNA translation. J. Virol. 66:1602-1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dyck, J. A., G. G. Maul, W. H. Miller, J. D. Chen, A. Kakizuka, and R. M. Evans. 1994. A novel macromolecular structure is a target of the promyelocyte-retinoic acid receptor oncoprotein. Cell 76:333-343. [DOI] [PubMed] [Google Scholar]
- 18.Eldin, P., L. Papon, A. Oteiza, E. Brocchi, T. G. Lawson, and N. Mechti. 2009. TRIM22 E3 ubiquitin ligase activity is required to mediate antiviral activity against encephalomyocarditis virus. J. Gen. Virol. 90:536-545. [DOI] [PubMed] [Google Scholar]
- 19.Escriou, N., C. Leclerc, S. Gerbard, M. Giraud, and S. van der Werf. 1995. Cytotoxic T cell response to Mengo virus in mice: effector cell phenotype and target proteins. J. Gen. Virol. 76:1999-2007. [DOI] [PubMed] [Google Scholar]
- 20.Everett, R. D. 2006. Interactions between DNA viruses, ND10 and the DNA damage response. Cell Microbiol. 8:365-374. [DOI] [PubMed] [Google Scholar]
- 21.Everett, R. D., and M. K. Chelbi-Alix. 2007. PML and PML nuclear bodies: implications in antiviral defence. Biochimie 89:819-830. [DOI] [PubMed] [Google Scholar]
- 22.Geoffroy, M. C., and R. T. Hay. 2009. An additional role for SUMO in ubiquitin-mediated proteolysis. Nat. Rev. Mol. Cell Biol. 10:564-568. [DOI] [PubMed] [Google Scholar]
- 23.Groppo, R., and A. C. Palmenberg. 2007. Cardiovirus 2A protein associates with 40S but not 80S ribosome subunits during infection. J. Virol. 81:13067-13074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hall, D. J., and A. C. Palmenberg. 1996. Mengo virus 3C proteinase: recombinant expression, intergenus substrate cleavage and localization in vivo. Virus Genes 13:99-110. [DOI] [PubMed] [Google Scholar]
- 25.Hato, S. V., C. Ricour, B. M. Schulte, K. H. Lanke, M. de Bruijni, J. Zoll, W. J. Melchers, T. Michiels, and F. J. van Kuppeveld. 2007. The mengovirus leader protein blocks interferon-alpha/beta gene transcription and inhibits activation of interferon regulatory factor 3. Cell Microbiol. 9:2921-2930. [DOI] [PubMed] [Google Scholar]
- 26.Hato, S. V., F. Sorgeloos, C. Ricour, J. Zoll, W. J. Melchers, T. Michiels, and F. J. van Kuppeveld. 2010. Differential IFN-alpha/beta production suppressing capacities of the leader proteins of mengovirus and foot-and-mouth disease virus. Cell Microbiol. 12:310-317. [DOI] [PubMed] [Google Scholar]
- 27.Hay, R. T. 2005. SUMO: a history of modification. Mol. Cell 18:1-12. [DOI] [PubMed] [Google Scholar]
- 28.Jensen, K., C. Shiels, and P. S. Freemont. 2001. PML protein isoforms and the RBCC/TRIM motif. Oncogene 20:7223-7233. [DOI] [PubMed] [Google Scholar]
- 29.Kamitani, T., K. Kito, H. P. Nguyen, H. Wada, T. Fukuda-Kamitani, and Y. E. T. 1998. Identification of three major sentrinization sites in PML. J. Biol. Chem. 273:26675-26682. [DOI] [PubMed] [Google Scholar]
- 30.Koken, M. H., F. Puvion-Dutilleul, M. C. Guillemin, A. Viron, G. Linares-Cruz, N. Stuurman, L. de Jong, C. Szostecki, F. Calvo, C. Chomienne, et. al. 1994. The t(15;17) translocation alters a nuclear body in a retinoic acid-reversible fashion. EMBO J. 13:1073-1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kubota, T., M. Matsuoka, T. H. Chang, P. Tailor, T. Sasaki, M. Tashiro, A. Kato, and K. Ozato. 2008. Virus infection triggers SUMOylation of IRF3 and IRF7, leading to the negative regulation of type I interferon gene expression. J. Biol. Chem. 283:25660-25670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lallemand-Breitenbach, V., J. Zhu, F. Puvion, M. Koken, N. Honore, A. Doubeikovsky, E. Duprez, P. P. Pandolfi, E. Puvion, P. Freemont, and H. de The. 2001. Role of promyelocytic leukemia (PML) sumolation in nuclear body formation, 11S proteasome recruitment, and As2O3-induced PML or PML/retinoic acid receptor alpha degradation. J. Exp. Med. 193:1361-1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lawrence, C., and R. E. Thach. 1974. Encephalomyocarditis virus infection of mouse plasmacytoma cells. I. Inhibition of cellular protein synthesis. J. Virol. 14:598-610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lidsky, P. V., S. Hato, M. V. Bardina, A. G. Aminev, A. C. Palmenberg, E. V. Sheval, V. Y. Polyakov, F. J. van Kuppeveld, and V. I. Agol. 2006. Nucleocytoplasmic traffic disorder induced by cardioviruses. J. Virol. 80:2705-2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Negorev, D., and G. G. Maul. 2001. Cellular proteins localized at and interacting within ND10/PML nuclear bodies/PODs suggest functions of a nuclear depot. Oncogene 20:7234-7242. [DOI] [PubMed] [Google Scholar]
- 36.Negorev, D. G., O. V. Vladimirova, and G. G. Maul. 2009. Differential functions of interferon-upregulated Sp100 isoforms: herpes simplex virus type 1 promoter-based immediate-early gene suppression and PML protection from ICP0-mediated degradation. J. Virol. 83:5168-5180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pallansch, M. A., O. M. Kew, A. C. Palmenberg, F. Golini, E. Wimmer, and R. R. Rueckert. 1980. Picornaviral VPg sequences are contained in the replicase precursor. J. Virol. 35:414-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pampin, M., Y. Simonin, B. Blondel, Y. Percherancier, and M. K. Chelbi-Alix. 2006. Cross talk between PML and p53 during poliovirus infection: implications for antiviral defense. J. Virol. 80:8582-8592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parks, G. D., J. C. Baker, and A. C. Palmenberg. 1989. Proteolytic cleavage of encephalomyocarditis virus capsid region substrates by precursors to the 3C enzyme. J. Virol. 63:1054-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Percherancier, Y., D. Germain-Desprez, F. Galisson, X. H. Mascle, L. Dianoux, P. Estephan, M. K. Chelbi-Alix, and M. Aubry. 2009. Role of SUMO in RNF4-mediated PML degradation: PML sumoylation and phospho-switch control of its SUMO binding domain dissected in living cells. J. Biol. Chem. 284:16595-16608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Porta, C., R. Hadj-Slimane, M. Nejmeddine, M. Pampin, M. G. Tovey, L. Espert, S. Alvarez, and M. K. Chelbi-Alix. 2005. Interferons alpha and gamma induce p53-dependent and p53-independent apoptosis, respectively. Oncogene 24:605-615. [DOI] [PubMed] [Google Scholar]
- 42.Porter, F. W., and A. C. Palmenberg. 2009. Leader-induced phosphorylation of nucleoporins correlates with nuclear trafficking inhibition by cardioviruses. J. Virol. 83:1941-1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Regad, T., and M. K. Chelbi-Alix. 2001. Role and fate of PML nuclear bodies in response to interferon and viral infections. Oncogene 20:7274-7286. [DOI] [PubMed] [Google Scholar]
- 44.Regad, T., A. Saib, V. Lallemand-Breitenbach, P. P. Pandolfi, H. de The, and M. K. Chelbi-Alix. 2001. PML mediates the interferon-induced antiviral state against a complex retrovirus via its association with the viral transactivator. EMBO J. 20:3495-3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sadler, A. J., and B. R. Williams. 2008. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 8:559-568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scaglioni, P. P., T. M. Yung, L. F. Cai, H. Erdjument-Bromage, A. J. Kaufman, B. Singh, J. Teruya-Feldstein, P. Tempst, and P. P. Pandolfi. 2006. A CK2-dependent mechanism for degradation of the PML tumor suppressor. Cell 126:269-283. [DOI] [PubMed] [Google Scholar]
- 47.Schlax, P. E., J. Zhang, E. Lewis, A. Planchart, and T. G. Lawson. 2007. Degradation of the encephalomyocarditis virus and hepatitis A virus 3C proteases by the ubiquitin/26S proteasome system in vivo. Virology 360:350-363. [DOI] [PubMed] [Google Scholar]
- 48.Shen, T. H., H. K. Lin, P. P. Scaglioni, T. M. Yung, and P. P. Pandolfi. 2006. The mechanisms of PML-nuclear body formation. Mol. Cell 24:331-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stadler, M., M. K. Chelbi-Alix, M. H. M. Koken, L. Venturini, C. Lee, A. Saïb, F. Quignon, L. Pelicano, M.-C. Guillemin, C. Schindler, and H. de Thé. 1995. Transcriptional induction of the PML growth suppressor gene by interferons is mediated through an ISRE and a GAS element. Oncogene 11:2565-2573. [PubMed] [Google Scholar]
- 50.Tatham, M. H., E. Jaffray, O. A. Vaughan, J. M. Desterro, C. H. Botting, J. H. Naismith, and R. T. Hay. 2001. Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J. Biol. Chem. 276:35368-35374. [DOI] [PubMed] [Google Scholar]
- 51.Van Dyke, T. A., and J. B. Flanegan. 1980. Identification of poliovirus polypeptide P63 as a soluble RNA-dependent RNA polymerase. J. Virol. 35:732-740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang, Z. G., D. Ruggero, S. Ronchetti, S. Zhong, M. Gaboli, R. Rivi, and P. P. Pandolfi. 1998. PML is essential for multiple apoptotic pathways. Nat. Genet. 20:266-272. [DOI] [PubMed] [Google Scholar]
- 53.Zennou, V., C. Petit, D. Guetard, U. Nerhbass, L. Montagnier, and P. Charneau. 2000. HIV-1 genome nuclear import is mediated by a central DNA flap. Cell 101:173-185. [DOI] [PubMed] [Google Scholar]
- 54.Zhu, J., M. H. Koken, F. Quignon, M. K. Chelbi-Alix, L. Degos, Z. Y. Wang, Z. Chen, and H. de The. 1997. Arsenic-induced PML targeting onto nuclear bodies: implications for the treatment of acute promyelocytic leukemia. Proc. Natl. Acad. Sci. U. S. A. 94:3978-3983. [DOI] [PMC free article] [PubMed] [Google Scholar]