

Abstract
The androgen receptor (AR) regulates growth and progression of androgen-dependent as well as androgen-independent prostate cancer cells. Peroxisome proliferator activated receptor gamma (PPARγ) agonists have been reported to reduce AR activation in androgen-dependent LNCaP prostate cancer cells. To determine whether PPARγ ligands are equally effective at inhibiting AR activity in androgen-independent prostate cancer, we examined the effect of the PPARγ ligands ciglitazone and rosiglitazone on C4-2 cells, an androgen- independent derivative of the LNCaP cell line. Luciferase-based reporter assays and Western blot analysis demonstrated PPARγ ligand reduced dihydrotestosterone (DHT)-induced increases in AR activity in LNCaP cells. However, in C4-2 cells these compounds increased DHT-induced AR driven luciferase activity. In addition, ciglitazone did not significantly alter DHT-mediated increases in prostate specific antigen (PSA) protein or mRNA levels within C4-2 cells. siRNA based experiments demonstrated that the ciglitazone-induced regulation of AR activity observed in C4-2 cells was dependent on the presence of PPARγ. Furthermore, overexpression of the AR corepressor cyclin D1 inhibited the ability of ciglitazone to induce AR luciferase activity in C4-2 cells. Thus, our data suggest both PPARγ and cyclin D1 levels influence the ability of ciglitazone to differentially regulate AR signaling in androgen-independent C4-2 prostate cancer cells.
Keywords: PPAR gamma, androgen receptor, androgen- independent prostate cancer
INTRODUCTION
Prostate cancer is the most common form of cancer and the second leading cause of cancer death in aging men of the United States [1]. Prostate cancer starts as an androgen-dependent disease, in that it requires the presence of androgens to proliferate and progress. Patients with metastatic cancer are primarily treated with androgen ablation therapy (AAT). In AAT, the level of circulating androgens in the body is reduced by treatment with androgen receptor antagonists and/or luteinizing hormone releasing hormone agonists or physical removal of the testes. Prostate tumor growth is initially retarded in response to AAT. This reduction in tumor growth is due in part to increases in tumor cell apoptosis as well as decreased cell proliferation. However, in the months following the initiation of AAT, some tumor cells become androgen-independent, and are able to survive and grow in the low androgen environment [2]. Currently, there are no effective treatments for advanced stage androgen-independent prostate cancers.
The biological actions of androgens are mediated by the androgen receptor (AR). The AR is a member of the nuclear receptor superfamily that functions as a ligand-activated transcription factor [3, 4]. Activation of the AR by androgens induces the growth and proliferation of the prostate. AR activation also leads to the transcription of prostate specific antigen (PSA) and many other androgen-dependent genes, such as FK506 binding protein 51 (FKBP51) and prostatic acid phosphatase (PAP). In advanced, androgen-independent prostate cancer the AR is still expressed [5] and appears to be transcriptionally active [3, 4]. Because the AR remains functional in androgen- independent prostate cancer, compounds that reduce AR activity are being explored as therapeutic options to reduce growth and progression of these advanced forms of prostate cancer.
One protein that has been reported to regulate activation of the AR is the peroxisome proliferator activated receptor gamma (PPARγ). Like AR, PPARγ is a transcription factor that belongs to the nuclear receptor superfamily. PPARγ is activated by both synthetic ligands and naturally occurring compounds. Thiazolidinediones (TZDs) are one group of synthetic PPARγ agonists. This class of drugs include the compounds ciglitazone (parent), troglitazone (2nd generation) and the 3rd generation compounds rosiglitazone and pioglitazone. Recent studies suggest that PPARγ may be a possible target for prostate cancer therapy [6]. The expression of PPARγ is significantly higher in prostate cancer cells than in normal epithelial prostate cells [7–9]. PPARγ ligands have also been shown to inhibit proliferation of both androgen-dependent and androgen-independent human prostate cancer cell lines [10, 11]. In addition, PPARγ ligands reduce the growth of primary cultures of human prostate cancer cells [9]. In vivo conditional gene knockout studies demonstrated that loss of PPARγ within the luminal epithelial cells of the mouse prostate produces lesions consistent with low grade prostatic intraepithelial neoplasia (PIN), the proposed precursor to prostate cancer [11]. Taken together, these data suggest that PPARγ plays a role in the development and progression of prostate cancer.
PPARγ ligands have been shown to inhibit AR transcriptional activity in the androgen-dependent LNCaP human prostate cancer cells [12, 13]. However, it is unclear whether PPARγ ligands also regulate AR activation in androgen-independent prostate cancer. Therefore, in this paper, we investigated how ligands that activate PPARγ regulate AR activity in C4-2 cells, an androgen-independent human prostate cancer cell line. Our data indicate that a synthetic ligand for PPARγ, ciglitazone, regulates the AR differently in androgen-dependent versus androgen-independent human prostate cancer cells. Additionally, we report that the level of functional PPARγ and down-regulation of cyclin D1, an AR corepressor, may be linked to the differential regulation of AR activity by ciglitazone.
MATERIALS AND METHODS
Materials
The tissue culture media (RPMI 1640, DMEM low glucose, DMEM/F12 and Hams F-12) and penicillin/streptomycin solution were purchased from Invitrogen (Carlsbad, CA). Adenine hemisulfate, d-biotin, insulin and apo-transferrin were purchased from Sigma Life Science (St. Louis, MO). Fetal bovine serum (FBS) was obtained from HyClone (Logan, UT). Zapoglobin and Isoton II were purchased from Beckman Coulter Inc. (Fullerton, CA). All tissue culture plasticware and additional chemicals were from Fisher Scientific (Suwanee, GA).
Cell Culture
The LNCaP cell line (ATCC, Manassas, VA) was grown in RPMI 1640 media supplemented with 10% FBS and 1% penicillin/streptomycin. The C4-2 cell line (ViroMed Laboratories, Minnetonka, MN) was grown in T media (80% DMEM low glucose media, 20% Hams’ F-12 media, 1% penicillin/streptomycin, 0.244 μg/ml d-biotin, 25 μg/ml adenine hemisulfate, 5 μg/ml insulin and 5 μg/ml apo-transferrin) supplemented with 5% heat inactivated FBS. The PC-3 cell line (ATCC) was grown in DMEM-F12 media supplemented with 10% FBS and 1% penicillin/streptomycin. Each cell line was maintained in a 37°C incubator in an atmosphere containing 5% CO2.
Cell Count Assays
Each cell line was plated in six-well plates at a density of 20,000 cells per well in either RPMI 1640 media with 10% FBS and 1% penicillin/streptomycin (LNCaP) or T medium containing 5% heat inactivated FBS and 1% penicillin/streptomycin (C4-2). The cells were allowed to adhere overnight. Then, the cells were treated with ethanol vehicle control (EtOH) or the PPARγ ligand ciglitazone (0–45 μM; EMD Bioscience, LaJolla, CA). The medium was changed and fresh drug was added after three days. After an additional three days (six days total treatment), the cells were removed from the wells with 0.05% trypsin-EDTA. The number of cells in each well was then counted using a Coulter Z1 cell counter (Beckman Coulter Inc., Fullerton, CA).
Transient Transfections
To measure PPARγ activation, both LNCaP and C4-2 cell lines (~ 10 million cells) were transfected with 20 μg of a PPARγ regulated reporter plasmid (PPRE3-luciferase) and 2 μg of CMV β-galactosidase plasmid via electroporation. Each cell line was then plated in six-well plates containing RPMI 1640 media supplemented with 10% FBS at a density of 540,000 cells per well. The next day the media was changed to RPMI 1640 media supplemented with 10% FBS. Cells were then treated with vehicle control (EtOH or DMSO), ciglitazone (0–45 μM), rosiglitazone (40 μM; Cayman Chemical, Ann Arbor, MI), or 15dPGJ2 (10 μM; EMD Bioscience) for 24 hours. Next the cells were harvested using Tris EDTA NaCl (TEN) buffer. The samples were then measured for luciferase activity using the Luciferase Assay System kit from Promega (Madison, WI). Luciferase activity in each sample was normalized to β-galactosidase activity.
To measure AR transcriptional activity, both cell lines (~ 10 million cells) were transfected with 20 μg of an AR responsive reporter plasmid (ARR2PB- luciferase or PSAE- luciferase) and 2 μg of CMV β-galactosidase in RPMI 1640 media supplemented with 10% FBS. The next day the cells were placed in RPMI 1640 media supplemented with 10% CSS for 24 hours. The cells were then treated with the EtOH or DMSO vehicle control; TZD (ciglitazone 15–45 μM or rosiglitazone 10–40 μM); dihydrotestosterone (DHT) 1 nM; or DHT 1 nM plus TZD) for 24 hours. Following treatment, the level of luciferase and β-galactosidase activity within the cells was measured.
Western Blot Analysis
Treated cells were lysed in RIPA buffer containing 1 mM sodium vanadate and 1 mM phenylmethylsulphonyl fluoride (PMSF). Next samples were centrifuged at 10,000 rpm at 4°C for 10 minutes. Protein concentration was determined using the Bradford reagent (BioRad, Hercules, CA). Equal amounts of protein were loaded on a SDS- PAGE gel, subjected to electrophoresis and transferred to a nitrocellulose membrane. Western blot analysis was performed using the following primary antibodies: α-tubulin (clone B- 7; Santa Cruz Biotechnology, Santa Cruz, CA), actin (Chemicon International, Temecula, CA), AR (clone AR 441; Lab Vision Corporation, Fremont, CA), PPARγ (clone H- 100; Santa Cruz Biotechnology), PSA (DakoCytomation, Carpinteria, CA) and topoisomerase I (clone H- 300; Santa Cruz Biotechnology). Following exposure to primary antibody, the blots were incubated with a secondary antibody conjugated to horseradish peroxidase. Proteins were then visualized using enzyme- linked chemiluminescence (ECL, Amersham Biosciences, Piscataway, NJ). Quantification of Western blot bands was performed using the UN-SCAN-IT gel 6.1 Program.
Quantitative Reverse Transcription PCR (qRT- PCR)
LNCaP and C4-2 cells were treated with either EtOH vehicle control; ciglitazone 45 μM; dihydrotestosterone (DHT) 1 nM; or DHT 1 nM plus ciglitazone over a 48 hour time period. Total RNA was extracted from treated cells using Trizol reagent (Invitrogen). Next, 1 μg of RNA was reverse transcribed at 42°C for 30 min using the iScript™ cDNA Synthesis Kit from BioRad and the PTC- 100 Peltier Thermal Cycler (MJ Research, Ramsey, Minnesota). The reaction was then heat inactivated at 85°C for 5 min. qPCR reactions were performed using the SYBR Green detection method and the iCYCLER iQ5 Real Time PCR machine from BioRad. DNA primers purchased from Integrated DNA Technologies, Inc. (Coralville, IA) were used to amplify regions of PSA and 18S cDNA. The sequence of the primers used in these reactions were as follows: PSA: 5′-GCA TCA GGA ACA AAA GCG TGA 3′ (sense), 5′-CCT GAG GAA TCG ATT CTT CAG 3′ (antisense); 18s: 5′-ATC AAC TTT CGA TGG TAG TCG 3′ (sense), 5′-TCC TTG GAT GTG GTA GCC 3′ (antisense). PCR conditions were 95°C for 3 min, followed by 40 cycles of 95°C for 30 sec, 58°C for 30 sec, and 72°C for 30 sec. The amount of PSA gene product in each sample was calculated using the ΔΔCt method and normalized to the amount of 18s RNA control.
Small Interference RNA (siRNA) Experiments
Cells were transfected with the PSAE- luciferase, CMV β-galactosidase plasmid, and 20 μM of either non- specific control SMARTpool siRNA or PPARγ SMARTpool siRNA (Dharmacon Inc., Lafayette, CO) via electroporation (~ 10 million cells per transfection). Following electroporation, the cells were plated in six-well plates at a density of 540,000 cells per well. Two days after transfection, the cells were treated as indicated in the figure legends for 24 hours. In one set of plates, luciferase activity was measured and normalized to β-galactosidase activity. In parallel plates, cells were harvested and Western blot analysis was performed as previously described to measure the level of PPARγ and actin protein.
PPARγ and CD1 Overexpression Experiments
To overexpress PPARγ, electroporation was used to transfect approximately 10 million LNCaP cells with the 2 μg PSAE- luciferase plasmid, 200 ng CMV β-galactosidase plasmid, and 100 ng of either pcDNA 3.1 plasmid (from K. Knudsen, Thomas Jefferson University) or human PPARγ cDNA expression vector clone (TC124177; OriGene Technologies Inc., Rockville, MD) expression vector. Two days following transfection, the cells were treated with EtOH vehicle control, ciglitazone 45 μM, DHT 1 nM, or DHT plus ciglitazone for 24 hours. In one set of plates, luciferase activity was measured and normalized to β-galactosidase activity. In parallel plates, cells were harvested by scraping. Western blot analysis was performed as previously described to measure the level of PPARγ and actin protein.
CD1 was overexpressed in C4-2 cells via electroporation with 2 μg PSAE- luciferase, 200 ng CMV β-galactosidase plasmid, and 1.5 μg of either pcDNA 3.1 plasmid or CD1-T286A expression vector (from K. Knudsen, Thomas Jefferson University and J. A. Diehl, University of Pennsylvania). Two days following transfection, the cells were treated with EtOH vehicle control, ciglitazone 45 μM, DHT 1 nM, or DHT plus ciglitazone for 24 hours. In one set of plates, luciferase activity was measured and normalized to β-galactosidase activity. In parallel plates, cells were harvested by scraping. Western blot analysis was performed as previously described to measure the level of cyclin D1 and actin protein.
Statistical Analysis
Each experiment was performed at least three times and representative data are shown. For proliferation assays and transient transfections, One Way Analysis of Variance (ANOVA) was used to detect differences between control and treated groups. These analyses were performed using the Sigma Stat 3.1 program (Systat Software Inc.). The standard for statistical significance was p< 0.05.
RESULTS
Functional PPARγ is present in the androgen- independent C4-2 cell line
The C4-2 cell line was derived from androgen- dependent LNCaP human prostate cancer cells grown in castrated mice and represent prostate cancer cells that have naturally become androgen- independent [14]. Previous studies suggest that levels of PPARγ vary between androgen- dependent and androgen- independent human prostate cancer cells [15]. To determine whether PPARγ levels vary between the androgen- dependent LNCaP and androgen- independent C4-2 cells, we measured the level of PPARγ protein in each cell line. Western blot analysis revealed that PPARγ is expressed in nuclear extracts of LNCaP and C4-2 cells. However, the level of PPARγ protein expressed in C4-2 cells is significantly higher than that present in the LNCaP cell line (Fig. 1A). To determine whether the PPARγ detected by Western blot analysis was transcriptionally active, we performed a series of reporter assays using the PPRE3- luciferase reporter plasmid [16]. In the C4-2 cells, the PPARγ ligands ciglitazone, rosiglitazone and 15dPGJ2 were able to activate PPARγ by at least 2-fold when compare to their vehicle control (Fig. 1B). However, there was no significant increase in luciferase activity in LNCaP cells following treatment with any of the PPARγ ligands. Although we did not see a PPARγ ligand- induced increase in luciferase activity, there was a substantial amount of β-galactosidase activity in the transfected LNCaP cells. This indicates that our inability to detect ligand- induced PPARγ activity was not due to problems with transiently introducing DNA within the LNCaP cell line. These data suggest that while PPARγ is expressed in both LNCaP and C4-2 cells, PPARγ is only transcriptionally active in the C4-2 cell line.
Fig 1. C4-2 cells express functional PPARγ.

(A) Nuclear extracts were prepared from the C4-2, LNCaP and PC-3 cell lines. The PPARγ protein present in each nuclear fraction was detected using Western blot analysis. The polyclonal PPARγ antibody used in these studies detects PPARγ (indicated by arrow) as well as a smaller, non- specific band. The blots were stripped and reprobed with an antibody against topoisomerase I (Topo I) to confirm purity of nuclear fractions. (B) LNCaP and C4-2 cells were transfected with the PPRE3-luciferase and CMV β-galactosidase plasmid constructs. Transfected cells were then treated with vehicle control (EtOH or DMSO) or PPARγ ligands at various concentrations for 24 hours. Luciferase activity was measured and normalized to β-galactosidase activity. Each bar represents the mean ± SD of three wells. *, P< 0.05 compared to vehicle control. A representative experiment is shown.
Ciglitazone inhibits the proliferation of androgen- independent human prostate cancer cells
Our laboratory and others have shown that ciglitazone as well as other PPARγ ligands reduce the proliferation of C4-2 cells ([17] and Fig. 2). LNCaP cells are also sensitive to the anti- proliferative effects of ciglitazone. Cell count assays revealed that concentrations of ciglitazone ≥ 15 μM significantly reduced proliferation of the LNCaP cell line (Fig. 2). There was a comparable growth inhibitory response to ciglitazone in the androgen- dependent and –independent cell lines. In both the C4-2 [17] and LNCaP cell lines (Fig. 2), we observed maximum growth inhibition with 45 μM ciglitazone.
Fig 2. Ciglitazone inhibits the growth of androgen- dependent and – independent human prostate cancer cells.
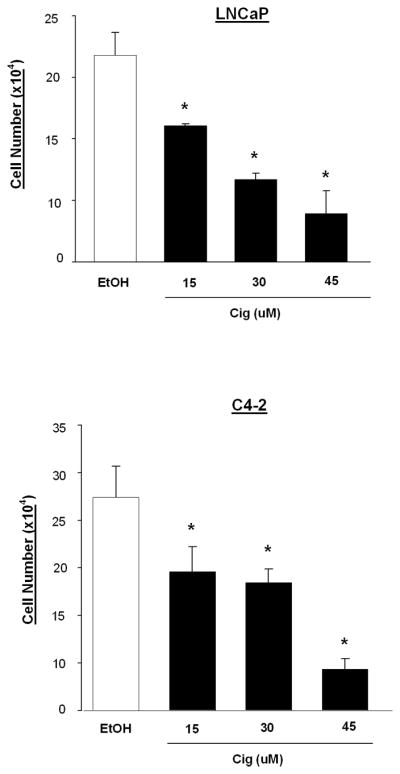
LNCaP and C4-2 cells were plated in 6 well plates and treated with ciglitazone (0 – 45 μM) for six days. Following treatment, cells were detached using Trypsin- EDTA. Cells were then counted using a Coulter Counter. Each bar represents the mean ± SD for three wells. *, P < 0.05 compared to EtOH control. A representative experiment is shown.
Ciglitazone regulates ligand- induced AR activation differently in LNCaP and C4-2 cells
There are comparable levels of AR protein expression in both LNCaP and C4-2 prostate cancer cell lines ([18] and Fig. 3A). The PPARγ ligands troglitazone, pioglitazone and 15dPGJ2 have been reported to decrease AR activation in LNCaP cells [12, 13, 19]. Since decreases in AR activity can result in reductions in cell proliferation [14], we decided to test whether PPARγ ligands were also effective at inhibiting AR activity in the C4-2 cell line. In these studies, we used a luciferase-based AR responsive reporter plasmid (PSAE-luciferase) to compare the effect of ciglitazone on AR transcriptional activation in LNCaP and C4-2 cells. The PSAE- luciferase plasmid construct contains the three AREs located in the promoter and enhancer regions of the human PSA gene [20]. As expected, treatment with the androgen dihydrotestosterone (DHT) induced AR- mediated transcription approximately 12 fold in LNCaP cells transfected with the PSAE- luciferase reporter. A concentration of ciglitazone that inhibits proliferation of LNCaP and C4-2 cells (45 μM) ([17] and Fig. 2) did not alter basal AR transcriptional activity. However, ciglitazone did significantly decrease DHT-induced increases in PSAE- luciferase reporter activity in the LNCaP cells (Fig. 3B). In addition, a concentrations of rosiglitazone that activated PPARγ (Fig. 1B) inhibited the DHT- induced PSAE- luciferase activity in LNCaP cells (Fig. 3B). Thus, both PPARγ agonists are effective at reducing DHT- induced AR activation. Similar to the LNCaP cells, DHT treatment increased AR- driven luciferase activity in C4-2 cells transfected with the PSAE- luciferase reporter. There was no alteration in basal activation of the AR with ciglitazone or rosiglitazone treatment. However, in the presence of DHT, neither ciglitazone nor rosiglitazone decreased AR activation. Instead, both ciglitazone (15 μM and 45 μM) and rosiglitazone (10 μM and 40 μM) enhanced DHT- induced AR activation in C4-2 cells, as indicated by an increase in the PSAE- luciferase reporter activity (Fig. 3B).
Fig 3. The ability of TZDs to regulate AR activation varies between LNCaP and C4-2 cells.

Panel (A): Whole cell lysates were prepared from PC-3, C4-2 and LNCaP cells. Total AR protein levels were assessed using Western blot analysis. The PC-3 cell lysate was used as a negative control for AR protein expression. Blots were stripped and reprobed for β-actin to confirm equal loading. Panels (B) and (C): Cells were transfected with CMV β-galactosidase and the AR- responsive reporter PSAE- luciferase (B) or ARR2PB- luciferase (C) and treated for 24 hours as indicated. Luciferase activity was measured and normalized against β-galactosidase activity. Each bar represents the mean ± SD of three wells. *, P< 0.05 compared to DHT alone (DHT+, Cig-). A representative experiment is shown.
To determine whether these results were unique to the PSAE-luciferase reporter, we repeated these experiments using another AR responsive promoter plasmid, ARR2PB-luciferase (Fig. 3C). The ARR2PB-luciferase plasmid construct contains two androgen response regions from the rat probasin promoter cloned upstream of the luciferase gene [21]. The results obtained in these experiments involving the ARR2PB-luciferase reporter were similar to those observed with the PSAE-luciferase reporter construct. Again, ciglitazone and rosiglitazone decreased DHT- induced AR transcriptional activation in the LNCaP cells, but did not decrease AR activation stimulated by DHT in the C4-2 cells.
To further explore the effect of PPARγ ligands on AR activation, we examined the ability of ciglitazone to regulate protein levels of prostate specific antigen (PSA), an endogenous AR responsive gene product. Ciglitazone inhibited the DHT-stimulated increase in PSA protein levels in the androgen-dependent LNCaP cells (Fig. 4A). A decrease could be detected as early as 12 hours, and PSA levels remained repressed after 48 hours. Additionally, ciglitazone inhibited DHT- induced increases in PSA mRNA expression in the LNCaP cell line (Fig. 4B). However, in C4-2 cells ciglitazone did not alter the increase in PSA protein expression induced by DHT (Fig. 4A). Furthermore, ciglitazone did not suppress DHT- mediated increases in PSA mRNA levels in the C4-2 cell line (Fig. 4B). Thus ciglitazone inhibits androgen- induced AR reporter activity as well as PSA protein and mRNA levels in LNCaP cells, whereas it does not lower these androgen- mediated responses in C4-2 cells.
Fig 4. Ciglitazone does not reduce DHT- mediated increases in PSA protein or mRNA within C4-2 cells.

(A) Whole cell lysates were prepared from LNCaP and C4-2 cells treated with ethanol (DHT-, Cig-), DHT (1 nM), Ciglitazone (45 μM), or DHT + Ciglitazone. PSA protein levels in treated cells were then detected using Western blot analysis. The blots were stripped and reprobed for β-actin to confirm equal loading of the gels. (B) Total RNA extracted from LNCaP and C4-2 cells treated with ethanol (DHT-, Cig-), DHT (1 nM), Ciglitazone (45 μM), or DHT + Ciglitazone. PSA mRNA levels were detected using qRT- PCR. The amount of PSA gene product in each sample was calculated using the ΔΔCt method and normalized to the amount of 18s control. Each bar represents the mean of two independent readings. A representative experiment is shown.
Ciglitazone does not alter AR protein expression
The fact that ciglitazone was ineffective at reducing AR activation in the C4-2 cells suggest that the regulation of AR signaling by ciglitazone varies between the LNCaP and C4-2 cell lines. Yang et al noted that high concentrations of the PPARγ ligand troglitazone (> 60μM) decreased AR protein levels in LNCaP cells [13]. Therefore, we compared regulation of AR protein levels by ciglitazone in the two cell lines. Western blot analysis on whole cell lysates was used to determine total AR protein levels following ciglitazone treatment. There was no change in total AR protein levels in LNCaP cells treated with ciglitazone after 12, 24, or 48 hours (Fig. 5). There was no alteration in AR levels in C4-2 cells exposed to ciglitazone for 12 or 24 hours. Ciglitazone did induce a 20% decrease in AR protein levels in the C4-2 cells at the later 48 hour time period. The changes we see in AR driven luciferase activity and PSA expression occur prior to this slight decrease. Therefore, our data suggest that ciglitazone does not alter AR transcriptional activity in LNCaP and C4-2 cells by reducing AR protein levels.
Fig 5. Ciglitazone does not alter total AR protein levels in androgen- dependent or -independent prostate cancer cells.

Whole cell lysates were prepared from LNCaP and C4-2 cells treated with ethanol or ciglitazone (45 μM). Total AR protein levels were detected using Western blot analysis. The blots were stripped and reprobed for β-actin to confirm equal loading of the gels. AR band density was quantified and normalized to the β-actin. The AR/actin ratio was determined and expressed as fold change relative to time matched EtOH controls.
PPARγ is required for ciglitazone-mediated increases in AR activation
Since the level of functional PPARγ varied between the LNCaP and C4-2 cells, we hypothesized that differences in PPARγ expression were responsible for the varying response to ciglitazone noted in these cell lines. To define the role of PPARγ in the ciglitazone- induced regulation of AR, we examined the effect of ciglitazone on AR activation in C4-2 cells lacking PPARγ. For these studies we measured PSAE- luciferase reporter activity in C4-2 cells transfected with either nonspecific control or PPARγ SMARTpool siRNA oligonucleotides. Western blot indicated that PPARγ protein levels were significantly reduced in C4-2 cells containing the PPARγ siRNA oligos (Fig. 6A inset). Ciglitazone enhanced DHT- induced AR activation in cells exposed to control siRNA oligos. However, the ability of DHT to enhance AR activity was not altered in C4-2 cells transfected with the PPARγ siRNA (Fig. 6A). These data suggest that ciglitazone functions through PPARγ to increase AR transcriptional activity in the androgen- independent C4-2 cell line.
Fig 6. PPARγ is required for ciglitazone- mediated increases in AR transcriptional activity.

In (A) C4-2 cells were transfected with the PSAE- luciferase reporter, the CMV β-galactosidase plasmid, and either a non- specific control or PPARγ SMARTpool siRNA. In (B) LNCaP cells were transfected with the PSAE- luciferase reporter, the CMV β-galactosidase plasmid, and either a pcDNA 3.1 empty vector or PPARγ cDNA expression vector. For both panels, following transfection, both C4-2 and LNCaP cells were treated for 24 hours with the indicated drugs. Luciferase activity in treated cells was measured and normalized to β-galactosidase activity. Each bar represents the mean ± SD of three wells. *, P< 0.05 compared to DHT alone. In parallel wells, PPARγ and actin protein levels were measured using Western blot analysis (insets). A representative experiment is shown.
To further characterize the role of PPARγ in the ciglitazone- mediated AR activity response, we overexpressed PPARγ in the LNCaP cells, which do not express a significant amount of functional PPARγ (Fig. 1). LNCaP cells were transiently transfected with either a pcDNA 3.1 vector control or a PPARγ cDNA expression plasmid, in addition to the AR responsive PSAE- luciferase reporter plasmid. As shown by Western blot, we were able to detect more PPARγ protein in the LNCaP cells transfected with the PPARγ cDNA (Fig. 6B inset). Ciglitazone treatment reduced the DHT- induced increase in AR activation in the LNCaP cells transfected with the control vector. In the LNCaP cells overexpressing PPARγ ciglitazone did not inhibit the DHT- mediated AR activity (Fig. 6B). These data suggest that the ability of ciglitazone to reduce AR activation in LNCaP cells is due in part to the lack of functional PPARγ within this cell line. In addition, it further supports the idea that ciglitazone, through activation of PPARγ, increases AR activity in androgen- independent C4-2 cells.
Regulation of cyclin D1 by ciglitazone appears to enhance AR transcriptional activity
Cyclin D1 (CD1) is a known corepressor for the AR in LNCaP prostate cancer cells [22]. Our laboratory has previously shown that ciglitazone down- regulates CD1 protein expression in the PC-3 prostate cancer cell line [17]. Western blots revealed ciglitazone also decreases CD1 protein expression in C4-2 cells (Fig. 7A and [17]). However, ciglitazone did not reduce CD1 protein levels in the LNCaP cells (Fig. 7A). Since reductions in CD1 can result in elevated AR signaling, it is possible that ciglitazone enhances AR activity in C4-2 cells by regulating the expression of the corepressor CD1. To test this hypothesis, we tested how overexpression of CD1 affects the ability of ciglitazone to increase AR activity in the C4-2 cell line. In PC-3 cells, ciglitazone decreases CD1 level by increasing proteasome- mediated degradation of CD1 protein [17]. Therefore, in these studies we transfected the C4-2 cells with a plasmid that encodes a mutant form of CD1 (CD1- T286A) that remains nuclear and is resistant to proteasomal degradation [23, 24]. Western blot analysis demonstrated that the level of CD1 protein expression in the C4-2 cells with the CD1- T286A construct is dramatically higher than that present in cells containing the pcDNA 3.1 vector (Fig. 7B inset). In C4-2 cells transfected with the control pcDNA 3.1 vector, ciglitazone enhanced DHT- induced AR transcriptional activity. However, ciglitazone did not enhance DHT- induced AR activity in C4-2 cells expressing the CD1- T286A mutant (Fig. 7B). Taken together, these data suggest ciglitazone- induced decreases in CD1 contribute to the enhanced AR transcriptional activation produced by ciglitazone in the C4-2 cell line.
Fig 7. Ciglitazone regulates the AR corepressor cyclin D1 in C4-2 but not LNCaP cells.

(A) Each cell line was treated with ethanol vehicle (-) or ciglitazone (30 uM) over a 24 hour time period. The amount of cyclin D1 (CD1) and actin expression was then measured by Western blot. (B) C4-2 cells were transfected with the PSAE- luciferase reporter, CMV β-galactosidase plasmid and either a pcDNA 3.1 control or cyclin D1 overexpressing vector (CD1- T286A). Following transfection, the cells were treated for 24 hours with the indicated compounds. Luciferase activity was measured and normalized to the level of β-galactosidase activity in each well. Western blot analysis was performed with the samples from parallel wells to assess the level of cyclin D1 expression in transfected cells (Fig. 6B inset). Each bar represents the mean ± SD of three wells. *, P< 0.05 compared to DHT alone. A representative experiment is shown.
DISCUSSION
In this report, we examined the regulation of AR activation by one class of PPARγ ligands, TZDs, in the androgen-dependent LNCaP cell line and an androgen-independent derivative of the LNCaP cells, the C4-2 cell line. Several reports indicate that PPARγ ligands troglitazone, pioglitazone and 15d PGJ2 inhibit AR transcriptional activation in the androgen- dependent LNCaP cell line [12, 19, 25, 26]. Consistent with these observations, our data indicate the PPARγ ligand ciglitazone also inhibits DHT-induced AR transcriptional activation in LNCaP cells. Like the parental LNCaP cell line, the androgen-independent C4-2 cells express comparable levels of the mutated T877A form of AR ([18] and Fig 3A). Ciglitazone also reduced proliferation of both the LNCaP and C4-2 cell lines. Therefore, we expected that ciglitazone and other TZDs would be equally effective at reducing AR activity in C4-2 cells. However, our data demonstrate that ciglitazone does not inhibit DHT-induced AR activation in the androgen-independent C4-2 human prostate cancer cell line. Ciglitazone increased AR activity in two luciferase reporter assays (PSAE- luciferase and ARR2PB- luciferase). It also did not suppress DHT- stimulated increases in PSA protein or mRNA expression in the C4-2 cells. It is important to note the PSAE- luciferase contains only a portion of the endogenous PSA promoter. This difference might contribute to the fact that ciglitazone increased DHT- induced AR activity in assays involving the PSAE- luciferase reporter, but did not increase DHT- induced expression of the endogenous PSA gene product. We do not know why the data obtained from the PSAE- luciferase assays does not exactly mimic the regulation of PSA mRNA and protein produced by ciglitazone. Nevertheless, the data from both types of AR activation assays suggest that ciglitazone does not inhibit AR activity in the C4-2 cell line as it does in the LNCaP cell line. Our findings suggest that ciglitazone regulates AR activity differently in the androgen-dependent LNCaP cells versus the androgen-independent C4-2 prostate cancer cell line.
Our study is the first to note that PPARγ is expressed and transcriptionally active in C4-2 cells. In addition, the amount of functional PPARγ expressed in C4-2 cells is greater than that found in the parental LNCaP cell line. Although PPARγ protein appeared to be expressed in the LNCaP cells, we were unable to detect any increase in PPARγ activation upon treatment with synthetic TZDs or proposed endogenous PPARγ ligands. This finding is in agreement with a previous report by Yang et al which suggested LNCaP cells express very little if any transcriptionally active PPARγ [19]. Previous reports have suggested that PPARγ expression increases as normal prostatic epithelial cells develop into malignant cells. PPARγ expression is increased in benign prostatic hyperplasia (BPH) and prostatic intraepithelial neoplasia (PIN) as compared to normal prostate tissue [15]. In addition, PPARγ mRNA and protein levels are expressed at higher levels in prostate cancer cells relative to normal prostate epithelial cells [8, 9]. It has been suggested that PPARγ expression also increases with the development of androgen-independent prostate cancer, for there are lower amounts of PPARγ present in the androgen-dependent LNCaP cells than in the independently derived androgen-independent PC-3 and DU145 cells [10]. Since PC-3 and DU145 cells lack AR, it was not known whether PPARγ levels are also elevated in AR positive, androgen- independent prostate cancers. Our data show that androgen- independent C4-2 cells express higher levels of PPARγ protein than the androgen- dependent LNCaP cell line. Additionally, we show that the minimal amount of PPARγ expressed in the LNCaP cells is not functional. Thus our data provide evidence that further suggests increased expression of PPARγ is associated with the development of androgen- independent prostate cancer.
Multiple reports from the C.S. Chen laboratory indicate that PPARγ ligands mediate AR transcriptional activity independent of PPARγ activation in the LNCaP cell line [13, 19, 27]. They have demonstrated PPARγ-inactive analogues of ciglitazone and troglitazone are still able to repress PSA protein levels and DHT- induced AR transcriptional activation in the LNCaP cell line [19]. In this paper, we were unable to detect significant levels of functional PPARγ in the LNCaP cells, but ciglitazone was effective at reducing AR activation in this cell line. Therefore, our data support the idea that the decrease in AR transcriptional activity induced by PPARγ ligands is independent of PPARγ in the LNCaP cell line. When we overexpressed PPARγ in the LNCaP cell line, ciglitazone was not able to inhibit the DHT- mediated increases in AR activity. However, ciglitazone enhances ligand- induced AR activation in the C4-2 cell line, which expresses PPARγ. In addition, when PPARγ levels were reduced in the C4-2 cell line, ciglitazone was not able to increase DHT- mediated AR transcriptional activity. Therefore, the presence of functional PPARγ may be linked to the differential regulation of AR activation by ciglitazone in the androgen- dependent versus androgen-independent prostate cancer cell lines.
The level of coactivator and corepressor expression regulates transcriptional activation of the AR and other nuclear receptors. Cyclin D1 (CD1) is a corepressor that inhibits AR transcriptional activation by interacting with the amino terminal region of the AR [22]. It has been shown that at concentrations greater than 40μM, the PPARγ ligand troglitazone decreases CD1 protein expression in LNCaP cells [13, 28]. However, under our experimental conditions, we did not see a decrease in CD1expression with ciglitazone in the LNCaP cell line. Our data show that there is differential regulation of CD1 by ciglitazone in LNCaP and C4-2 cells. While ciglitazone did not modulate CD1 expression in the LNCaP cells, it dramatically reduced CD1 levels in the C4-2 cell line. In addition, overexpression of CD1 prevented ciglitazone- stimulated increases in DHT- dependent AR activity. Thus ciglitazone- induced increases in AR activity appear to be linked to the down- regulation of the corepressor CD1 in C4-2 androgen- independent prostate cancer cells. SMRT, NCoR [29–31] and SIRT1 [32] are other proteins that have been shown to function as AR corepressors. siRNA- mediated knockdown of NCoR and SMRT has been shown to increase ligand- mediated AR signaling in prostate cancer cells [31, 33]. DHT- induced activation of AR is also enhanced in the presence of nicotinamide, a chemical inhibitor of SIRT1 [32]. Thus altered expression or chemically induced inhibition of corepressors can also influence the level of AR signaling within cells. We do not know if ciglitazone regulates expression and/or activity of other corepressors. However, it is possible that the increase in AR activity induced by ciglitazone in androgen- independent prostate cancer cells may be due not only to the regulation of cyclin D1 but also other corepressors.
In summary, our findings suggest a novel mechanism of action for ciglitazone in the regulation of AR activation in C4-2 human prostate cancer cells. While ciglitazone inhibits AR activation in androgen-dependent LNCaP human prostate cancer cells, it does not lower AR activity in the androgen-independent C4-2 cell line. The regulation of AR activity by ciglitazone in C4-2 cells appears to require PPARγ and is linked to ciglitazone- induced down- regulation of the corepressor CD1. Although ciglitazone did not suppress AR activity, it effectively decreased the proliferation of androgen- independent C4-2 human prostate cancer cells. This would suggest that inhibition of AR is not the primary mechanism by which ciglitazone reduces the growth of androgen- independent C4-2 cells. It still needs to be determined whether ciglitazone- induced reductions in AR activity are critical for the anti- proliferative effect of ciglitazone in the androgen- dependent LNCaP cells. Nonetheless, our data support the idea that PPARγ plays little if any role in the ability of ciglitazone to reduce proliferation and suppress ligand- activated AR activity in LNCaP cells. Additional studies need to be conducted in order to fully understand the signaling pathways mediated by ciglitazone, as well as other PPARγ agonists, to prevent tumor growth in androgen- dependent and advanced stage, androgen- independent forms of prostate cancer.
Acknowledgments
We thank Dr. Robert Matusik (Vanderbilt University) for the ARR2PB-luciferase and PSAE-luciferase reporter plasmids; Dr. Stephen Safe (Texas A&M University) for the PPRE- luciferase reporter plasmid; Dr. Nancy Weigel (Baylor College of Medicine) for the CMV-β-galactosidase construct; and Drs. J. Alan Diehl (University of Pennsylvania) and Karen Knudsen (Thomas Jefferson University) for the CD1- T286A overexpression and pcDNA 3.1 vectors, respectively. This work was supported by a NCI K01 Career Development Award (CA114253) and a Department of Defense PCRP New Investigator Award (W81XWH-05-1-0605).
Abbreviations
- AR
androgen receptor
- PPARγ
peroxisome proliferator activated receptor gamma
- DHT
dihydrotestosterone
- PSA
prostate specific antigen
- AAT
androgen ablation therapy
- TZD
thiazolidinedione
- CD1
cyclin D1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.American Cancer Society. cancer facts and figures, 2009. www.acsevents.org.
- 2.Westin P, Stattin P, Damber JE, Bergh A. Castration therapy rapidly induces apoptosis in a minority and decreases cell proliferation in a majority of human prostatic tumors. Am J Pathol. 1995;146:1368–1375. [PMC free article] [PubMed] [Google Scholar]
- 3.Balk SP. Androgen receptor as a target in androgen-independent prostate cancer. Urology. 2002;60:132–138. doi: 10.1016/s0090-4295(02)01593-5. discussion 138–139. [DOI] [PubMed] [Google Scholar]
- 4.Burnstein KL. Regulation of androgen receptor levels: implications for prostate cancer progression and therapy. J Cell Biochem. 2005;95:657–669. doi: 10.1002/jcb.20460. [DOI] [PubMed] [Google Scholar]
- 5.Arnold JT, Isaacs JT. Mechanisms involved in the progression of androgen-independent prostate cancers: it is not only the cancer cell’s fault. Endocr Relat Cancer. 2002;9:61–73. doi: 10.1677/erc.0.0090061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peehl DM, Feldman D. Interaction of nuclear receptor ligands with the vitamin D signaling pathway in prostate cancer. J Steroid Biochem Mol Biol. 2004;92:307–315. doi: 10.1016/j.jsbmb.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 7.Smith MR, Kantoff PW. Peroxisome proliferator-activated receptor gamma (PPargamma) as a novel target for prostate cancer. Invest New Drugs. 2002;20:195–200. doi: 10.1023/a:1015670126203. [DOI] [PubMed] [Google Scholar]
- 8.Nwankwo JO, Robbins ME. Peroxisome proliferator-activated receptor- gamma expression in human malignant and normal brain, breast and prostate-derived cells. Prostaglandins Leukot Essent Fatty Acids. 2001;64:241–245. doi: 10.1054/plef.2001.0266. [DOI] [PubMed] [Google Scholar]
- 9.Xu Y, Iyengar S, Roberts RL, Shappell SB, Peehl DM. Primary culture model of peroxisome proliferator-activated receptor gamma activity in prostate cancer cells. J Cell Physiol. 2003;196:131–143. doi: 10.1002/jcp.10281. [DOI] [PubMed] [Google Scholar]
- 10.Chaffer CL, Thomas DM, Thompson EW, Williams ED. PPARgamma-independent induction of growth arrest and apoptosis in prostate and bladder carcinoma. BMC Cancer. 2006;6:53. doi: 10.1186/1471-2407-6-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang M, Shappell SB, Hayward SW. Approaches to understanding the importance and clinical implications of peroxisome proliferator-activated receptor gamma (PPARgamma) signaling in prostate cancer. J Cell Biochem. 2004;91:513–527. doi: 10.1002/jcb.10770. [DOI] [PubMed] [Google Scholar]
- 12.Hisatake JI, Ikezoe T, Carey M, Holden S, Tomoyasu S, Koeffler HP. Down-regulation of prostate-specific antigen expression by ligands for peroxisome proliferator-activated receptor gamma in human prostate cancer. Cancer Res. 2000;60:5494–5498. [PubMed] [Google Scholar]
- 13.Yang CC, Wang YC, Wei S, Lin LF, Chen CS, Lee CC, Lin CC. Peroxisome proliferator-activated receptor gamma-independent suppression of androgen receptor expression by troglitazone mechanism and pharmacologic exploitation. Cancer Res. 2007;67:3229–3238. doi: 10.1158/0008-5472.CAN-06-2759. [DOI] [PubMed] [Google Scholar]
- 14.Wu HC, Hsieh JT, Gleave ME, Brown NM, Pathak S, Chung LW. Derivation of androgen-independent human LNCaP prostatic cancer cell sublines: role of bone stromal cells. Int J Cancer. 1994;57:406–412. doi: 10.1002/ijc.2910570319. [DOI] [PubMed] [Google Scholar]
- 15.Segawa Y, Yoshimura R, Hase T, Nakatani T, Wada S, Kawahito Y, Kishimoto T, Sano H. Expression of peroxisome proliferator-activated receptor (PPAR) in human prostate cancer. Prostate. 2002;51:108–116. doi: 10.1002/pros.10058. [DOI] [PubMed] [Google Scholar]
- 16.Qin C, Burghardt R, Smith R, Wormke M, Stewart J, Safe S. Peroxisome proliferator-activated receptor gamma agonists induce proteasome-dependent degradation of cyclin D1 and estrogen receptor alpha in MCF-7 breast cancer cells. Cancer Res. 2003;63:958–964. [PubMed] [Google Scholar]
- 17.Lyles BE, Akinyeke TO, Moss PE, Stewart LV. Thiazolidinediones regulate expression of cell cycle proteins in human prostate cancer cells via PPARgamma-dependent and PPARgamma-independent pathways. Cell Cycle. 2009;8:268–277. doi: 10.4161/cc.8.2.7584. [DOI] [PubMed] [Google Scholar]
- 18.Yeung F, Li X, Ellett J, Trapman J, Kao C, Chung LW. Regions of prostate-specific antigen (PSA) promoter confer androgen-independent expression of PSA in prostate cancer cells. J Biol Chem. 2000;275:40846–40855. doi: 10.1074/jbc.M002755200. [DOI] [PubMed] [Google Scholar]
- 19.Yang CC, Ku CY, Wei S, Shiau CW, Chen CS, Pinzone JJ, Ringel MD. Peroxisome Proliferator-Activated Receptor {gamma}-Independent Repression of Prostate-Specific Antigen Expression by Thiazolidinediones in Prostate Cancer Cells. Mol Pharmacol. 2006 doi: 10.1124/mol.105.018333. [DOI] [PubMed] [Google Scholar]
- 20.Lee SE, Jin RJ, Lee SG, Yoon SJ, Park MS, Heo DS, Choi H. Development of a new plasmid vector with PSA-promoter and enhancer expressing tissue-specificity in prostate carcinoma cell lines. Anticancer Res. 2000;20:417–422. [PubMed] [Google Scholar]
- 21.Zhang J, Thomas TZ, Kasper S, Matusik RJ. A small composite probasin promoter confers high levels of prostate-specific gene expression through regulation by androgens and glucocorticoids in vitro and in vivo. Endocrinology. 2000;141:4698–4710. doi: 10.1210/endo.141.12.7837. [DOI] [PubMed] [Google Scholar]
- 22.Knudsen KE, Cavenee WK, Arden KC. D-type cyclins complex with the androgen receptor and inhibit its transcriptional transactivation ability. Cancer Res. 1999;59:2297–2301. [PubMed] [Google Scholar]
- 23.Alt JR, Cleveland JL, Hannink M, Diehl JA. Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation. Genes Dev. 2000;14:3102–3114. doi: 10.1101/gad.854900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petre CE, Wetherill YB, Danielsen M, Knudsen KE. Cyclin D1: mechanism and consequence of androgen receptor co-repressor activity. J Biol Chem. 2002;277:2207–2215. doi: 10.1074/jbc.M106399200. [DOI] [PubMed] [Google Scholar]
- 25.Mueller E, Smith M, Sarraf P, Kroll T, Aiyer A, Kaufman DS, Oh W, Demetri G, Figg WD, Zhou XP, Eng C, Spiegelman BM, Kantoff PW. Effects of ligand activation of peroxisome proliferator-activated receptor gamma in human prostate cancer. Proc Natl Acad Sci U S A. 2000;97:10990–10995. doi: 10.1073/pnas.180329197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kubota T, Koshizuka K, Williamson EA, Asou H, Said JW, Holden S, Miyoshi I, Koeffler HP. Ligand for peroxisome proliferator-activated receptor gamma (troglitazone) has potent antitumor effect against human prostate cancer both in vitro and in vivo. Cancer Res. 1998;58:3344–3352. [PubMed] [Google Scholar]
- 27.Yang J, Wei S, Wang DS, Wang YC, Kulp SK, Chen CS. Pharmacological exploitation of the peroxisome proliferator-activated receptor gamma agonist ciglitazone to develop a novel class of androgen receptor-ablative agents. J Med Chem. 2008;51:2100–2107. doi: 10.1021/jm701212m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei S, Yang HC, Chuang HC, Yang J, Kulp SK, Lu PJ, Lai MD, Chen CS. A novel mechanism by which thiazolidinediones facilitate the proteasomal degradation of cyclin D1 in cancer cells. J Biol Chem. 2008;283:26759–26770. doi: 10.1074/jbc.M802160200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liao G, Chen LY, Zhang A, Godavarthy A, Xia F, Ghosh JC, Li H, Chen JD. Regulation of androgen receptor activity by the nuclear receptor corepressor SMRT. J Biol Chem. 2003;278:5052–5061. doi: 10.1074/jbc.M206374200. [DOI] [PubMed] [Google Scholar]
- 30.Shang Y, Myers M, Brown M. Formation of the androgen receptor transcription complex. Mol Cell. 2002;9:601–610. doi: 10.1016/s1097-2765(02)00471-9. [DOI] [PubMed] [Google Scholar]
- 31.Yoon HG, Wong J. The corepressors silencing mediator of retinoid and thyroid hormone receptor and nuclear receptor corepressor are involved in agonist- and antagonist-regulated transcription by androgen receptor. Mol Endocrinol. 2006;20:1048–1060. doi: 10.1210/me.2005-0324. [DOI] [PubMed] [Google Scholar]
- 32.Dai Y, Ngo D, Forman LW, Qin DC, Jacob J, Faller DV. Sirtuin 1 is required for antagonist-induced transcriptional repression of androgen-responsive genes by the androgen receptor. Mol Endocrinol. 2007;21:1807–1821. doi: 10.1210/me.2006-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hodgson MC, Astapova I, Hollenberg AN, Balk SP. Activity of androgen receptor antagonist bicalutamide in prostate cancer cells is independent of NCoR and SMRT corepressors. Cancer Res. 2007;67:8388–8395. doi: 10.1158/0008-5472.CAN-07-0617. [DOI] [PubMed] [Google Scholar]