

Abstract
Purpose
Recent studies have shown that the DNA damage response (DDR) is activated in precancerous lesions, suggesting that neoplastic cells may avoid apoptosis by impairing the DDR which acts as a barrier against tumor progression. To define the role of the DDR pathway in human colorectal carcinoma, we investigated the level of phosphorylated proteins of the DDR pathway.
Results
Immunostaining for pATM, γH2AX and pChk2 revealed that all were significantly expressed during tumor progression in advanced carcinoma (vs. normal tissue for pATM [p < 0.05]; vs. normal and adenoma for γH2AX [p < 0.05]; and vs. normal tissue for pChk2 [p < 0.05]. Western blot analysis of γH2AX and pChk2 revealed that their level increased gradually during tumor progression and was maximal in advanced carcinoma (vs. normal tissue; p < 0.05). No apoptotic cells were found in any tissue sample.
Experimental design
Colorectal tissue samples were obtained at the time of surgery, from 55 patients at two hospitals. The tissues were classified into four groups according to pathology: normal mucosa, adenoma, early carcinoma and advanced carcinoma. We evaluated phosphorylated ataxia telangiectasia mutated (pATM), phosphorylated H2AX (γH2AX) and Chk2 (pChk2) protein levels by immunohistochemistry and western blot analysis. We also evaluated apoptosis by the TUNEL assay.
Conclusions
The DDR pathway was activated during cancer progression, but no apoptosis was detected, even among the cells with activated DDR. It is likely that activation of DDR was induced by stress signaling as a consequence of oxidative, replication and mechanical stresses occurring during growth and expansion of the colorectal cancer.
Keywords: DNA damage response, H2AX, ATM, Chk2, colorectal carcinoma, cancer progression, apoptosis
Introduction
Colorectal carcinoma is one of the most common malignancies in the world and its treatment is based on surgical removal of the tumor. Although the prognosis associated with colorectal cancer is better than that of many other solid tumors, its morbidity and mortality remain high.
Genomes of tumor cells exhibit multiple alterations compared with the parent cells, and it is now established that tumor progression is associated with a multistep process of genetic alterations. These alterations allow the cell to acquire characteristics that are universal among tumors, and have been referred to as the hallmarks of cancer.1 The loss of systems to ensure genomic integrity allows the cell to acquire genetic changes more easily. Cells respond to DNA damage by launching a set of actions known as DNA damage response (DDR).2 Two related protein kinases, ataxia telangiectasia mutated (ATM) and ataxia telangiectasia related (ATR), play a central role in the DDR mechanism in human cells. The DDR is initiated following recognition of DNA damage by the recruitment of ATM and ATR to the site of DNA damage. ATM and ATR can modify chromatin at the site of DNA damage, by phosphorylating Ser-139 of histone H2AX (γH2AX), an event that allows for DDR enforcement. After their recruitment to the site of DNA damage, ATM and ATR then phosphorylate a number of protein substrates,3–5 including the protein kinases Chk1 and Chk2, which, in turn, target other proteins to induce cell cycle arrest and DNA repair.6
According to recent reports, the DNA damage response pathway is activated during the earliest stages of carcinogenesis in human solid tumor samples.7,8 This has been attributed to the DNA damage caused by increased replicative stress in rapidly dividing pre-neoplastic lesions. These findings suggest that DDR pathway activation plays an integral role in creating a barrier against tumor progression and genetic instability.7 However, another question that has arisen from previous reports is this: Is the DDR pathway always almost completely impaired in advanced carcinoma? The alteration of the DDR pathway may occur at any site of this pathway. If the upstream components of this pathway, such as ATM or Chk2, are impaired, the pathway would be totally impaired. However, if a downstream component is impaired, the upstream protein would be phosphorylated, but the DDR still might not work efficiently. It would also account for obtaining the new characteristics in the advanced carcinoma. There is a report stating that the final decision on survival or apoptosis for DNA damaged cells is being made by the downstream components of the DDR pathway; namely, cyclin-dependent kinase 2 (CDK2) and forkhead box O transcription factor 1 (FOXO1).9 The activation of ATM, H2AX and Chk2 thus may be unaffected while the effectiveness of DNA repair and signaling along the apoptotic pathway is impaired. We examined the expression of markers of the DDR pathway, ATM, H2AX and Chk2, as well as of apoptosis to reflect the progression of colorectal carcinogenesis, using surgically resected human colorectal carcinomas.
Results
Patient’s characteristics
The patient’s baseline characteristics are summarized in Table 1. Colorectal tissue samples were obtained from 55 patients who underwent surgery at the two hospitals during the study period. For immunohistochemistry, 48 tissue samples of advanced carcinoma (T2-4), 6 of early carcinoma (Tis-1), and 1 of adenoma were obtained from each patient and 7 tissue samples of adenoma were obtained from four patients with carcinoma. Normal mucosal tissue from all patients was also examined. For western blotting analysis, 45 tissue samples of advanced carcinoma, 6 of early carcinoma, and 1 of adenoma were obtained from 52 patients and 4 tissue samples of adenoma were obtained from two patients with carcinoma. Normal mucosal tissue from 20 patients was also evaluated. There was no significant correlation between the activation of the DDR pathway and clinico-pathological factors (Table 1).
Table 1.
Clinicopathological characteristics of the patients
| Gender (no.) | |
| Male | 36 |
| Female | 19 |
| Median age (yrs) | 73.6 |
| Tumor site (no.) | |
| Colon | 36 |
| Rectum | 19 |
| Cancer stage (no.) | |
| Adenoma | 1 |
| Tis | 2 |
| T1 | 4 |
| T2 | 6 |
| T3 | 31 |
| T4 | 11 |
| Differentiation (no.) | |
| Well | 10 |
| Moderately | 41 |
| Poorly | 3 |
| Nodal status (no.) | |
| Positive | 22 |
| Negative | 32 |
| Duke’s classification (no.) | |
| A | 28 |
| B | 2 |
| C | 18 |
| D | 6 |
| Distant metastasis (no.) | |
| Positive | 6 |
| Negative | 48 |
Immunohistochemistry for DNA damage response
Immunostaining was performed with antibodies to pATM, γH2AX and pChk2 for human colorectal tissues, which included normal, adenoma, early carcinoma and advanced carcinoma, and HL-60 cells for the control. Normal colon tissues had very low, immunocytochemically undetectable and hardly detectable by western blotting, background level of activated (phosphorylated) ATM, H2AX or Chk2 reflecting the constitutive activation of these proteins, reported to be partially due to oxidative DNA damage by the endogenous, metabolically generated, oxidants.10,11 The increase in ATM phosphorylation above the control level was seen in both in adenoma and cancer tissues while the level of γH2AX and p-Chk2 above that of the control colon was seen only in carcinoma tissues. In the positive cells, all three markers were well co-localized in the nuclei (Fig. 1). The levels of all three phosphorylated markers increased during tumor progression, and they were significantly elevated in advanced carcinoma (vs. normal tissue for pATM: p < 0.05, vs. normal and adenoma for γH2AX: p < 0.05, vs. normal tissue for pChk2: p < 0.05; Fig. 2).
Figure 1.
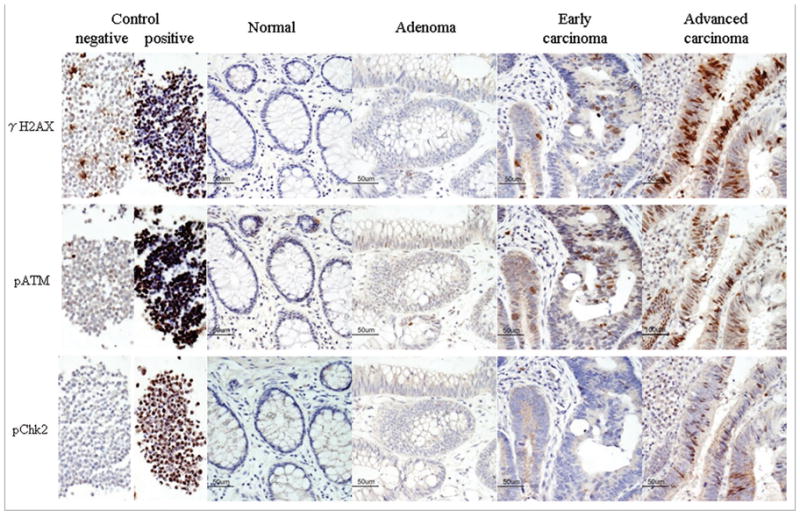
Representative images stained with phosphorylated ataxia telangiectasia mutated (pATM), phosphorylated H2AX (γH2AX) and phosphorylated Chk2 (pChk2). HL60 cells treated with 100 nM camptothecin for positive control, untreated for negative control, normal mucosa, adenoma, early carcinoma (Tis-1) and advanced carcinoma (T2-4) are shown. The normal mucosa and adenoma did not express any of the markers. The staining appears weak for each marker in early carcinoma and strong for each marker in advanced carcinoma. (Magnification ×400).
Figure 2.

The percentage of positive nuclei was calculated for each specimen. All three markers were expressed more strongly with tumor progression. The expression was significantly increased in advanced carcinoma [(A) vs. normal tissue for pATM (B) vs. normal and adenoma for γH2AX (C) vs. normal tissue for pChk2] *p < 0.05.
It should be noted that the technical aspects of tissue fixation also influence immunohistochemistry. We noticed that old tissue samples did not stain well even when antigens were retrieved. This unstable staining could be caused by the uneven penetration of formaldehyde and different fixation times. Therefore, we obtained fresh carcinoma tissue, from which one 5 mm thick sample was sliced, and the formaldehyde penetrated this tissue sample evenly. The duration of the fixation ranged from 24 h to 1 week, but we found 24–48 h fixation to be the best duration for our immunohistochemical staining (data not shown). After using these techniques, immunohistochemical staining was very stable. These observations caution about interpreting the data of immunohistochemical staining using formaldehyde-fixed tissue samples.
Western blot analysis for DNA damage response
Western blot analysis was performed on colorectal carcinoma surgical specimens and the corresponding normal tissues, using γH2AX and pChk2 antibodies. The lysates from HL60 cells treated with camptothecin were used as a positive control (Fig. 3). The γH2AX and pChk2 levels were lowest in normal mucosa and increased gradually with tumor progression, but varied widely in the adenoma and early/advanced carcinoma tissues. The γH2AX and pChk2 level differed significantly between normal mucosa and advance carcinoma (p < 0.05; Fig. 4).
Figure 3.

Western blot analysis using γH2AX and pChk2 antibodies in normal mucosa, adenoma, early carcinoma and advanced carcinoma. The lysates from HL60 cells treated with camptothecin were used as a positive control. γH2AX and pChk2 expression levels were lowest in normal mucosa and gradually increased with tumor progression. β-actin served as a control for the amount of protein loading. PC: positive control.
Figure 4.

Quantitative analysis by western blotting of γH2AX (A) and pChk2 (B). The protein level normalized to β-actin in normal mucosa, adenoma, early carcinoma and advanced carcinoma. Both expression levels gradually increased with tumor progression. There was a significant difference between normal mucosa and advanced carcinoma in both γH2AX and pChk2 expression. *p < 0.05.
Apoptosis detection by TUNEL
All tissue slides and the camptothecin-treated HL-60 cells slide were examined by TUNEL for apoptosis. Although many apoptotic cells were found in camptothecin-treated HL-60 cells, none were seen in any tissue samples, even if there were pATM-, γH2AX- and pChk2-positive cells.
Discussion
Activation of the DNA damage checkpoint induces p53-dependent cell cycle arrest, apoptosis or senescence.12 ATM plays a major role in the surveillance of the genome’s integrity and is the key signal transducer in response to the induction of DNA double-strand breaks (DSBs).13,14 Activated ATM leads to the phosphorylation of histone H2AX, and the phosphorylation of mediators and adaptor proteins such as 53BP1 and BRCA1, known to assist in the assembly of multiprotein complexes at the sites of DSBs.15,16 ATM activates the transcription factors, p53 and E2F1, and the downstream effecter kinases, Chk2 and Chk1, located at the nexus of the DNA repair and cell cycle progression pathways.17 Histone H2AX, one of the variants of the nucleosome core histone H2A,18 undergoes phosphorylation on Ser-139 in response to the induction of DSBs.19,20 The phosphorylation is mediated by ATM-,3 ATR-,21 and DNA-dependent protein kinase.22 After the induction of DSBs, the Ser-139-phosphorylated H2AX (γH2AX) can be detected immnocytochemically in the form of discrete nuclear foci, with each focus presumed to represent a single DSB.20 DNA damage engages a checkpoint pathway to arrest progression through the cell cycle until DNA integrity is restored during the repair process.23 Phosphorylation of Cdc25A and Cdc25C phosphatases mediated by ATM-activated Chk2 and Chk1 leads to the sequestration of Cdc25C by a 14-3-3 protein in the cytoplasm, or degeneration of Cdc25A through the ubiquitin/proteasome pathway. Consequently, that leaves Cdk2/cyclin E, Cdk2/cyclin A and Cdk1/cyclin B kinases in their phosphorylated, inactive state, thereby preventing cell cycle progression.15–17
Recent studies have shown that DDR is highly activated in precancerous lesions, suggesting that neoplastic cells may avoid apoptosis by impairing DDR which works as a barrier against tumor progression.8,9,12,24,25 However, it is still not known how DDR is activated in advanced carcinoma, nor can we explain the sensitivity to oxidative and carcinogenic stress. According to certain experimental reports, DDR was activated in cancer cell lines by oxidative stress or cytotoxic agents,26–29 which means that some advanced carcinomas may have at least an intact upstream pathway of DDR. Concerning sensitivity, normal cells should tolerate oxidative or carcinogenic stress in the colorectal lumen whereas advanced cancer cells should be more sensitive because of instability of their genome during early growth phase under the same conditions. Therefore, we investigated how the DDR pathway works during the progression of human colorectal cancer.
Contrary to other recent reports, we observed low levels of pATM, γH2AX and pChk2 in normal and adenoma tissues, gradually increasing in cancer tissues, and reaching maximal levels in advanced carcinoma. This shows that DDR was activated increasingly throughout tumor progression and peaked in advanced carcinoma. These results may be explained by our hypothesis that the cell’s sensitivity to surrounding stress is different individually. Whereas normal mucosal cells because of effectiveness of their DDR would tolerate the stress in the colorectal lumen, neoplastic cells having defective DDR would be easily damaged under the same conditions due to instability of their genome and continuous growth and proliferation. Thus, the elevated levels of pATM, γH2AX and pChk2 would be the markers of DNA damage induced e.g., by the replicative stress under conditions of hypoxia within the cancer tissue.
Genomic instability itself may induce DDR activation. Some report showed that normal cell line with a mutated succinate dehydrogenase subunit C (SDHC) gene overproduced superoxide anion from mitochondria, and some cells caused benign tumors when injected under the epithelium of nude mice.30,31 These results indicate that genomic instability itself induces oxidative stress which causes DNA damage, and oxidative stress induces genomic instability as a malignant cycle. This may be a reason that DDR pathway is activated throughout cancer progression.
Our data also showed no evidence that the DDR activation led to apoptosis. Several explanations can be advanced to explain this observation: (i) the DNA damage itself was not severe enough and was repaired; (ii) the DDR pathway was impaired downstream and DNA damage was sensed but the apoptotic pathway was not activated; (iii) the DDR pathway was intact but some components of the apoptotic pathway were ineffective; (iv) apoptosis is a stochastic process and apoptotic cells are rapidly removed from the tissue by the macrophages. The “time window” to detect apoptosis is short and therefore frequency of apoptotic cells in tissue may be very low;32 (v) other forms of cell death, such as necrosis and permanent cell arrest with phenotype characteristics of senescence.33
The barrier to cancer progression associated with progressive increase of genome instability is provided by an effective DDR pathway that ensures complete DNA repair. Also effective apoptotic mechanisms remove cells with damaged DNA. However, if the damaged cells cannot trigger apoptosis because of impairment of the downstream pathway of DDR or apoptosis, the increasing DNA damage may cause new characteristics, further enhancing the chance of malignant transformation. It was recently demonstrated that CDK2-mediated phosphorylation of FOXO1, a member of the forkhead box O (FOXO) family of transcription factors, plays an important role in DNA damage-induced apoptosis.9,34 Phosphorylation of FOXO1 was abrogated upon DNA damage through the cell cycle checkpoint pathway that is dependent on the protein kinases, Chk1 and Chk2. Chk1 and Chk2 inhibit the activity of CDK2 and activated FOXO1 promotes apoptosis of damaged cells. However, if the Chk1- and Chk2-CDK2 correlation is abrogated, which is our hypothesis about impairment of DDR pathway downstream, Chk1 and Chk2 activated by DNA damage response cannot inhibit CDK2 activity, which might cause phosphorylation of FOXO1 and prohibit apoptosis in severely damaged cells. Therefore, DDR impairment may enhance the chance of malignant transformation. These speculations are reinforced by a report which showed that the tumor biology in advanced carcinoma after treatment with cytotoxic drugs is altered as evidenced by characteristics of tumor markers detected clinically.35
One of the reasons of the lack of apoptosis may be the way of detection of apoptosis. Apoptosis is a transient, kinetic event, so that the entire apoptotic process from onset to ultimate cell disintegration is often short and of variable duration which depends on the cellular environment.36 It is possible that immunohistochemical staining assay can just provide a “snap-shot” of apoptotic process, which may be a reason why we could see few apoptotic cells in each tissue sample.
We also focused on the necrosis and cell senescence for the lack of apoptosis. Necrosis might be a reason of no apoptotic cells in the tissues. Generally, necrosis is occurred in the inside of the tumor because of hypoxia due to low blood perfusion. However, all slides were stained by Hematoxylin-eosin and there were few necrotic cells morphologically in any area of the samples. Senescent cells which undergo permanent growth arrest with metabolically active and production of secreted proteins with potential tumor-promoting activities do not represent detectable DNA breaks. Nevertheless, senescent cells displayed components of DNA damage response (DDR) such as γH2AX foci and uniform nuclear staining for p-ATM. Importantly, there was no accumulation of 53BP1 in γH2AX foci of senescent cells.37,38 It is possible explanation that our data showed no evidence of apoptosis in spite of activated DDR pathway. We are following up on the clinical outcomes of our patients whose tumors were presently examined to assess how the altered DDR pathway correlated with the clinical markers and tumor prognosis.
In conclusion, we observed that the DDR pathway was activated with colorectal cancer progression. The activation was not related to DNA damage known to occur during apoptosis and in fact the frequency of apoptosis was minimal. It is likely that the activation was triggered by the stress signaling pathways that may be related to oxidative stress,39,40 replication stress,41 and/or mechanical stress due to the cancer cells microenvironment,42 the factors associated with tumor progression. Further investigations are needed to link the observed DDR changes with other markers of cell signaling and with clinical prognostic markers.
Materials and Methods
Patients and tissue samples
This prospective clinical trial was conducted at two hospitals: Yamaguchi University Hospital, Ube, Japan and Saiseikai Shimonoseki General Hospital, Shimonoseki, Japan. The inclusion criteria were as follows: the subjects were at least 18 years old, they had preoperative biopsy-proven colorectal carcinoma, they had not received neoadjuvant therapy, and they had undergone complete resection. The study was approved by the institutional review boards of the Yamaguchi University and Saiseikai Shimonoseki General Hospitals.
Colorectal tissue samples were obtained from consecutive operations performed at the two hospitals between April and December, 2008. Historical diagnoses were established by senior pathologists. The clinical and pathological characteristics of the patients were obtained from our database and supplemented by review of the medical records. Informed consent was obtained from each patient according to institutional guidelines.
Antibodies
For immunohistochemistry and immunocytochemistry, we used anti-phospho-histone H2AX (Ser-139) monoclonal antibody (mAb) (γH2AX, Cell Signaling Technology, Danvers, MA, USA), anti-phospho-ATM (Ser-1981) mAb (pATM, Rockland, Gilbertsville, PA, USA) and anti-phospho-Chk2 (Thr-68) mAb (pChk2, Abcam, Cambridge, UK). For western blotting, we used γH2AX mAb (Millipore, Billerica, MA, USA), pChk2 (Thr-68) mAb (Abcam) and anti-beta-actin (Alpha diagnostic international, San Antonio, TX, USA).
Treatment and fixation of the cells for control
HL60 cells for antibody validation were incubated in RPMI medium supplemented with 10% fetal bovine serum (FBS) and 50 units/ml penicillin (all from GIBCO/BRL Life Technologies, Inc., Grand Island, NY, USA) at 37°C in an atmosphere of 5% CO2 in air. The cells were then treated with 100 nM camptothecin (Sigma Chemical Co., St. Louis, USA) for 30 min. Control cultures were treated with dimethylsulfoxide (DMSO, Sigma) for 30 min. After treatment, the cells were washed with phosphate-buffered saline (PBS), fixed in suspension in 1% methanol-free formaldehyde (Polysciences, Inc., Warrington, PA, USA) in PBS at 0°C for 15 min, and post-fixed with 80% ethanol at −20°C. The cells were rinsed in PBS (200 g, 5 min) and resuspended in 1% bovine serum albumin (BSA, Sigma) in PBS to suppress nonspecific antibody binding. After centrifugation, the pellet was embedded in paraffin.
Immunohistochemistry
After resection of the colon or rectum, 5 mm thick tissue sections were made and each was fixed immediately in Ufix™ containing 18.5% formaldehyde, methanol and cineole (Sakura Finetek Japan Co., Ltd., Tokyo, Japan) for 24–48 h while swinging, and then embedded in paraffin. Consecutive 3-μm thick paraffin tissue sections were mounted on polylysine-coated slides. The slides were deparaffinized in xylene, cleared in a graded ethanol series and heat-treated for 30 min in 10 mM citrate buffer ph 7.0 in a microwave at 98°C for antigen retrieval. Primary antibodies, pATM (1:400), γH2AX (1:100) and pChk2 (1:100), were incubated with the sections overnight at 4°C. After incubation, the slides were washed several times with PBS containing 0.1% Tween-20 (PBST). A chromogen reaction was carried out with 0.2 mg/ml diaminobenzidine for 4 min, and counterstaining was done with Mayer’s hematoxylin for 20 min. The immunostaining evaluation was performed by two people and confirmed by two pathologists. We counted the number of nuclei with positive staining in three randomly chosen high-power microscopic fields within the sections, and calculated the percentage of positively stained nuclei.
Western blotting analysis
Part of each tissue sample was frozen immediately in liquid nitrogen and stored at −80°C for western blotting. Approximately 300 mg of each sample was homogenized in 1.8 ml of lysis buffer [50 mM Tris (pH 7.4), 150 mM NaCl, 20 mM MgCl2 and 5% Triton X-100 (Wako, Osaka, Japan)] with 1% protease inhibitor cocktail (EDTA free) (Nacalai Tesque, Kyoto, Japan) and 1% phosphatase inhibitor cocktail (EDTA free) (Nacalai Tesque). The homogenate was centrifuged at 4,000 rpm for 10 min at 4°C. The supernatant was collected, and the total protein concentration was measured with a BCA protein assay reagent kit (Thermo Fisher Scientific Inc., Rockford, IL, USA). After subjecting 3 μg γH2AX and 6 μg Chk2 of the total protein to 15% and 7.5% PAGE, respectively, electroblotting was done onto a polyvinylidene difluoride membrane. Then, after blocking in 5% BSA in TBST (TBS and 0.1% Tween 20), the membrane was incubated with H2AX (1:2,000), Chk2 (1:2,000) and beta-actin (1:8,000) antibody, followed by incubation with the secondary antibody at a dilution of 1:5,000. Enhanced chemiluminescence western blotting detection reagents (GE Healthcare, Buckinghamshire, UK) were used to detect the immunocomplex. Membranes were scanned and quantitated by the Fujifilm LAS-1000 Luminescence Image Analyzer and the chemiluminescence detection system (Fuji Photo Film Co., Ltd., Tokyo, Japan), and they were then normalized to β-actin.
Apoptosis detection by TUNEL
Slides were deparaffinized by heating at 57°C for 5 min to melt the wax, then immersed in xylene cleared in a graded ethanol series. The TACS TdT Kit (R&D system, Inc., Minneapolis, MN, USA) was used for this experiment. The slides were first incubated with proteinase K digestion at 37°C for 1 h, and then with quenching solution at room temperature for 5 min. For a positive control, TACS-Nuclease treatment was used to generate DNA breaks in the majority of cells before quenching. TdT-labeling was performed, and the slides were stained by diaminobenzidine (DAB) to detect apoptosis. Nucleus counterstaining was done with methyl green. The slides were evaluated and scored by the same methods as used for immunohistochemistry.
Statistical analysis
Statistical analysis was performed using the Statview J-5.0 program (Abacus Concepts, Inc., Berkeley, CA, USA). Data distribution was analyzed and statistical differences for pathological stages were evaluated by analysis of variance and the Tukey-Kramer test.
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Shiloh Y. The ATM-mediated DNA-damage response: taking shape. Trends Biochem Sci. 2006;31:402–10. doi: 10.1016/j.tibs.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 3.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462–7. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 4.Lee J-H, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–4. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 5.Paull TT, Lee JH. The Mre11/Rad50/Nbs1 complex and its role as a DNA-double strand break sensor for ATM. Cell Cycle. 2005;4:737–40. doi: 10.4161/cc.4.6.1715. [DOI] [PubMed] [Google Scholar]
- 6.Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer cell. 2003;3:421–9. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 7.Bartkova J, Horejsí Z, Koed K, Kramer A, Tort F, Zieger K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–70. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 8.Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–13. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 9.Huang H, Regan KM, Lou Z, Chen J, Tindall DJ. CDK2-dependent phosphorylation of FOXO1 as an apoptotic response to DNA damage. Science. 2006;314:294–7. doi: 10.1126/science.1130512. [DOI] [PubMed] [Google Scholar]
- 10.Tanaka T, Halicka HD, Huang X, Traganos F, Darzynkiewicz Z. Constitutive histone H2AX phosphorylation and ATM activation, the reporters of DNA damage by endogenous oxidants. Cell Cycle. 2006;5:1940–5. doi: 10.4161/cc.5.17.3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao H, Tanaka T, Halicka HD, Traganos F, Zarebski M, Dobrucki J, et al. Cytometric assessment of DNA damage by exogenous and endogenous oxidants reports the aging-related processes. Cytometry A. 2007;71:905–14. doi: 10.1002/cyto.a.20469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kastan MB, Bartek J. Cell cycle checkpoints and cancer. Nature. 2004;432:316. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 13.Bakkenist CJ, Kasten MB. Initiating cellular stress response. Cell. 2004;118:9–17. doi: 10.1016/j.cell.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 14.Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–7. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- 15.Kasten MB, Lim DS. The many substrates and functions of ATM. Nat Rev Mol Cell Biol. 2000;1:179–86. doi: 10.1038/35043058. [DOI] [PubMed] [Google Scholar]
- 16.Shiloh Y. ATM and related protein kinases safeguarding genome integrity. Nat Rev Cancer. 2003;3:155–68. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 17.Kristjansdottir K, Rudolph J. Cdc 25 phosphatases and cancer. Chem Biol. 2004;11:1043–51. doi: 10.1016/j.chembiol.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 18.Pehrson JR, Fuji RN. Evolutionary conservation of histone H2A macro-H2A subtypes and domains. Nucleic Acids Res. 1998;26:2837–42. doi: 10.1093/nar/26.12.2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 20.Sedelnikova OA, Rogakou EP, Panuytin IG, Bonner W. Quantitive detection of 125IUdr-induced DNA double-strand breaks with γH2AX antibody. Radiat Res. 2002;158:486–92. doi: 10.1667/0033-7587(2002)158[0486:qdoiid]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 21.Furuta T, Takemura H, Liao ZY, Aune GJ, Redon C, Sedelnikova OA, et al. Phosphorylation of histone H2AX and activation of Mre11, Rad50 and Nbs1 in response to replication-dependent DNA double-strand breaks induced by mammalian topoisomerase I cleavage complexes. J Biol Chem. 2003;278:20303–12. doi: 10.1074/jbc.M300198200. [DOI] [PubMed] [Google Scholar]
- 22.Park EJ, Chan DW, Park JH, Oettinger MA, Kwon J. DNA-PK is activated by nucleosomes and phosphorylates H2AX within the nucleosomes in an acetylation-dependent manner. Nucleic Acids Res. 2003;31:6819–27. doi: 10.1093/nar/gkg921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ahn J, Urist M, Prives C. The Chk2 protein kinase. DNA repair. 2004;3:1039–47. doi: 10.1016/j.dnarep.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 24.Bartkova J, Bakkenist CJ, Rajpert-De Meyts E, Skakkebaek NE, Sehested M, Lukas J, et al. ATM activation in normal human tissues and testicular cancer. Cell Cycle. 2005;4:838–45. doi: 10.4161/cc.4.6.1742. [DOI] [PubMed] [Google Scholar]
- 25.Raynaud CM, Jang SJ, Nuciforo P, Lantuejoul S, Brambilla E, Mounier N, et al. Telomere shortening is correlated with DNA damage response and telomeric protein downregulation in colorectal preneoplastic lesions. Ann Oncol. 2008;19:1875–81. doi: 10.1093/annonc/mdn405. [DOI] [PubMed] [Google Scholar]
- 26.Tanaka T, Huang X, Halicka HD, Zhao H, Traganos F, Albino AP, et al. Cytometry of ATM activation and histone H2AX phosphorylation to estimate extent of DNA damage induced by exogenous agents. Cytometry A. 2007;71:648–61. doi: 10.1002/cyto.a.20426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanaka T, Huang X, Jorgensen E, Gietl E, Traganos F, Darzynkiewicz Z, et al. ATM activation accompanies histone H2AX phosphorylation in A549 cells upon exposure to tobacco smoke. BMC Cell Biol. 2007;8:26. doi: 10.1186/1471-2121-8-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hong Z, Traganos F, Darzynkiewicz Z. Phosphorylation of p53 on Ser15 during cell cycle caused by Topo I and Topo II inhibitors in relation to ATM and Chk2 activation. Cell Cycle. 2008;7:1–8. doi: 10.4161/cc.7.19.6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong Z, Traganos F, Albino AP, Darzynkiewicz Z. Oxidative stress induces cell cycle-depentent Mre11 recruitment, ATM and Chk2 activation and histome H2AX phosphorylation. Cell Cycle. 2008;7:1490–5. doi: 10.4161/cc.7.10.5963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishii T, Yasuda K, Akatsuka A, Hino O, Hartman PS, Ishii N. A mutation in the SDHC gene of complex II increases oxidative stress, resulting in apoptosis and tumorigenesis. Cancer Res. 2005;65:203–9. [PubMed] [Google Scholar]
- 31.Slane BG, Aykin-Burns N, Smith BJ, Kalen AL, Goswami PC, Domann FE, et al. Mutation of succinate dehydrogenase subunit C results in increased O2•−, oxidative stress and genomic instability. Cancer Res. 2006;66:7615–20. doi: 10.1158/0008-5472.CAN-06-0833. [DOI] [PubMed] [Google Scholar]
- 32.Darzynkiewicz Z, Bedner E, Traganos F. Difficulties and pitfalls in analysis of apoptosis. Meth Cell Biol. 2001;63:527–59. doi: 10.1016/s0091-679x(01)63028-0. [DOI] [PubMed] [Google Scholar]
- 33.Portugal J, Bataller M, Mansilla S. Cell death pathway in response to antitumor therapy. Tumori. 2009;95:409–21. doi: 10.1177/030089160909500401. [DOI] [PubMed] [Google Scholar]
- 34.Huang H, Tindall DJ. CDK2 and FOXO1. Cell Cycle. 2007;6:902–6. doi: 10.4161/cc.6.8.4122. [DOI] [PubMed] [Google Scholar]
- 35.Modrak DE, Gold DV, Goldenberg DM, Blumenthal RD. Colonic tumor CEA, CSAp and MUC-1 expression following radioimmunotherapy or chemotherapy. Tumor Biol. 2003;24:32–9. doi: 10.1159/000070658. [DOI] [PubMed] [Google Scholar]
- 36.Smolewski P, Grabarek J, Lee BW, Johnson GL, Darzynkiewicz Z. Kinetics of HL-60 cell entry to apoptosis during treatment with TNFα or camptothecin assayed by the Stathmo-Apoptosis method. Cytometry. 2002;47:143–9. doi: 10.1002/cyto.10062. [DOI] [PubMed] [Google Scholar]
- 37.Roninson IB, Broude EV, Chang BD. If not apoptosis, then what? Treatment-induiced senescence and mitotic catastrophe in tumor cells. Drug Resist Updat. 2001;4:303–13. doi: 10.1054/drup.2001.0213. [DOI] [PubMed] [Google Scholar]
- 38.Pospelova TV, Demidenko ZN, Bukreeva EI, Pospelov VA, Gudkov AV, Blagosklonny MV. Pseudo-DNA damage response in senescent cells. Cell Cycle. 2009;8 doi: 10.4161/cc.8.24.10215. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Visconti R, Grieco D. New insights on oxidative stress in cancer. Curr Opin Drug Discov Devel. 2009;12:240–5. [PubMed] [Google Scholar]
- 40.Inokuma T, Haraguch M, Fujita F, Tajima Y, Kanematsu T. Oxidative stress and tumor progression in colorectal cancer. Hepatogastroenterology. 2009;56:343–7. [PubMed] [Google Scholar]
- 41.Ruzankina Y, Asara A, Brown EJ. Replicative stress, stem cells and aging. Mech Aging Dev. 2008;129:460–6. doi: 10.1016/j.mad.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Butcher DT, Alliston T, Weaver VM. A tense situation: forcing tumour progression. Nat Rev Cancer. 2009;9:108–22. doi: 10.1038/nrc2544. [DOI] [PMC free article] [PubMed] [Google Scholar]