

Abstract
Intrinsic optical emissions, such as autofluorescence and second harmonic generation (SHG), are potentially useful for functional fluorescence imaging and biomedical disease diagnosis for neurodegenerative diseases such as Alzheimer’s disease (AD). Here, using multiphoton and SHG microscopy, we identified sources of intrinsic emissions in ex vivo, acute brain slices from AD transgenic mouse models. We observed autofluorescence and SHG at senile plaques as well as characterized their emission spectra. The utility of intrinsic emissions was demonstrated by imaging senile plaque autofluorescence in conjunction with SHG from microtubule arrays to assess the polarity of microtubules near pathological lesions. Our results suggest that tissues from AD transgenic models contain distinct intrinsic emissions, which can provide valuable information about the disease mechanisms.
1. Introduction
Alzheimer’s disease (AD) is a prevalent neurodegenerative disease for which there is no known cure. Patients usually suffer from dementia, along with other behavioral changes, so examinations on memory and cognitive abilities are standard tests for diagnosing AD. However, confirming the initial diagnosis of the actual cause of dementia is often difficult. Definitive confirmation is possible with post-mortem autopsy, where histology on brain tissues reveals senile plaques and neurofibrillary tangles (NFT), the two hallmark pathological lesions in AD. As a result, there are strong incentives to develop reliable methods for early diagnosis of AD. To address this need, new approaches are being developed. Positron emission tomography (PET) imaging of Pittsburgh Compound-B has been shown to bind amyloid-beta deposits in human patients in vivo [1]. Optical imaging with amyloid-beta deposit-specific near-infrared optical probes is being developed and has generated some successes in transgenic mouse models [2]. These new imaging approaches use exogenous compounds that can selectively label pathological lesions and therefore enhance signal-to-noise. Alternatively, endogenous optical signals may also provide contrast between AD and normal tissues. For example, one recent study [3] reports that transmission and reflectance spectra in near-infrared region from unstained in vitro tissue slab samples may be useful in distinguishing AD from normal tissues, although the molecular origin of this observation was not specified. Interpreting this type of studies is difficult due to a lack of information about intrinsic emissions from AD tissues, which has only been recently investigated in detail in normal tissues [4].
It is generally known that brain tissues contain various endogenous proteins that can generate autofluorescence and/or second harmonic emissions. It is possible that these endogenous optical signals can provide contrast between normal and AD tissues. Moreover, if applied to AD tissues, intrinsic emissions may provide functional information that is useful in understanding the disease mechanisms. For example, fluorescence from NAD(P)H, an electron carrier that is crucial in metabolic pathways, has been used to investigate the dynamics of functional brain energy metabolism [5, 6]. Second harmonic generation (SHG) emitted from microtubules has been used to map the distribution of polarized microtubule arrays in acute brain tissues from mice of various ages [7, 8].
There have been several reports on autofluorescence from AD-related lesions. An early paper from Dowson [9] described autofluorescence from senile plaque in the brain tissues from human patients. Using tissues embedded in paraffin wax and sectioned thinly, he described a blue autofluorescence (>430nm) from senile plaques that were excited with ultraviolet light. This observation was confirmed by a more recent study [10], which also studied human AD tissues. For transgenic mouse models, one study [11] has reported that a particular mouse model that overexpresses human mutant amyloid precursor protein (APP) contain intrinsically fluorescent senile plaques. Lastly, several years ago, our group and collaborators have reported that NFTs in human AD brain tissues can generate a blue autofluorescence that has an emission peak at ~460nm [4].
Because most of the previous studies [9–11] used wide-field or confocal microscopy on post-mortem or fixed brain tissues, they are restricted to superficial areas or thinly-sectioned preparations. Furthermore, dead or fixed tissues lose intrinsic emissions that could originate from live cells, such as autofluorescence from NAD(P)H and SHG from microtubules. Here, we use multiphoton microscopy [12, 13] to investigate the various sources of intrinsic emissions, including from senile plaques, from transgenic AD mouse models. We demonstrate the utility of these intrinsic emissions by assessing the polarity and morphology of dendritic microtubule arrays near senile plaques and finally, discuss how endogenous signals may be useful for AD diagnosis.
2. Materials and methods
2.1. Multiphoton and second harmonic microscope
Imaging was performed on a custom-built multiphoton microscope based on a commercial laser beam scanning unit (Bio-Rad MRC 1024) and an inverted microscope (Olympus, IX-70). Trains of laser pulses at 80MHz and ~100fs duration were generated by a mode-locked Ti:Sapphire laser (Spectra-Physics Tsunami), which was pumped by a 5W diode laser (Spectra-Physics Millennia). Intensity and polarization were controlled by a Pockels cell (Conoptics 350–50) and a Berek compensator (New Focus 5540). The beam was focused onto the sample by an Olympus UApo/340 20X/NA 0.7 water immersion objective, which also collected the epi-fluorescence. The transmitted SHG was collected by an Olympus XLUMPlanFl 20X/NA 0.95 dipping objective. The average power after the objective was ~70–130mW. For imaging, samples were excited at 774nm and signals were detected with bi-alkali photomultiplier tubes (Hamamatsu HC125-02). SHG was collected behind a focusing lens, an IR-blocking short-pass dichroic and a narrowband emission filter, centered at 387nm (Semrock FF720 and FF01-387/11). Autofluorescence was collected from 400–550nm behind a blue glass filter (Chroma BGG22).
Emission spectra were obtained using a liquid nitrogen-cooled CCD spectrometer (Jobin Yvon Spex270M). The spectrometer was coupled to the microscope via an optical fiber replacing the usual bi-alkali photomultiplier in the transmission-direction. To collect one spectrum, a small square area ~50 μm wide was continuously scanned at high zoom for 10 seconds. To reduce photodamage, excitation wavelength was set at 830nm and average power was reduced to ~25mW. Dark counts were subtracted by acquiring a blank spectrum with shutter closed. The spectrometer was calibrated with collagen SHG spectra collected from a rat tendon sample.
2.2. Tissue preparation
Acute brain slices were prepared from 12 mice, including one with APP Swedish mutation [14] (obtained from G. K. Gouras), two with APPSwe/PS1 double mutations [15] (ordered from Jackson Laboratory), two with APPSwe/TauJNPL3 double mutations [16] (obtained from K. Duff), and three with APPSwe/PS1/Tau triple mutations [17] (obtained from G. K. Gouras), and four wild-type mice. All mice were at least 1-year old. All preparations were performed in accordance with Cornell University animal use regulations (IACUC protocol 00-46-03).
To prepare acute slices, we removed the brain following CO2 euthanasia. Immediately, the brain was dipped in iced artificial cerebrospinal fluid (ACSF) composed of (in mM): NaCl, 120; KCl, 2.5; NaH2PO4, 1; MgSO4, 1.3, NaHCO3, 25; D-glucose, 10; CaCl2, 2.5, that was saturated with 95% O2 and 5% CO2. Transverse hippocampal slices 300–400μm thick were cut using a vibratome (Campden Instruments). Slices were then incubated in oxygenated ACSF at 35°C for 1 hour. During imaging, slices were held under nylon grid anchors in a flow chamber (Warner Instruments) and perfused with oxygenated ACSF at room temperature.
After imaging the intrinsic emissions from unstained brain slices, AD pathology was verified by Thioflavin-S or BTA-1 staining of senile plaques. Brain slices were fixed in 4% paraformaldehyde in PBS in the refrigerator overnight. Next the slices were incubated for 1–2 hours in Thioflavin-S (Sigma, 0.0005–0.001% by weight in PBS), or 40μM BTA-1 (Sigma, 40mM in DMSO then diluted 1:1000 in PBS), and then rinsed three times in PBS. During imaging, the slices were placed in 35mm glass-bottom dishes (Warner Instruments).
3. Results
3.1. Autofluorescence and SHG in acute brain slices of AD mouse models
Acute hippocampal slices from transgenic AD mouse models show a distinct pattern of autofluorescence and SHG emissions (Fig. 1). Four sources of emissions can be identified by morphology: senile plaques, lipofuscins, microtubules, and blood vessels. Senile plaques appear as spherical objects with diameter ~30–70μm that emit a diffuse autofluorescence. Lipofuscin is small, bright, and densely packed along the pyramidal cell layers. Microtubules are only visible in the SHG channel and are most visible in regions adjacent to the cell layers [7, 8]. Blood vessels are sparse and tend to branch with Y-shaped junctions [8]. When comparing AD mouse models to wild-type mice, autofluorescence from senile plaques is unique to the transgenic models.
Fig. 1.

Autofluorescence and second harmonic emissions from acute hippocampal brain slice of transgenic Alzheimer’s disease mouse models. (a) Autofluorescence, (b) second harmonic emissions, (c) and their overlay of the hippocampus of a 17-month old APPSwe/TauJNPL3 mouse. Senile plaques emit autofluorescence (white arrows) that were morphologically distinct from other sources of intrinsic emissions and were missing from wild-type animals, as shown in (d). Each panel is a mosaic of z-projections of a 50μm thick image stack acquired in 10μm steps. Multiphoton excitation wavelength = 774nm, circular polarization.
Previous studies have reported senile plaque autofluorescence in thin tissue slices, including from both native and fixed “smear preparations” from human brain [10] or 10–20μm-thick sections prepared for immunohistochemistry [9, 11]. Here we found autofluorescence from senile plaques in 400μm-thick native brain slices, a much thicker sample, by using multiphoton microscopy, which is more suited for imaging in highly scattering tissues [13]. We used acute slices that were prepared fresh from transgenic animal models, perfused continuously with oxygenated ringer, and used within ~5 hrs; therefore, this preparation represents an ex vivo tissue model that most accurately resembles in vivo tissues. Because acute slices preserve healthy neurons, they are the standard ex vivo preparation for experiments such as patch-clamp recordings of neurons [18]. Intrinsically fluorescent senile plaques were observed in acute slices from all transgenic mouse models examined. Moreover, this type of autofluorescence can be observed in acute slices >100μm below the surface.
To confirm the identity of the intrinsically fluorescent structures that morphologically resemble senile plaques, after imaging we fixed the brain slices and stained with plaque-specific dyes Thioflavin-S or BTA-1. Figure 2 compares unstained and stained plaque-burdened regions to show that the autofluorescence originates from senile plaques. Interestingly, a small amount of SHG emission was also seen to be generated by senile plaques.
Fig. 2.
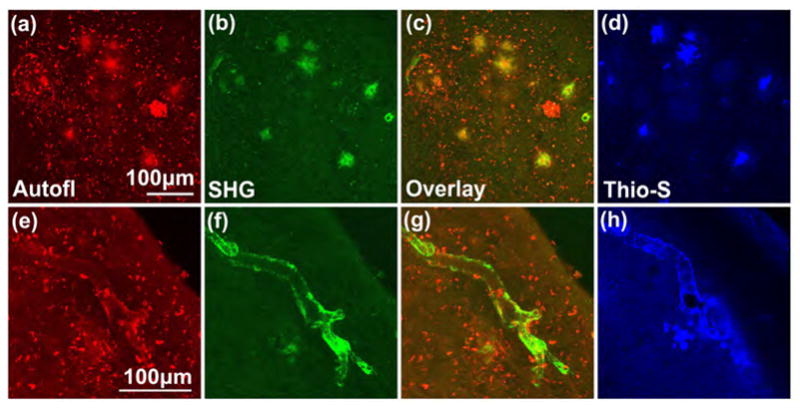
Senile plaques emit autofluorescence and second harmonic signal. (a) Autofluorescence and (b) second harmonic emissions detected in the entorhinal cortex in acute slices of a 22-month old APPSwe/TauJNPL3 mouse. (c) Overlay of (a) and (b) shows the same large, round structures (appears as yellow in the overlay) emit both the autofluorescence and second harmonic signals, which were then identified to be senile plaques when (d) this brain slice was subsequently fixed and stained with the plaque-specific dye Thioflavin-S. The small, bright specks were lipofuscins, which did not generate second harmonic emission. (e-h) Autofluorescence and second harmonic signal, of unknown molecular origin, were also detected near a blood vessel that was affected by cerebral amyloid angiopathy. Signal could be emitted from a source similar to that in senile plaque or from collagen. Each panel is a z-projection of a 40–60μm thick image stack acquired in 3 or 4μm steps. Multiphoton excitation wavelength = 774nm, circular polarization.
The number density of intrinsically fluorescent senile plaques is highly variable between different mice, possibly due to the variable stages of disease progression. All mice showed substantial numbers of intrinsically fluorescent senile plaques in the entorhinal cortex and a lesser amount in the neocortex. Senile plaques tend to be sparse or absent in area CA1 in the hippocampus, although a fair number of plaques couldbe seen in an 18-month-old APPSwe2756/PS1 mouse that was probably in an advanced disease state.
We tested whether our preparation methods could affect the quality of autofluorescence. Senile plaque autofluorescence was seen in slices that were imaged immediately following dissection, i.e. without the 1-hour incubation period. Moreover, the autofluorescence persists after overnight fixation in formaldehyde.
Lipofuscins appeared as small, bright specks in all areas of the brain, but were especially numerous along the CA1 and CA3 layers in the hippocampus. We compared the number density of lipofuscins between transgenic and wild-type mice at similar ages, but found no statistically significant differences (data not shown), which agrees with a published study [19]. Finally, our group has previously reported [3] that NFTs in human AD brain tissues are intrinsically fluorescent, but we have not been able to find such examples in our APPSwe/PS1/Tau transgenic models that have been reported to contain NFTs [17]. The lack of autofluorescence here does not necessarily imply a negative result: the mice examined in this work could be at relatively early stages of disease, or they may lack particular tangle pathologies when compared to human tissues. These ideas are addressed in detail in the Discussions section.
3.2. Emission spectrum of autofluorescence from individual senile plaques
The emission spectrum of senile plaque autofluorescence has not been determined before, presumably because the structures are relatively small and bulk tissues contain other sources of intrinsic emissions. We overcome these problems by relying on multiphoton microscopy’s micron-scale axial and lateral spatial resolution, which enable us to scan a single plaque and obtain a clean emission spectrum of the autofluorescence (Fig. 3). The emission spectrum is broad and peaks at ~525nm. This measurement agrees with general statements about “blue autofluorescence” from previous reports [9–11]. Qualitatively, we observed that the senile plaque autofluorescence can be multiphoton-excited from ~720–860nm. Moreover, a second emission peak can be seen at exactly half of the excitation wavelength. The wavelength and the narrow width imply that this second emission peak is due to SHG. The second harmonic nature of this emission peak was confirmed by a corresponding blue shift when the excitation wavelength was lowered.
Fig. 3.
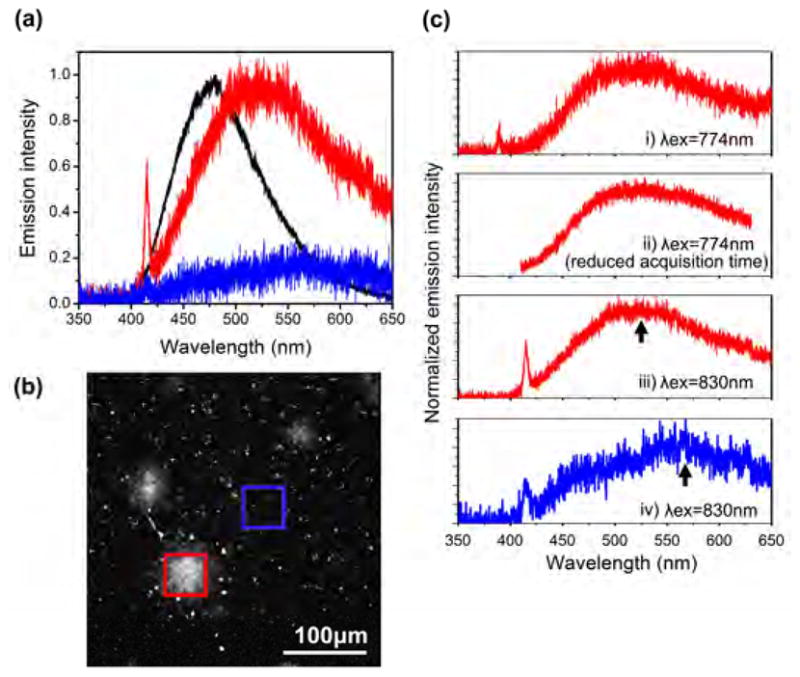
Typical emission spectra of senile plaque autofluorescence. (a) The emission spectra of a senile plaque (red trace) and of an adjacent plaque-free region (blue trace) were measured 50μm beneath the surface of an acute slice from an 18-month-old APPSwe mouse. A typical emission spectrum of the senile plaque autofluorescence, excited at 830nm, peaked at ~520nm and showed detectable SHG emissions at 415nm. After acquiring the spectra, this brain slice was fixed and stained with Thioflavin-S. Emission spectrum of one Thioflavin-S-stained senile plaque, obtained at ~10 times less power and 10 times less acquisition time as the autofluorescence, is shown (black trace, normalized) for comparison. (b) The region in the neocortex of the acute slice where the spectra (red and blue boxes) were taken for (a). (c) The emission spectra for four different conditions recorded from the same mouse: i) emission spectrum at senile plaque, excited at 774nm; ii) emission spectrum at a different senile plaque, excited at 774nm, but with quick acquisition to reduce photodamage; iii) emission spectrum at another senile plaque, excited at 830nm; iv) emission spectrum at a plaque-less location, dominated by autofluorescence, presumably from lipofuscin because of the high emission wavelength. Second harmonic peak in iv) is exaggerated because emission intensity was re-scaled. The actual recorded second harmonic peak was weak, see panel (a), and it may be extraneous signal generated when the spectrometer gratings diffract the intense excitation beam. Arrows indicate the locations of the peak emission wavelengths for the autofluorescence.
In an adjacent region with no plaque, the detected emission is significantly weaker and seems to contain multiple broad peaks. It is likely that lipofuscins contribute to emissions at higher wavelengths and possibly NAD(P)H or flavins contribute to emissions below 500nm (Cf. [4]). Comparing the spectra obtained at senile plaques and from adjacent regions showed that autofluorescence can be distinguished from the background by the emission spectrum. We also obtained the emission spectra of senile plaques that were stained with Thioflavin-S following formaldehyde fixation. The Thioflavin-S fluorescence has a relatively narrow spectrum that peaked at ~480nm, which is distinct from the autofluorescence emission obtained from unstained brain slices. Thioflavin-S fluorescence in stained brain slices was significantly brighter than the autofluorescence in unstained brain slices. There was significant difference between the peak emission wavelength and spectral shape of the emissions recorded at plaques from Thioflavin-S-stained and unstained brain slices. This difference confirms that the autofluorescence in unstained brain slices is not fluorescence resulting from Thioflavin-S contamination.
3.3. Functional imaging of intrinsic emissions in AD mouse models
To demonstrate the utility of intrinsic emissions, we imaged SHG emissions from polarized microtubules and autofluorescence from senile plaques to investigate whether the polarity or the morphology of microtubule arrays are affected in AD mouse models. Tau, a microtubule-associated protein, has been implicated as a possible cause for neurodegeneration [20]. Electron microscopy studies have shown that tau can accumulate in postsynaptic locations [21, 22], and therefore can possibly affect microtubule organization in the dendrites. In neuronal culture models of AD, manipulation of tau expressions or mutations show that microtubules can be depolymerized [23, 24], or be forming a star-shaped aster array after reversing polarity [25]. Such modifications to the microtubule arrays as formation of a star-shaped structure should drastically alter morphology and be directly observable by our imaging technique because SHG emission is a good qualitative indicator of the polarity and number density of microtubules in single neurites [7, 8].
We initially tested whether it is possible to observe SHG emissions from microtubules near senile plaques stained with Thioflavin-S or BTA-1. We found that even a small amount of plaque-specific dye would emit too much fluorescence, and therefore obscures intrinsic emissions. Therefore, in all of the following imaging experiments, we chose to use unstained brain slices. In unstained acute slices, we were able to image simultaneously intrinsic emissions from polarized microtubules in apical dendrites in area CA1 and senile plaques (Fig. 4). We measured the length and number density of polarized microtubules in this region, using a previously described procedure [8]. We found that the length of polarized microtubule arrays is 77±13μm (n=6 transgenic AD models) versus 89±9μm (n=4 wild-types) and the number density is 0.053±0.010 m−2 versus 0.062±0.011μm−2. These differences are not statistically significant, although our sample size is small. Interestingly, the three mice that showed the shortest microtubule arrays were the mice with the triple APPSwe2756/PS1/Tau mutations. However, those same mice were also older than most of the mice examined and there were only data from one wild-type littermate of similar age.
Fig. 4.
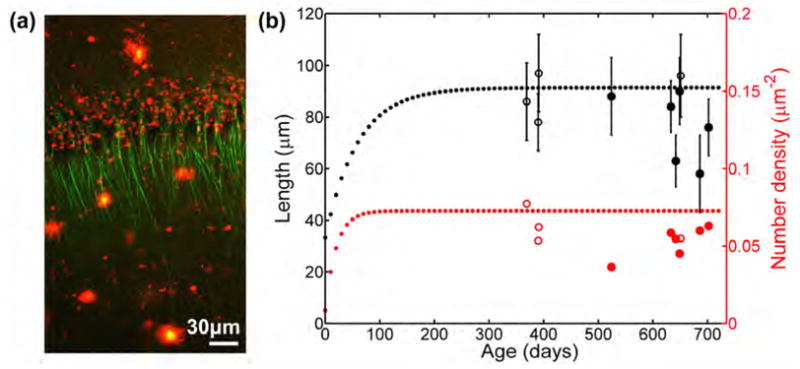
Length and number density of polarized microtubule arrays in area CA1 in Alzheimer’s disease mouse models. (a) Typical autofluorescence (red) and second harmonic emissions (green) in area CA1 from an acute slice of a 21-month-old APPSwe/PS1 mouse. This image is a z-projection of an 18μm thick image stack acquired in 3μm steps. Multiphoton excitation wavelength = 774nm, linear polarization. (b) The length and number density of polarized microtubules in area CA1 of Alzheimer’s disease mouse models (solid dots) and of wild-type mice studied that were over 1 year old (hollow dots). The dotted lines show the trend based on data from a larger set of wild-type mice from our previous study [8].
When focusing on a single senile plaque, we can resolve the polarized microtubule arrays within individual apical dendrites of CA1 pyramidal neurons (Fig. 5). There have previously been studies [26–29] on the morphology of axons and dendrites near a senile plaque. Reported abnormal features in neurite morphology include loss of dendritic spines [26–28], shaft atrophy [28], varicosity and sprouting [27, 28]. Axons and dendrites seem to be affected differently such that axons frequently have varicosities, which are not seen as often in dendrites [28]. Moreover, there have been conflicting observations as to whether the length and diameter of dendrite shafts are affected [27, 29]. One common theme is that the amount of abnormal features tends to increase dramatically within a ~15μm-diameter region around the senile plaque [26, 27].
Fig. 5.

Polarized microtubules in apical dendrites near a senile plaque. A series of images shows second harmonic emissions from polarized microtubules of apical dendrites (green) near an autofluorescent senile plaque (red). The z value denotes the distance to the slice surface. The senile plaque was located in the area CA1 in the hippocampus in an acute slice from a 17-month old APPSwe/TauJNPL3 mouse. Multiphoton excitation wavelength = 774nm, circular polarization.
Our observations found that SHG emissions from polarized microtubule arrays in apical dendrites are mostly unaffected by the presence of a senile plaque. We did not find polarized microtubules that traverse through the central core of the plaque, but most apical dendrites were unaffected at the periphery of the autofluorescence. In contrast, unhealthy dendrites, with a zigzag trajectory, could be found near the tissue surface due to damage during sample preparation (z=−20μm in Fig. 5). We attempted to look at SHG emissions from axonal microtubules, which have been observed before in the mossy fiber [7, 8], but in all of our experiments, we failed to find a senile plaque in that region. Moreover, we found no significant difference in the intensity of dendritic SHG emissions near or far from the senile plaques. Taken all together, our observations suggest that the polarity and the morphology of dendritic microtubule arrays do not seem to be affected adversely in these transgenic AD mouse models. However, apical dendrites are relatively thick (~3μm in diameter), so they may be more tolerant to the kind of damages that were seen in previous studies.
4. Discussion
We have shown that autofluorescence and SHG emissions can be excited and imaged by multiphoton microscopy in acute brain slices of transgenic AD mouse models. More specifically, we confirmed that senile plaques exhibit autofluorescence with a distinct emission spectrum and also weakly generate SHG. This autofluorescence was seen in all four of the transgenic mouse models examined; therefore suggesting this type of intrinsic emission is a general property. This ability to identify senile plaques by their autofluorescence enables an array of possible functional studies using other intrinsic emissions, which we demonstrated by imaging SHG from dendritic microtubule arrays near senile plaques.
Previous studies that use multiphoton microscopy in AD mouse models focus on imaging dye-stained senile plaques. In a pioneering study [30], Christie et al. developed a thinned skull preparation in mouse that allows for imaging of senile plaques over several months. This success was followed by other in vivo time-lapse studies, which use other complementary techniques such as neurite tracing [26, 27], calcium imaging [31], astrocyte stains [32], or direct application of drugs and antibodies [33] to obtain additional insights on the disease. Here, we showed that weaker intrinsic emissions can also be imaged effectively in AD mouse tissues. This opens the door for new approaches for functional imaging of other intrinsic emissions, such as NAD(P)H metabolic imaging, to be used to study the pathological mechanisms of AD.
Although not demonstrated here, it may be possible to use autofluorescence as a diagnostic tool for in vivo studies in transgenic mouse models. Our study shows that autofluorescence from senile plaques is weak but clearly detectable and has a characteristic multiphoton-excited emission spectrum. One possible diagnostic method deep within the brain is to use gradient-index (GRIN) lenses [34, 35] or optical fiber bundles [36] to excite and collect autofluorescence from a small volume within the brain. Imaging may not be required, since spectral information will be the key for distinguishing AD versus normal tissues.
In our imaging-only results, senile plaques and lipofuscin have broad emission spectra so we distinguish them by morphology. In terms of size, senile plaques have diameters ~10 times larger than lipofuscin. This size difference can be clearly resolved in multiphoton microscopy images, which have submicron spatial resolution within scattering tissues. In some cases, lipofuscin can “clump” together to form larger structures. However, in those cases, multiphoton microscopy images can still clearly resolve individual, smaller-diameter lipofuscins (see Fig. 4(a) in [4]). On the other hand, it is unlikely to find small, nascent plaques. Plaques grow quickly in vivo within 1–3 days to their full size [30, 37]. Therefore, finding nascent plaques is a rare occurrence even in animal models with significant plaque load [37]. Although morphology may be a good correlate, identification of senile plaques versus other autofluorescent materials would be more accurate if spectral information were obtained.
We have characterized the emission spectrum of senile plaque autofluorescence. Previous studies [9–11] have used excitation and detection at a variety of wavelengths, so knowledge of the emission spectrum will be useful for more sensitive detection. As a quick test to find the molecular origin of this autofluorescence, we tested solutions of A-beta fibrils, in which fibrils were verified with transmission electron microscopy, and saw no one-photon-excited fluorescence in a standard fluorimeter. This initial observation suggests that senile plaque autofluorescence is unlikely to originate from A-beta, which is the principal component of senile plaques [38]. Many other materials are known to exist within senile plaque [38] and can contribute to the intrinsic emission.
Autofluorescence from NFTs has been described before in post-mortem human tissues [4], but was not observed in the APPSwe/PS1/Tau models in this study. A previous report [16] has shown that these mice are capable of generating Thioflavin-S-positive tau-related lesions. The absence of autofluorescence from NFTs in our work could be due to two reasons: One, the previous report [16] observed Thioflavin-S-positive lesions in 12-month old homozygous mice, which may be at a more advanced stage of AD than our ~20-month old heterozygous mice. This argument is supported by the observation that Thioflavin-S did not stain any structures that resemble tau-related lesions in our acute slices. Two, it is possible that the only types of human tau pathology that exhibit autofluorescence have no analogs in the APPSwe/PS1/Tau transgenic mouse model. To distinguish between these possibilities, more research would be required on characterizing autofluorescence and immunoreactivity from various kinds of tau-related lesions in human and transgenic mouse model tissues.
Intrinsic emissions such as autofluorescence and SHG are useful indicators for detecting the presence of pathological lesions. Changes in the intensity and spectrum of intrinsic emissions have previously been studied as possible diagnosis methods for tumors [39] or skin pathology [40], and can potentially also be useful for neurodegenerative diseases. Here we have demonstrated that intrinsic emissions, particularly that of senile plaques, can be detected from relatively thick, native tissues. Furthermore, the senile plaque emission spectrum is distinct from the background to possibly further enhance detection sensitivity. We anticipate that this work will be useful for interpreting future studies that aim to use endogenous optical signals as a diagnostic tool or as functional fluorescent indicators.
Acknowledgments
We would like to thank Huizhong Xu for help on the spectrometer, Valerie Anderson for amyloid fibrils and Mark Williams for editorial assistance. This research was supported by NIH grant 9-P41-EB001976 for A.C.K. and W.W.W., by NIH grant 1-R21-AG026650 for W.W.W., and by the Nanobiotechnology Center (NBTC), an STC Program of the National Science Foundation under Agreement No. ECS-9876771 for A.C.K.
Footnotes
OCIS codes: (170.0110) Medical optics and biotechnology: Imaging systems; (170.2520) Medical optics and biotechnology: Fluorescence microscopy; (170.3880) Medical optics and biotechnology: Medical and biological imaging; (170.6510) Medical optics and biotechnology: Spectroscopy, tissue diagnostics; (170.6930) Medical optics and biotechnology: Tissue
References and links
- 1.Ikonomovic MD, Klunk WE, Abrahamson EE, Mathis CA, Price JC, Tsopelas ND, Lopresti BJ, Ziolko S, Bi W, Paljug WR, Debnath ML, Hope CE, Isanski BA, Hamilton RL, DeKosky ST. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain. 2008;131:1630–1645. doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hintersteiner M, Enz A, Frey P, Jaton AL, Kinzy W, Kneuer R, Neumann U, Rudin M, Staufenbiel M, Stoeckli M, Wiederhold KH, Gremlich HU. In vivo detection of amyloid-beta deposits by near-infrared imaging using an oxazine-derivative probe. Nat Biotechnol. 2005;23:577–583. doi: 10.1038/nbt1085. [DOI] [PubMed] [Google Scholar]
- 3.Hanlon EB, Perelman LT, Vitkin EI, Greco FA, McKee AC, Kowall NW. Scattering differentiates Alzheimer disease in vitro. Opt Lett. 2008;33:624–626. doi: 10.1364/ol.33.000624. [DOI] [PubMed] [Google Scholar]
- 4.Zipfel WR, Williams RM, Christie R, Nikitin AY, Hyman BT, Webb WW. Live tissue intrinsic emission microscopy using multiphoton-excited native fluorescence and second harmonic generation. Proc Natl Acad Sci. 2003;100:7075–7080. doi: 10.1073/pnas.0832308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kasischke KA, Vishwasrao HD, Fisher PJ, Zipfel WR, Webb WW. Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science. 2004;305:99–103. doi: 10.1126/science.1096485. [DOI] [PubMed] [Google Scholar]
- 6.Vishwasrao HD, Heikal AA, Kasischke KA, Webb WW. Conformational dependence of intracellular NADH on metabolic state revealed by associated fluorescence anisotropy. J Biol Chem. 2005;280:25119–25126. doi: 10.1074/jbc.M502475200. [DOI] [PubMed] [Google Scholar]
- 7.Dombeck DA, Kasischke KA, Vishwasrao HD, Ingelsson M, Hyman BT, Webb WW. Uniform polarity microtubule assemblies imaged in native brain tissue by second-harmonic generation microscopy. Proc Natl Acad Sci. 2003;100:7081–7086. doi: 10.1073/pnas.0731953100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kwan AC, Dombeck DA, Webb WW. Polarized microtubule arrays in apical dendrites and axons. Proc Natl Acad Sci. 2008;105:11370–11375. doi: 10.1073/pnas.0805199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dowson JH. A sensitive method for demonstration of senile plaques in the dementing brain. Histopathology. 1981;5:305–310. doi: 10.1111/j.1365-2559.1981.tb01789.x. [DOI] [PubMed] [Google Scholar]
- 10.Thal DR, Ghebremedhin E, Haass C, Schultz C. UV light-induced autofluorescence of full-length abeta-protein deposits in the human brain. Clin Neuropathol. 2002;21:35–40. [PubMed] [Google Scholar]
- 11.Diez M, Koistinaho J, Kahn K, Games D, Hökfelt T. Neuropeptides in hippocampus and cortex in transgenic mice overexpressing V717F beta-amyloid precursor protein – initial observations. Neuroscience. 2003;100:259–286. doi: 10.1016/s0306-4522(00)00261-x. [DOI] [PubMed] [Google Scholar]
- 12.Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248:73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- 13.Zipfel WR, Williams RM, Webb WW. Nonlinear magic: multiphoton microscopy in the biosciences. Nat Biotechnol. 2003;21:1369–1377. doi: 10.1038/nbt899. [DOI] [PubMed] [Google Scholar]
- 14.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 15.Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- 16.Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 17.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 18.Sakmann B, Stuart G. Patch-pipette recordings from the soma, dendrites and, and axon of neurons in brain slices. In: Sakmann B, Neher E, editors. Single-Channel Recording. 2. Plenum; 1983. pp. 199–211. [Google Scholar]
- 19.Drach LM, Bohl J, Goebel HH. The lipofuscin content of nerve cells of the inferior olivary nucleus in Alzheimer’s disease. Dementia. 1994;5:234–239. doi: 10.1159/000106729. [DOI] [PubMed] [Google Scholar]
- 20.Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 21.Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002;161:1869–1879. doi: 10.1016/s0002-9440(10)64463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takahashi RH, Capetillo-Zarate E, Lin MT, Milner TA, Gouras GK. Co-occurrence of Alzheimer’s disease beta-amyloid and tau pathologies at synapses. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.07.021. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li B, Chohan MO, Grundke-Iqbal I, Iqbal K. Disruption of microtubule network by Alzheimer abnormally hyperphosphorylated tau. Acta Neuropathol. 2007;113:501–511. doi: 10.1007/s00401-007-0207-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qiang L, Yu W, Andreadis A, Luo M, Baas PW. Tau protects microtubules in the axon from severing by katanin. J Neurosci. 2006;26:3120–3129. doi: 10.1523/JNEUROSCI.5392-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shemesh OA, Erez H, Ginzburg I, Spira ME. Tau-induced traffic jam reflect organelles accumulation at points of microtubule polar mismatching. Traffic. 2008;9:458–471. doi: 10.1111/j.1600-0854.2007.00695.x. [DOI] [PubMed] [Google Scholar]
- 26.Spires TL, Meyer-Leuhmann M, Stern EA, McLean PJ, Skoch J, Nguyen PT, Bacskai BJ, Hyman BT. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005;25:7278–7287. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsai J, Grutzendler J, Duff K, Gan WB. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal brances. Nat Neurosci. 2004;7:1181–1183. doi: 10.1038/nn1335. [DOI] [PubMed] [Google Scholar]
- 28.Grutzendler J, Helmin K, Tsai J, Gan WB. Various dendritic abnormalities are associated with fibrillar amyloid deposits in Alzheimer’s disease. Ann N Y Acad Sci. 2007;1097:30–39. doi: 10.1196/annals.1379.003. [DOI] [PubMed] [Google Scholar]
- 29.Alpár A, Ueberham U, Brückner MK, Seeger G, Arendt T, Gärtner U. Different dendrite and dendritic spine alterations in basal and apical arbors in mutant human amyloid precursor protein transgenic mice. Brain Res. 2006;1099:189–198. doi: 10.1016/j.brainres.2006.04.109. [DOI] [PubMed] [Google Scholar]
- 30.Christie RH, Bacskai BJ, Zipfel WR, Williams RM, Kajdasz ST, Webb WW, Hyman BT. Growth arrest of individual senile plaques in a model of Alzheimer’s disease observed by in vivo multiphoton microscopy. J Neurosci. 2001;21:858–864. doi: 10.1523/JNEUROSCI.21-03-00858.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eichhoff G, Busche MA, Garaschuk O. In vivo calcium imaging of the aging and diseased brain. Eur J Nucl Med Mol Imaging. 2008;35:S99–S106. doi: 10.1007/s00259-007-0709-6. [DOI] [PubMed] [Google Scholar]
- 32.Takano T, Han X, Deane R, Zlokovic B, Nedergaard M. Two-photon imaging of astrocytic Ca2+ signaling and the microvasculature in experimental mice models of Alzheimer’s disease. Ann NY Acad Sci. 2007;1097:40–50. doi: 10.1196/annals.1379.004. [DOI] [PubMed] [Google Scholar]
- 33.Bacskai BJ, Kajdasz ST, Christie RH, Carter C, Games D, Seubert P, Schenk D, Hyman BT. Imaging of amyloid-beta deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat Med. 2001;7:369–372. doi: 10.1038/85525. [DOI] [PubMed] [Google Scholar]
- 34.Levene MJ, Dombeck DA, Kasischke KA, Molloy RP, Webb WW. In vivo multiphoton microscopy of deep brain tissue. J Neurophysiol. 2004;91:1908–1912. doi: 10.1152/jn.01007.2003. [DOI] [PubMed] [Google Scholar]
- 35.Jung JC, Schnitzer MJ. Multiphoton endoscopy. Opt Lett. 2003;28:902–904. doi: 10.1364/ol.28.000902. [DOI] [PubMed] [Google Scholar]
- 36.Göbel W, Kerr JN, Nimmerjahn A, Helmchen F. Miniaturized two-photon microscope based on a flexible coherent fiber and a gradient-index lens objective. Opt Lett. 2004;29:2521–2523. doi: 10.1364/ol.29.002521. [DOI] [PubMed] [Google Scholar]
- 37.Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature. 2008;451:720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Atwood CS, Martins RN, Smith MA, Perry G. Senile plaque composition and posttranslational modification of amyloid-beta peptide and associated proteins. Peptides. 2002;23:1343–1350. doi: 10.1016/s0196-9781(02)00070-0. [DOI] [PubMed] [Google Scholar]
- 39.Zhu C, Breslin TM, Harter J, Ramanujam N. Model based and empirical spectral analysis for the diagnosis of breast cancer. Opt Express. 2008;16:14961–14978. doi: 10.1364/oe.16.014961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Palero JA, de Bruijn HS, van der Ploeg-van den Heuvel A, Sterenborg HJCM, Gerritsen HC. In vivo nonlinear spectral imaging in mouse skin. Opt Express. 2006;14:4395–4402. doi: 10.1364/oe.14.004395. [DOI] [PubMed] [Google Scholar]