

Abstract
A widened DNA base pair architecture is studied in an effort to explore the possibility of whether new genetic system designs might possess some of the functions of natural DNA. In the “yDNA” system, pairs are homologated by addition of a benzene ring, yielding (in the present study), benzopyrimidines that are correctly paired with purines. Here we report initial tests of ability of the benzopyrimidines yT and yC to store and transfer biochemical and biological information in vitro and in bacterial cells. In vitro primer extension studies with two polymerases showed that the enzymes could insert the correct nucleotides opposite these yDNA bases, but with low selectivity. PCR amplifications with a thermostable polymerase resulted in correct pairings in 15–20% of the cases, and more successfully when yT or yC were situated within the primers. Segments of DNA containing one or two yDNA bases were then ligated into a plasmid and tested for their ability to successfully lead the expression of an active protein in vivo. Although active at only a fraction of the activity of fully natural DNA, the unnatural bases encoded the correct codon bases in the majority of cases when singly substituted, yielding functioning green fluorescent protein. Although the activities with native polymerases are modest with these large base pairs, this is the first example of encoding protein in vivo by an unnatural DNA base pair architecture.
Keywords: Wide DNA, benzopyrimidine, PCR, Vent polymerase, replication, mispair, wobble
Introduction
A long-standing challenge for chemists inspired by natural systems has been to find new chemical designs for DNA base pairs.[1–6] One goal of such work is to evaluate whether such designs can mimic natural physical properties of DNA such as helix formation. Fewer studies have been directed to biochemical and biological properties of such new pairs. However, some unnatural base pairs have recently been shown to be replicated by polymerase enzymes in vitro,[2,7–12] suggesting future applications in expansion of the genetic code.[13]
Until recently, designed base pairs have been constructed exclusively for function in the context of the natural DNA genetic system. However, one can ask whether other genetic systems could exist that operate with an entirely different base pairing architecture from that of natural DNA. In this light we recently reported the design and synthesis of size-expanded nucleosides (xDNA and yDNA) capable of forming base pairs and helices larger than those of natural DNA.[14–16] In these designs, the dimension of the natural nucleobases was expanded by 2.4 Å by addition of a benzene ring to the natural heterocyclic framework.[17] While the yDNA analogues studied here (Figure 1) differ from the xDNA analogues in the extension vector orientation, both could be synthetically incorporated into oligonucleotide strands and form stable expanded double helices. These expanded nucleotide analogues may serve as useful tools in better understanding the self assembly and replication of DNA, and its recognition by proteins. They also have the potential to lead to new applications in biotechnology as a result of their ability to bind DNA with high affinity,[14] and the fact that all the expanded bases are fluorescent.[18]
Figure 1.
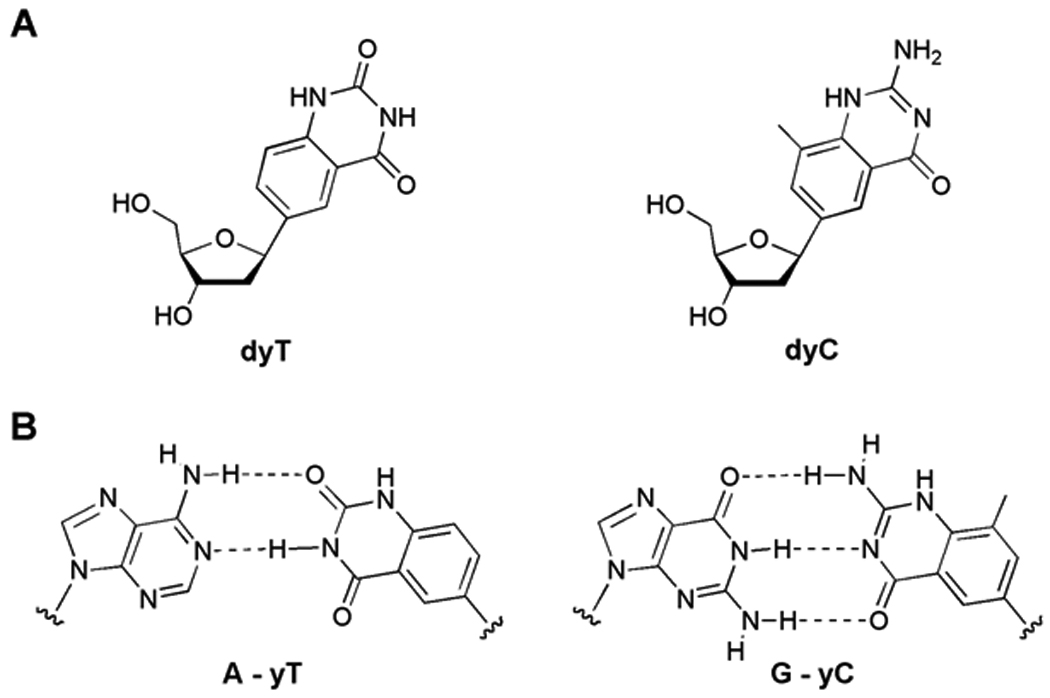
Widened (yDNA) nucleoside and base pair structures. (A) Structures of benzopyrimidine nucleosides dyT and dyC. (B) Proposed structures of widened base pairs in this study.
If designed genetic systems are to function like natural ones, they must be able to encode and transfer sequence information to copies. However, this presents a serious challenge for the widened yDNA pairs, because DNA polymerases can be highly sensitive to the sizes of base pairs being synthesized.[19] While our long-term goal is to discover and develop new enzyme variants that can adapt to the large size of expanded pairs, we have begun to examine whether naturally occurring enzymes and replication systems have any ability to process the information encoded by expanded pairs. Here we report on information encoding in the yDNA system. We find that although such pairs are processed with relatively low efficiency and fidelity in vitro, native polymerases can in fact replicate such pairs successfully in some cases. Moreover, we demonstrate that single yDNA bases can correctly encode genetic information in living cells.
Results and Discussion
Our initial experiments explored the ability of the benzopyrimidine C-nucleosides[15b] dyT and dyC (Figure 1A) to template the addition of natural nucleotides opposite them, using native polymerases. We studied the Klenow fragment of E. coli DNA polymerase I (KF exo−), and a commonly used thermostable polymerase, the DNA polymerase from Thermococcus litoralis (Vent exo−)[20] to recognize dyT and dyC bases in a synthetic 28mer DNA template. If insertion of dATP and dGTP (respectively) were successful, the resulting A-yT and G-yC pairs would be ca. 2.4 Å larger than natural Watson-Crick pairs. The single nucleotide insertion studies were performed under non-forcing conditions such that inefficiently formed base pairs, such as mismatches, would not be formed in the case of the natural base system. The data showed (Fig. 2) that both the polymerases tested incorporated the natural nucleotides of dA opposite yT and dG opposite yC with qualitatively moderate efficiencies. The selectivity with both enzymes was low, as dTTP was also incorporated across from both yC and yT with comparable efficiencies. We hypothesize that this T-yT and T-yC mispairing is due to a wobble-type pair geometry (Fig. 3). Interestingly, such a T-yT pair is reasonably close in size and orientation to a natural A-T pair. A similar structure can be drawn with a possible tautomer of yC.[21,22]
Figure 2.
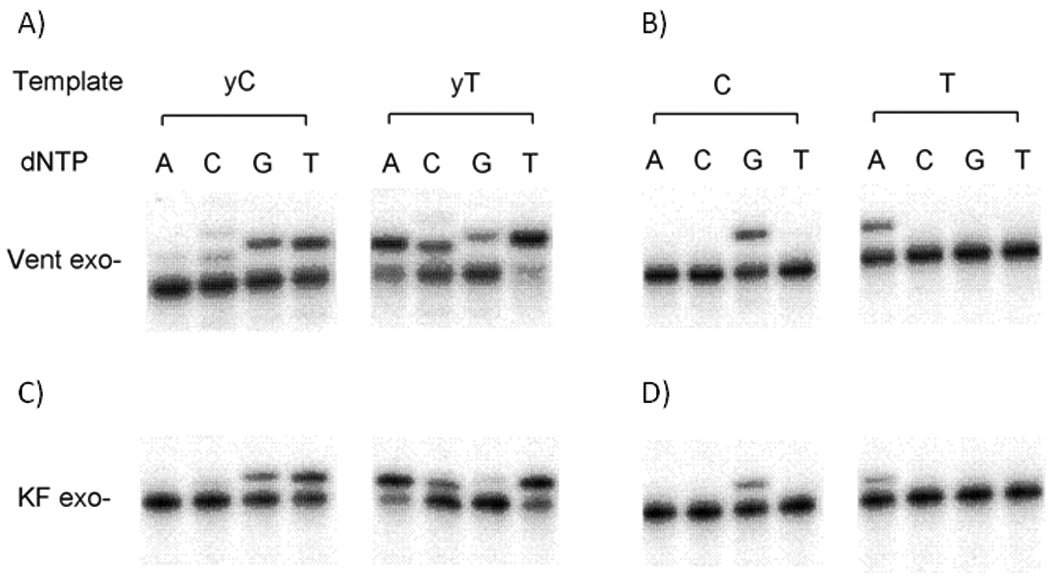
Survey of selectivity of enzymatic nucleotide incorporation opposite yDNA bases. A and C show yDNA template bases; B and D show natural bases as controls. Enzymes are Thermococcus litoralis DNA polymerase (Vent exo−) and Klenow fragment of DNA pol I (KF exo−). Standard 10 µL reactions were performed at 37 °C (KF exo−) or 68 °C (Vent exo−) and contained 25 nM radiolabeled primer-template (primer, 5'-TAA TAC AAC TCA CTA TAG GGA GA-3'; template, 5'-ACT GXT CTC CCT ATA GTG AGT CGT ATT A-3', where X=yT,T,yC, or C). (A) Reaction solution contained 0.4 unit/µL Vent exo−, 500 µM dNTP; reaction time 20 min. (B) Reaction contained 0.05 unit/µL Vent exo−, 50 µM dNTP; reaction time 3 min. (C) Reaction contained 0.02 unit/µL KF exo−, 50 µM dNTP; reaction time 4 min. (D) Reaction containe2 0.005 unit/µL KF exo−,5 µM dNTP; reaction time 3 min. Products were resolved by denaturing polyacrylamide gel electrophoresis (7.6 M urea, 20% acrylamide) and visualized by phosphorimaging.
Figure 3.
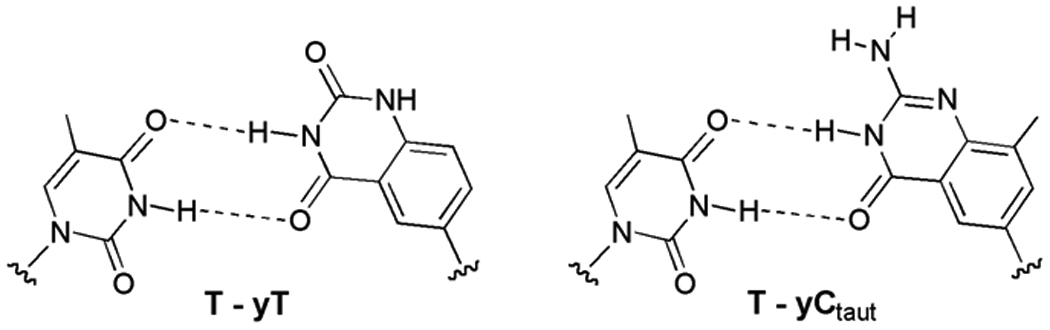
Proposed structures of mismatched T-yT and T-yC wobble-type pairs. The similarity in geometry to standard Watson-Crick pairs may explain the common T-yT and T-yC mispairing observed in vitro.
Encouraged by the ability of the thermostable Vent exo− polymerase to incorporate nucleotides opposite the expanded bases in a DNA template, we next explored the fidelity of this activity in a PCR-based assay. Two oligonucleotides were synthesized and annealed, each containing two modifications (one dyT and one dyC), and having a 16nt region of overlap at their 3’ ends (Table 1). PCR was performed using this template and natural dNTPs. Note that the product after amplification in these reactions will be natural DNA, since no modified nucleotides were used in the PCR. The resulting 79 base pair (bp) PCR products were directly cloned into a PCR cloning vector, and transformed into E. coli. DNA was then extracted from a small population of the transformed clones, and the resulting sequences analyzed. The obtained sequences (Table 1) provide information regarding the fidelity of replication of yT and yC bases in the initial template DNA. We observed that in a majority of the clones, the obtained sequence corresponded to the misincorporation of dT opposite both yC and yT in the template, consistent with the above single nucleotide incorporation experiments. The expanded pairs were formed relatively infrequently, with yC being read correctly (i.e., incorporation of G) in 15% of clones at one of the two modified positions and similarly, yT being read correctly (incorporation of A) in 20% of clones at one of the two positions. Any other mispairings were rare.
Table 1.
PCR amplification of templates containing yT and yC bases with natural dNTPs
 |
Observed sequences were obtained after bacterial cloning and sequencing of amplified duplexes. The yDNA template bases are underlined. Expected bases replacing these are shown in blue; mutated results are shown in red. Represented percentages of sequences cloned are as shown.
To test yDNA base pair replication in a second context, we next performed PCR using primers that each contained two yDNA bases. The modified primers were designed such that the y-bases were incorporated towards the 5’ end, so that their ability to anneal with the template would not be compromised (Table 2). In these experiments, the replication is more challenging since the polymerase would have to continually use DNA containing yDNA bases as the template during each step of PCR. As in the previous assay, the PCR products were directly cloned and sequenced. We observed again that in the region downstream from the primer, the observed sequence corresponded to the misincorporation of T opposite both yC and yT in the template in the majority of cases. “Correct” incorporation of A opposite yT occurred in 20% of cases on average (10% at one position and 30% at the second position), and of G opposite yC, in 10% of cases on average (15% at one position and 5% at the second position) (Table 2). However, in the primer binding regions, we observed far fewer misincorporations. All cases tested (at 2 sites in each of 20 clones) showed correct replacement of yT by T, indicating A-yT pairing. Similarly, yC was replaced by C on an average in 88% of the cases (75% at one position, 100% at the second position. In this experiment, it should be noted that the products from each PCR cycle are modified DNAs containing yC and yT modifications near the ends. This could potentially pose an additional difficulty for ligation and cloning into the TA vector. Thus in this experiment we cannot rule out some possible bias of the bacterial replication machinery during plasmid replication for certain sequence outcomes over others, especially near the ends of the amplified duplexes. Nevertheless, the results show that a native polymerase can read and replicate the information stored by yDNA pairs to a substantial extent.
Table 2.
PCR amplification with both primers and templates containing yT and yC bases (using natural dNTPs).
 |
The yDNA template bases are underlined. Expected bases replacing these are shown in blue; mutated results are shown in red. Represented percentages of sequences cloned are as shown. Template sequence is given in Table 1. Observed sequences were obtained after bacterial cloning and sequencing of amplified duplexes.
The above experiments demonstrated that small numbers of yDNA base pairs can be amplified in vitro and replicated in vivo. However, the cloned segments were in noncoding parts of the DNA, and the yDNA bases were processed prior to reaching cells. Thus the question remained as to whether yDNA bases could encode genetic information (as observed by a phenotype) to any extent in a living system. To directly probe this question, we examined the in vivo replication of synthetic DNAs containing yDNA bases without amplification. A 46 base pair segment within the green fluorescent protein (GFP) gene in pGFPuv plasmid vector, a plasmid with a high copy number of expression in cells, (see Experimental methods) was replaced with an identical synthetically prepared DNA containing yDNA modifications (Table 3). Two oligonucleotides, each containing increasing numbers of yT and yC bases within the gene segment, were synthesized such that they have complementary sequence, and upon hybridization, yield a double stranded DNA fragment with sticky ends similar to those generated by two restriction enzymes on the plasmid vector. Genetically silent mutations were also introduced within the synthetic gene to distinguish the input unnatural DNA from possible unmodified vector contamination. These “artificial gene” segments were cloned into the vector, replacing the original sequence, and then transformed into E. coli. Here, only if the bacterial DNA replication machinery is capable of correctly propagating the genetic information provided in the form of modified DNA content, would it form colonies with green fluorescence on an LB plate. Note that the bacteria only needs to make a few faithful copies of the modified plasmid input initially to foster further natural plasmid replication. A high copy number yielding plasmid would also help maintain the continued replication as compared with a low copy number plasmid.
Table 3.
In vivo replication of gene segments containing yDNA base pairs.
 |
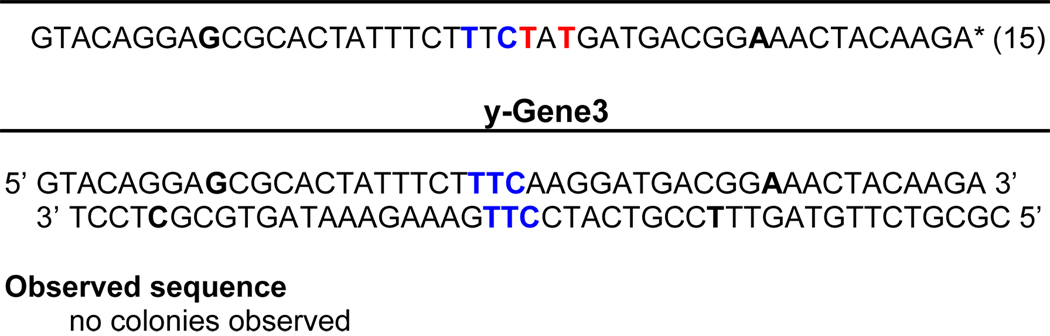 |
The yDNA bases are underlined in the synthetic GFP gene segments. Expected bases replacing these are shown in blue; mutated results are shown in red. Percentages of clones are given in brackets. Observed sequences were obtained after bacterial cloning and sequencing of synthetic duplexes in a green fluorescent protein-encoding plasmid.
Silent mutation sites (used as markers) are bold.
We found that green colonies were in fact obtained when gene segments containing one yDNA base and two yDNA bases in each strand were present in the vector. Transformation efficiency was lowered by ca. 200-fold compared to a control reaction where unmodified DNA was used for ligation and transformation. However, upon sequencing the resulting plasmids extracted from the fluorescent colonies from modified DNAs, we observed that there were many clones where the genetic information was preserved by correct replication of the unnatural pairs. When there were two yDNA modifications in the cloned gene segment, 60% of the sequences obtained had the expected fidelity of replication with dA added opposite yT. The mutational tendency for the misinsertion of T opposite yT was also observed again, with 40% of the clones exhibiting the corresponding mutation (Table 3). In the more stringent case where there were two yDNA modifications in each strand, the sequences obtained were more diverse, with only 15% of the clones showing complete fidelity; a small population also showed a preference for the addition of A opposite yC. Interestingly, no misincorporations were observed while replicating the yDNA modifications in the top strand, thus showing complete fidelity in the clones tested. In the case where there were three consecutive yDNA base modifications in each strand, we did not observe any green transformants, thus indicating the limits of the E. coli replication machinery for yDNA replication.
Conclusion
Overall, our experiments show that despite the large size of yDNA base pairs, DNA partially modified with yDNA bases can be recognized and replicated to some extent in vitro even by natural polymerases. This suggests the future possibility of finding or developing enzymes that are more accepting of the large size of these pairs. More surprisingly, yDNA pairs could be successfully replicated in living bacterial cells to a limited extent, and we demonstrate for the first time that yDNA base pairs can help store and transfer information leading to an observable phenotype. The findings suggest that, with further development of replicases that can better adapt to altered geometries, genetic systems that use unnatural information-encoding architecture may one day be viable.
Experimental Section
Modified oligodeoxynucleotides
Modified nucleoside analogues dyT and dyC were synthesized and characterized following the methods described previously.[21] Suitably protected cyanoethyl phosphoramidite derivatives were prepared following literature procedures.[21] DNA sequences containing modified bases were prepared on an Applied Biosystems 394 instrument following the published methods.[15b] Unmodified oligodeoxynucleotides were purchased from Integrated DNA technologies (IDT), Coralville, IA.
Polymerase reactions
28mer / 23mer template-primer duplexes having the sequence (5′-ACT GXT CTC CCT ATA GTG AGT CGT ATT A) • (5′- TAA TAC GAC TCA CTA TAG GGA GA), where X is dyT, dyC, dT, or dC, were used as polymerase substrates. The enzymes used were the exonuclease-deficient Klenow fragment of DNA polymerase I (Cat. No. M0212L from New England Biolabs) and the exonuclease-deficient Vent DNA polymerase from Thermococcus litoralis (Cat. No. M0257S from New England Biolabs. Primer 5’ termini were radio-labeled by using 5’-[γ-32P]-ATP (Amersham Biosciences) and T4 Polynucleotide Kinase (Invitrogen). Labeled primers were purified by MicroSpin™ G-25 columns (Amersham Biosciences). The single nucleotide insertion reactions were performed at 37°C (for KF exo−) or 68 °C (for Vent exo−). 10X reaction buffer contained 500 mM Tris-HCl(pH 7.4), 100 mM MgCl2, 10 mM DTT, 0.5 mg/mL BSA (for KF exo−); or 100 mM KCl, 100 mM (NH4)2SO4, 200 mM Tris-HCl(pH 8.8), 20 mM MgSO4, 1% Triton X-100 (for Vent exo−). 2.5 µL duplex solution containing 5 µM non-radiolabeled duplex, 25 nM radiolabeled duplex and 2X reaction buffer was mixed with 2.5 µL enzyme solution containing varied concentrations of enzyme and 2X reaction buffer, and was incubated for one minute. 5.0 µL dNTP solution at various concentrations was then added and the mixture reacted for various times (see Figure 2 legend). Reactions were stopped by addition of 15 µL stop buffer containing 40% formamide, 0.05% xylene cyanol and 0.05 % bromophenol blue. Products were resolved by denaturing polyacrylamide gel electrophoresis (7.6 M urea, 20% acrylamide) and visualized by phosphorimager (Molecular Dynamics) and ImageQuant program.
Polymerase amplification
For the PCR amplification experiments, primers (0.4 µM each), templates (50ng of annealed double stranded DNA, Table 1), and dNTPs (200 µM each), were mixed with reaction buffer and Vent exo− polymerase (both from New England Biolabs, Ipswich, MA; Cat No: M0254S) and adjusted to a final reaction volume of 50 µL with water. The reaction conditions consisted of an initial denaturation step of 95 °C for 1 min, followed by 20 cycles of 94 °C for 30 sec, 50 °C for 30 sec, and 68 °C for 40 sec. Products after PCR were visualized on an agarose gel. The conditions for the PCR reactions were initially optimized to determine the optimal annealing and extension temperatures for amplification using modified yDNA templates and Vent DNA polymerase. In general, with modified DNA as templates, it was observed that higher elongation temperature provided more robust amplifications.
Plasmid construction and cloning
The products from PCR assays using yDNA templates were directly cloned into the pCR2.1 vector using TA cloning kit (Invitrogen, Carlsbad, CA; cat no: K4575) and transformed into E.coli. Plasmid DNA was isolated from the transformants using Miniprep DNA isolation kit (Qiagen, Valencia, CA) and the amplified region was sequenced using appropriate primers. All DNA sequencing was done using the standard automated fluorescent sequencing techniques.
For cloning the GFP gene segment containing the yDNA bases to pGFPuv plasmid (Clontech, Mountain View, CA; Cat No:632312), which is a high copy number plasmid yielding ~500 copies/cell, the vector was first prepared by digesting the plasmid with the restriction enzymes, BsrGI and MluI (New England Biolabs); this removes a 46 base pair region from within the GFP gene. The resulting linearized vector with cohesive ends was ligated with the annealed duplex DNA containing the yDNA bases (Table 3) using T4 DNA ligase (New England Biolabs). The ligated product was transformed to BL21 (DE3) cells (Stratagene, La Jolla, CA) and plated on LB agar plates containing ampicillin and isopropyl thiogalactosidase (IPTG). The green fluorescent colonies obtained after transformation, identified by viewing under uv transilluminator, were picked, plasmid DNA isolated, and the cloned region within the gene was sequenced. Transformation efficiencies were compared with that of a control ligation reaction under identical conditions using unmodified duplex DNA, which was otherwise identical in sequence to the yDNA containing segment.
Acknowledgements
This work was supported by the U.S. National Institutes of Health (GM63587). A.H.F.L. acknowledges a Croucher Foundation fellowship.
References
- 1.a) Piccirilli JA, Krauch T, Moroney SE, Benner SA. Nature. 1990;343:33. doi: 10.1038/343033a0. [DOI] [PubMed] [Google Scholar]; b) Switzer CY, Moroney SE, Benner SA. Biochemistry. 1993;32:10489. doi: 10.1021/bi00090a027. [DOI] [PubMed] [Google Scholar]; (c) Horlacher J, Hottiger M, Podust VN, Hubscher U, Benner SA. Proc. Natl. Acad. Sci. USA. 1995;92:6329. doi: 10.1073/pnas.92.14.6329. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Benner SA. Acc. Chem. Res. 2004;37:784. doi: 10.1021/ar040004z. [DOI] [PubMed] [Google Scholar]
- 2.Rappaport HP. Nucleic Acids Res. 1988;16:7253. doi: 10.1093/nar/16.15.7253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Schweitzer BA, Kool ET. J. Am. Chem. Soc. 1995;117:1863. doi: 10.1021/ja00112a001. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Matray TJ, Kool ET. J. Am. Chem. Soc. 1998;120:6191. doi: 10.1021/ja9803310. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lai J, Qu J, Kool ET. J. Am. Chem. Soc. 2004;126:3040. doi: 10.1021/ja039571s. [DOI] [PubMed] [Google Scholar]; d) Kool ET. Acc. Chem. Res. 2002;35:936. doi: 10.1021/ar000183u. [DOI] [PubMed] [Google Scholar]
- 4.a) Tae EL, Wu Y, Xia G, Schultz PG, Romesberg FE. J. Am. Chem. Soc. 2001;123:7439. doi: 10.1021/ja010731e. [DOI] [PubMed] [Google Scholar]; b) Henry AA, Romesberg FE. Curr. Opin. Chem. Biol. 2003;6:727. doi: 10.1016/j.cbpa.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 5.a) Mitsui T, Kimoto M, Sato A, Yokoyama S, Hirao I. Bioorg. Med. Chem. Lett. 2003;13:4515. doi: 10.1016/j.bmcl.2003.09.059. [DOI] [PubMed] [Google Scholar]; b) Mitsui T, Kitamura A, Kimoto M, To T, Sato A, Hirao I, Yokoyama S. J. Am. Chem. Soc. 2003;125:5298. doi: 10.1021/ja028806h. [DOI] [PubMed] [Google Scholar]; c) Hirao I. Curr. Opin. Chem. Biol. 2006;10:622. doi: 10.1016/j.cbpa.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 6.a) Tanaka K, Tengeiji A, Kato T, Toyama N, Shiro M, Shionoya M. J. Am. Chem. Soc. 2002;124:12494. doi: 10.1021/ja027175o. [DOI] [PubMed] [Google Scholar]; b) Shionoya M, Tanaka K. Curr. Opin. Chem. Biol. 2004;8:592. doi: 10.1016/j.cbpa.2004.09.006. [DOI] [PubMed] [Google Scholar]; c) Zimmermann N, Meggers E, Schultz PG. J. Am. Chem. Soc. 2002;124:13684. doi: 10.1021/ja0279951. [DOI] [PubMed] [Google Scholar]; d) Parsch J, Engels JW. Nucleosides Nucleotides Nucleic Acids. 2001;20:815. doi: 10.1081/NCN-100002436. [DOI] [PubMed] [Google Scholar]; e) Hikishima S, Minakawa N, Kuramoto K, Fujisawa Y, Ogawa M, Matsuda A. Angew. Chem. Int. Ed. 2005;44:596. doi: 10.1002/anie.200461857. [DOI] [PubMed] [Google Scholar]; f) Johar Z, Zahn A, Leumann CJ, Jaun B. Chemistry. 2008;14:1080. doi: 10.1002/chem.200701304. [DOI] [PubMed] [Google Scholar]; g) Heuberger BD, Shin D, Switzer C. Org. Lett. 2008;10:1091. doi: 10.1021/ol703029d. [DOI] [PubMed] [Google Scholar]
- 7.a) Sismour AM, Benner SA. Nucleic Acids Res. 2005;33:5640. doi: 10.1093/nar/gki873. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yang Z, Hutter D, Sheng P, Sismour AM, Benner SA. Nucleic Acids Res. 2006;34:6095. doi: 10.1093/nar/gkl633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Morales JC, Kool ET. Nature Struct. Biol. 1998;5:950. doi: 10.1038/2925. [DOI] [PubMed] [Google Scholar]; b) Matray TJ, Kool ET. Nature. 1999;399:704. doi: 10.1038/21453. [DOI] [PubMed] [Google Scholar]; c) Lai JS, Kool ET. Chemistry. 2005;11:2966. doi: 10.1002/chem.200401151. [DOI] [PubMed] [Google Scholar]
- 9.a) Henry AA, Yu C, Romesberg FE. J. Am. Chem. Soc. 2003;125:9638. doi: 10.1021/ja035398o. [DOI] [PubMed] [Google Scholar]; b) Henry AA, Olsen AG, Matsuda S, Yu C, Geierstanger BH, Romesberg FE. J. Am. Chem. Soc. 2004;126:6923. doi: 10.1021/ja049961u. [DOI] [PubMed] [Google Scholar]; c) Matsuda S, Henry AA, Romesberg FE. J. Am. Chem. Soc. 2006;128:6369. doi: 10.1021/ja057575m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hwang GT, Romesberg FE. Nucleic Acids Res. 2006;34:2037. doi: 10.1093/nar/gkl049. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Matsuda S, Fillo JD, Henry AA, Rai P, Wilkens SJ, Dwyer TJ, Geierstanger BH, Wemmer DE, Schultz PG, Spraggon G, Romesberg FE. J. Am. Chem. Soc. 2007;129:10466. doi: 10.1021/ja072276d. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Leconte AM, Hwang GT, Matsuda S, Capek P, Hari Y, Romesberg FE. J. Am. Chem. Soc. 2008;130:2336. doi: 10.1021/ja078223d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Hirao I, Kimoto M, Mitsui T, Fujiwara T, Kawai R, Sato A, Harada Y, Yokoyama S. Nat. Methods. 2006;9:729. doi: 10.1038/nmeth915. [DOI] [PubMed] [Google Scholar]; b) Hirao I, Harada Y, Kimoto M, Mitsui T, Fujiwara T, Yokoyama S. J. Am. Chem. Soc. 2004;126:13298. doi: 10.1021/ja047201d. [DOI] [PubMed] [Google Scholar]
- 11.a) Paul N, Nashine VC, Hoops G, Zhang P, Zhou J, Bergstrom DE, Davisson VJ. Chem. Biol. 2003;9:815. doi: 10.1016/j.chembiol.2003.08.008. [DOI] [PubMed] [Google Scholar]; b) Adelfinskaya O, Nashine VC, Bergstrom DE, Davisson VJ. J. Am. Chem. Soc. 2005;127:16000. doi: 10.1021/ja054226j. [DOI] [PubMed] [Google Scholar]
- 12.a) Rappaport HP. Biochem. J. 2004;381:709. doi: 10.1042/BJ20031776. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Johnson SC, Sherrill CB, Marshall DJ, Moser MJ, Prudent JR. Nucleic Acids Res. 2004;32:1937. doi: 10.1093/nar/gkh522. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kincaid K, Beckman J, Zivkovic A, Halcomb RL, Engels JW, Kuchta RD. Nucleic Acids Res. 2005;33:2620. doi: 10.1093/nar/gki563. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhang X, Lee I, Berdis A. J. Biochem. 2005;44:13101. doi: 10.1021/bi050585f. [DOI] [PubMed] [Google Scholar]
- 13.Bain JD, Switzer C, Chamberlin AR, Benner SA. Nature. 1992;356:537. doi: 10.1038/356537a0. [DOI] [PubMed] [Google Scholar]
- 14.a) Liu H, Gao J, Lynch S, Maynard L, Saito D, Kool ET. Science. 2003;302:868. doi: 10.1126/science.1088334. [DOI] [PubMed] [Google Scholar]; b) Liu H, Gao J, Kool ET. J. Am. Chem. Soc. 2005;127:1396. doi: 10.1021/ja046305l. [DOI] [PubMed] [Google Scholar]; c) Gao J, Liu H, Kool ET. Angew. Chem. Int. Ed. 2005;44:3118. doi: 10.1002/anie.200500069. [DOI] [PubMed] [Google Scholar]; d) Lynch SR, Liu H, Gao J, Kool ET. J. Am. Chem. Soc. 2006;128:14704. doi: 10.1021/ja065606n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Lu H, He K, Kool ET. Angew. Chem. Int. Ed. 2004;43:5834. doi: 10.1002/anie.200461036. [DOI] [PubMed] [Google Scholar]; b) Lee AHF, Kool ET. J. Am. Chem. Soc. 2005;127:3332. doi: 10.1021/ja0430604. [DOI] [PubMed] [Google Scholar]
- 16.Krueger A, Lu H, Lee AHF, Kool ET. Accts. Chem. Rev. 2007;40:141. [Google Scholar]
- 17.a) Scopes DI, Barrio JR, Leonard NJ. Science. 1977;195:296. doi: 10.1126/science.188137. [DOI] [PubMed] [Google Scholar]; b) Leonard NJ. Acc. Chem. Res. 1982;15:128. [Google Scholar]
- 18.a) Liu H, Gao J, Saito D, Maynard L, Kool ET. J. Am. Chem. Soc. 2004;126:1102. doi: 10.1021/ja038384r. [DOI] [PubMed] [Google Scholar]; b) Lee AHF, Kool ET. J. Org. Chem. 2004;70:132. doi: 10.1021/jo0483973. [DOI] [PubMed] [Google Scholar]; c) Liu H, Gao J, Kool ET. J. Org. Chem. 2004;70:639. doi: 10.1021/jo048357z. [DOI] [PubMed] [Google Scholar]
- 19.a) Kim TW, Delaney JC, Essigmann JM, Kool ET. Proc. Natl. Acad. Sci. USA. 2005;102:15803. doi: 10.1073/pnas.0505113102. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kim TW, Brieba LG, Ellenberger T, Kool ET. J. Biol. Chem. 2006;281:2289. doi: 10.1074/jbc.M510744200. [DOI] [PubMed] [Google Scholar]; c) Sintim HO, Kool ET. Angew. Chem. Int. Ed. 2006;45:1974. doi: 10.1002/anie.200504296. [DOI] [PubMed] [Google Scholar]
- 20.Mattila P, Korpela J, Tenkanen T, Pitkanen K. Nucleic Acids Res. 1991;19:4967. doi: 10.1093/nar/19.18.4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee AHF, Kool ET. J. Org. Chem. 2005;70:132. doi: 10.1021/jo0483973. [DOI] [PubMed] [Google Scholar]
- 22.Cabrera MF, Sumpter BG, Lipkowski P, Wells JC. J. Phys. Chem. B. 2006;110:6379. doi: 10.1021/jp057356n. [DOI] [PubMed] [Google Scholar]