

Abstract
Recent studies are beginning to paint a clear and consistent picture of the impairments in psychological and cognitive competencies that are associated with microdeletions in chromosome 22q11.2. These studies have highlighted a strong link between this genetic lesion and schizophrenia. Parallel studies in humans and animal models are starting to uncover the complex genetic and neural substrates altered by the microdeletion. In addition to offering a deeper understanding of the effects of this genetic lesion, these findings may guide analysis of other copy-number variants associated with cognitive dysfunction and psychiatric disorders.
The 22q11.2 deletion syndrome (22q11.2DS; also known as velocardiofacial syndrome1 or DiGeorge syndrome2) is caused by a microdeletion in chromosome 22 and has an incidence of 1 in 2,000–4,000 live births3-5. Although atypical microdeletions have been described6, most of the microdeletions are either 3 megabases (Mb) in size7,8 (including approximately 60 known genes) or 1.5 Mb in size7,8 (including approximately 35 known genes), and most of the genes affected are expressed in the brain9 (Supplementary information S1 (table)). The phenotype of the disorder is highly variable10 and can affect multiple organs and tissues, but the severity is unrelated to the size of the deletion11, which suggests that genes within the 1.5 Mb region are crucial for the etiology of the syndrome. Phenotypic variability may be due to breakpoint heterogeneity as well as other genetic, environmental and stochastic factors. Common physical manifestations of the disorder include craniofacial and cardiovascular anomalies, immunodeficiency, short stature and hypocalcaemia3,10,12.
Individuals with 22q11.2DS also have cognitive and behavioural impairments and a high risk for developing schizophrenia. Long-term medical care and prenatal screening are increasingly being directed towards the treatment and recognition of these symptoms. Recent studies in humans and animal models have shed light on the neuroanatomical and cognitive manifestations of 22q11.2DS. In addition, there is growing evidence for a widespread role of copy-number variants (CNVs) — which include chromosomal microdeletions and microduplications — in determining susceptibility to cognitive disorders and schizophrenia13.
In this Review, we describe the cognitive, psychiatric and neuroanatomical phenotypes associated with 22q11.2DS. We summarize the emerging principles that underlie the genetic architecture of the syndrome and outline recent advances in animal model studies. Finally, we attempt to integrate existing knowledge on the disease and define future goals for the identification of the genetic and neural elements of the cognitive and psychiatric phenotypes. A detailed discussion of human genetic association studies in this locus is not within the scope of this Review.
Human 22q11.2 deletion syndrome phenotype
Cognitive impairments
Most school-aged children with 22q11.2DS have lower than typical full scale IQ (FSIQ). Borderline intellectual function (FSIQ 70–75) is most common, mild intellectual disability (FSIQ 55–75) is slightly less frequent and a small percentage of children fall into the low average intelligence range (FSIQ >85)14. Most children with 22q11.2DS achieve higher scores in verbal tasks than in nonverbal tasks, although this pattern of dysfunction is not universal14-17. Whether this profile of impairments changes as children age is unclear.
Not all functional domains have been studied equally, and a clear pattern of cognitive strengths and weaknesses in children with 22q11.2DS has not yet been established. However, standardized neuropsychological tests have revealed some consistencies. Although receptive language (the ability to understand what is being said) is generally strong in preschool children, by school age general language delays are apparent17-21. Nevertheless, during school years, reading, spelling and rote verbal memory scores are in the low average to superior range16,22,23 and verbal memory is consistently stronger than visual–spatial memory24-26. The domains of attention and executive function have not received detailed psychometric study, although impairments have been reported in both23,27. Overall, the profile of stronger verbal abilities and weaker nonverbal, spatiotemporal and numerical competence in most individuals is similar to that of other neurogenetic disorders, such as fragile X, Williams and Turner syndromes28.
The analytical power of standardized tests is limited because each one generally encompasses several cognitive functions. Analyses of more specific aspects of cognitive function in 22q11.2DS have been initiated (TABLE 1); however, this effort remains far from comprehensive. Much of the work attempts to explain the nonverbal cognitive impairments in 22q11.2DS in terms of changes in lower-level cognitive functions and the associated brain systems. Its hypothesis-driven nature has focused many studies on the attention system, which functions to select task- or goal-relevant information from the environment and to inhibit the processing of irrelevant information29-31. Attention seems to be essential for many nonverbal cognitive functions, which suggests that impairments in attention may be a major cause of the cognitive profile in 22q11.2DS. Studies suggest that children with 22q11.2DS have difficulty in finding and interpreting salient spatial and temporal information32-34, which indicates that their attention is engaged less efficiently in terms of navigating space, guiding vision and selecting and integrating goal-relevant information. Although the developing neural circuitry of children with 22q11.2DS has not been examined, these impairments indicate dysfunction in parietal and frontal cortical circuitry31,35.
Table 1.
Experimental cognitive task descriptions and pattern of findings in children with 22q11.2 deletion syndrome
| Name | Function(s) | Task | Findings | Refs |
|---|---|---|---|---|
| Cueing | Visual–spatial selective attention |
Detect a target stimulus on screen following spatial cues that facilitate detection (valid cues) or hinder detection (invalid cues) |
Impaired response to invalid cues |
175,176 |
| Attention networks test |
Selective, alerting and executive attention |
Identify the direction of an arrow or similar stimulus with the presence or absence of spatial cues and distracting flankers |
Executive system impairments mainly reported |
30,177 |
| Object/space selection |
Visual–spatial selective attention |
Detect a target stimulus on screen while valid or invalid cues direct attention to locations in or outside bounded objects |
Greater impairment of spatial attention compared with object attention |
178 |
| Inhibition of return | Visual–spatial attention control |
Detect a target stimulus on screen in a cued or non-cued location after a short or long delay |
Delayed relative to TD controls |
179,180 |
| Enumeration | Quantification, attention |
Enumerate small to large sets of visually presented objects on screen while being timed |
Impaired only in counting range |
181,182 |
| Magnitude comparison |
Quantitative judgment |
Identify when stimuli of varying magnitudes are different from each other |
Impairment greater with smaller differences |
183,184 |
| Prepulse inhibition | Inhibitory function | Suppress startle response following an almost imperceptible warning tone before an aversive noise |
Impaired relative to TD controls |
185 |
| Mismatch negativity, oddball |
Executive function, attention |
Detect unusual stimuli in a stream of serially presented items | Reduced frontal lobe signal during detection |
186,187 |
| N-back | Working memory | Identify whether the current letter matches one that was presented N spots earlier in the stream of items. For example, in a 2-back task the participant tries to remember whether the current letter matches one that was presented 2 letters earlier in the stream of stimuli |
Accuracy and frontal lobe activation reduced |
188 |
| Go–NoGo | Inhibitory, executive function |
Maintain memory rule and inhibit response to a rare non-target item in a stream of common target items |
More parietal activation than TD controls |
189,190 |
Basic numerical cognition also depends on spatial attention32,36. Counting more than three or four items requires spatial attention, whereas subitizing — the rapid enumeration of fewer than three or four items — does not37-39. As predicted, counting but not subitizing performance is impaired in children with 22q11.2DS32. Children and adults with 22q11.2DS demonstrate impairments in ‘magnitude comparison’ and ‘time duration comparison’32,36,40, further strengthening the connections among spatial, temporal and numerical impairments and implicating dysfunction in the parietal and frontal circuitry that underlies attentional and numerical cognition41-44.
Attention is also important for inhibiting the processing of irrelevant information. Impairments in this ability have been reported in children with 22q11.2DS27,45-47. Other inhibitory impairments, such as reduced sensorimotor gating (usually in the form of inhibition of a startle response using the ‘prepulse inhibition’ (PPI) paradigm), have been reported and are associated with impairments in tests of attention and with high-risk schizophrenia symptoms27,48. Related functional impairments have been reported in children and young adults with 22q11.2DS49,50, using the ‘mismatch negativity’ (MMN) paradigm. This task, which is often associated with executive dysfunction and psychiatric symptoms, measures the ability to detect a novel stimulus within the context of many similar items.
Despite the importance of working memory (WM) as a core component of executive function and its association with the dorsolateral prefrontal cortex (PFC), there has been relatively little study of WM in 22q11.2DS. However, a functional MRI (fMRI) study of non-spatial WM51 in children and psychometric studies of children25 and adults52 have reported WM impairments in patients with 22q11.2DS.
Susceptibility to schizophrenia
A strong and specific relationship exists between the presence of the 22q11.2 microdeletion and schizophrenia53,54. Individuals with the 22q11.2 deletion are sometimes given other diagnoses early in life, including attention-deficit hyperactivity disorder (ADHD), generalized anxiety disorder, obsessive compulsive disorder and autism spectrum disorders55-59. However, with the exception of schizophrenia, most of these diagnoses may not meet the criteria set forth in the literature60,61 for a behavioural phenotype that is specifically associated with a syndrome. Specifically, most of these conditions are not found at a higher frequency among individuals with the 22q11.2 microdeletion than in cohorts with other developmental disorders associated with learning disabilities. In addition, no significant enrichment of 22q11.2 deletions has been identified in cohorts of children diagnosed with some of these conditions62-64. This indicates that the behavioural diagnoses other than schizophrenia represent nonspecific expressions of factors that affect brain development and function or, alternatively, that their diagnostic criteria are not designed for children with developmental disabilities and so may lead to the reporting of erroneously high rates of these phenotypes. Additional complications may arise owing to the association with schizophrenia. For example, diagnoses of autism spectrum disorders may reflect misdiagnosis of social impairments associated with premorbidity to schizophrenia65.
In late adolescence and early adulthood, up to one-third of all individuals carrying the 22q11.2 deletion develop schizophrenia or schizoaffective disorder as defined strictly in the Diagnostic and Statistical Manual of Mental Disorders66-70. This inordinately high risk of developing schizophrenia is not associated with any other neurogenetic syndrome. 22q11.2 microdeletions account for up to 1–2% of schizophrenia cases53,54,71,72 and are the only confirmed recurrent structural mutations that are responsible for introducing sporadic cases of schizophrenia into the population. The 22q11.2 microdeletion is therefore one of the highest known risk factors for schizophrenia (BOX 1). Importantly, there are no major clinical differences in the core schizophrenia pheno type between individuals with schizophrenia who are 22q11.2 microdeletion carriers and those who are not73,74. Moreover, due to high phenotypic variability, many individuals with 22q11.2DS who develop schizophrenia have no serious intellectual disability and their congenital abnormalities (such as facial dysmorphologies) can be so subtle that they may be missed on clinical evaluation, making these individuals indistinguishable from other schizophrenia patients. Whether any neuroanatomical abnormalities distinguish those patients carrying the 22q11.2 deletion who develop schizophrenia from those who do not is unclear (see below). The few completed studies aimed at defining the risk factors in 22q11.2DS that predict the development of psychosis have hinted at the relationships to focus on68 (BOX 2), but further exploration is needed.
Box 1. Rare mutations in psychiatric disorders.
Psychiatric disorders, like other common disorders, are multifactorial in nature with complex genetic etiologies. The genetic architecture underlying disease susceptibility is characterized by both the frequency and penetrance of risk alleles162. Frequency is defined here as the proportion of the chromosomes carrying the risk allele, and penetrance is defined as the proportion of individuals carrying a risk allele that actually exhibit the disease symptoms. The common disease–common allele (CDCA) hypothesis emphasizes the importance of relatively common alleles, each of low penetrance, acting together to increase disease risk. Conversely, the common disease–rare allele (CDRA) hypothesis emphasizes the impact of individually rare yet highly penetrant alleles. The reproducible observation of rare and highly penetrant de novo structural mutations at the 22q11.2 locus in sporadic (non-familial) cases of schizophrenia provided the first evidence to support the importance of rare recurrent mutations in schizophrenia vulnerability53. This association seems to be specific, as 22q11.2 microdeletions are not enriched in, for example, cases with autism64. A number of recent studies, made possible by technological advances, have extended and complemented the initial observation in the 22q11.2 locus54,71,72. Although the pattern of bidirectional association described for the 22q11.2 microdeletion has yet to be demonstrated for any of the recently discovered structural mutations, it is now widely recognized that rare structural mutations (both inherited and de novo) collectively have a substantial etiological role and account for a considerable portion of sporadic and familial cases of the disease. Notably, de novo mutations, such as the 22q11.2 microdeletions, can at least in part explain how schizophrenia persists in the population despite the low fecundity of affected individuals54.
Box 2. Prodromal symptoms predictive of schizophrenia.
Children with 22q11.2 microdeletions exhibit impairments in a range of cognitive functions that are also seen in those diagnosed with schizophrenia49,163,164. The same is true for some of the impairments in temporal processing165,166. It is unlikely that a simple relationship exists between individual cognitive dysfunctions or sets of cognitive dysfunctions and high risk for schizophrenia. Similarly, the predictive relevance for future psychosis of many structural, functional and connective brain findings remains equally unclear. Several studies49,56,57,68 have identified patterns of psychotic, prodromal and associated symptoms in young people with 22q11.2 deletion syndrome. The most stable symptoms seem to be attention deficit, mood and anxiety disorders and impaired social adaptive skills. The only known prospective study68 found that in children, the development of psychotic symptoms between being studied at age 12 and at age 18 was best predicted in part by the presence of psychotic symptoms at the time of the baseline study and in part by anxiety and depression scores. When predicting follow-up psychosis rating-scale scores, lower IQ at baseline was a further predictor. Attention-deficit hyperactivity disorder was not a predictor of psychotic outcomes. A related study167 found that intracranial and cerebellar white matter and superior temporal gyrus and caudate volumes increased with age but that amygdalar volumes decreased. Declining verbal IQ was associated with reductions in left cerebral grey matter.
Neuroanatomical changes
Given the well-established links between specific brain regions and circuits, and the cognitive functions that are impaired in 22q11.2DS, it is reasonable to suggest that there may be specific neural changes in individuals with 22q11.2DS75. Given that many physical anomalies associated with 22q11.2DS affect midline structures, there is good reason to expect that this is also true for the brain.
As in other neurodevelopmental disorders, reduced brain volumes relative to typical developing (TD) controls are well documented in children with 22q11.2DS, with slightly greater reductions in posterior compared with anterior regional volumes and in total white matter compared with grey matter76-80. Despite an extensive literature on neuroimaging81, studies have been less consistent regarding changes in specific neural regions or structures. Therefore, to be conservative, we discuss here (BOX 3) only the differences that have been reported by at least two independent groups in separate studies involving mostly or exclusively children. Direct functional correlates of these structural changes have not yet been firmly established, largely due to the use of different measurement protocols in few studies with relatively small but differing samples and with diverse covariates.
Box 3. The neuroanatomy of brain regions in children with 22q11.2 deletion syndrome.
Some brain structures are either volumetrically enlarged or reduced in children with 22q11.2 deletion syndrome relative to typical developing (TD) controls. Enlarged structures include the lateral ventricles, the caudate and the insula. However, both the hippocampus and medial cerebellum – including the vermis, anterior lobes and neocerebellum – are reduced relative to controls. The figure shows the location of some of these structures in the human brain. The table summarizes the findings of several different studies for each brain region.
Cerebellar differences are summarized in the table but were omitted from the figure because the findings were generated by taking into account the area derived from a single, mid-sagittal slice. Therefore, the extent of the difference in cerebellar size is not known. The figure was created by mapping regional parcellations onto a brain template using the Harvard–Oxford atlas (for the caudate, lateral ventricles and hippocampi) and the Montreal Neurological Institute (MNI) label atlas (for the cerebellum and insula). These atlases are provided in the brain atlas tools distributed with the FMRIB Software Library (FSL). The regions were rendered by in-house scripts created by S. Srivastava at the University of California, Davis, USA, using Matlab and OpenGL.
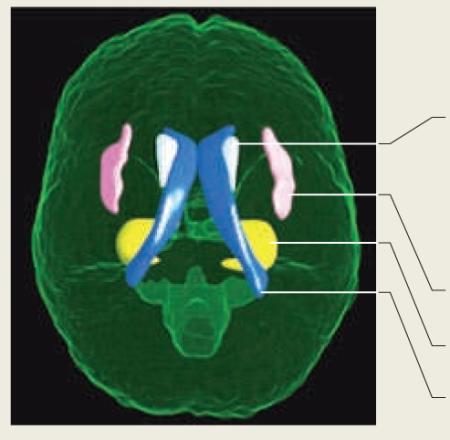
| Structure/region | Finding (variable) | Refs. |
|---|---|---|
| Caudate | 22q11.2 > TD control (volume) |
76,79 168,169 |
| Medial cerebellum (including vermis, anterior lobes and neocerebellum) |
22q11.2 < TD control (area) | 170,171 |
| Insula | 22q11.2 > TD control (volume) | 32,76 |
| Hippocampus | 22q11.2 < TD control (volume) | 32,76-79 |
| Lateral ventricles | 22q11.2 > TD control (volume) | 32,76,88 |
Recently, more advanced methods have replicated and extended these findings. Children with 22q11.2DS and TD children have similar overall cortical thickness. However, lateral thinning in parieto-occipital, occipital pole and inferior prefrontal regions, and medial thinning in anterior cingulate, medial frontal gyrus, subgenual prefrontal, posterior cingulate gyrus, cuneus and lingual gyrus regions is found in 22q11.2DS. Furthermore, the correlation between cortical thinning and increasing age was greater in the 22q11.2DS group82,83. Reduced cortical gyral complexity was found in the frontal and parietal cortices of those with 22q11.2DS, with localized reductions overlapping many of the thinner cortical regions84,85. These differences in gyral complexity, might be associated with changes in neural connectivity, as it has been suggested that tensile influences created by developing connective tracts underlie cortical folding86.
Diffusion tensor imaging (DTI) has enabled the connective patterns of large axonal fibre tracts to be examined more directly, revealing changes in the posterior corpus callosum80,87 (FIG. 1 Supplementary information S2 (table)). Functional relationships have been reported between counting ability88, spatial attention36 and arithmetic89 and frontal and parietal DTI measurements or callosal area, which suggests that this altered connectivity might contribute to these phenotypes in the 22q11.2DS.
Figure 1. Altered connectivity in children with 22q11.2 deletion syndrome.
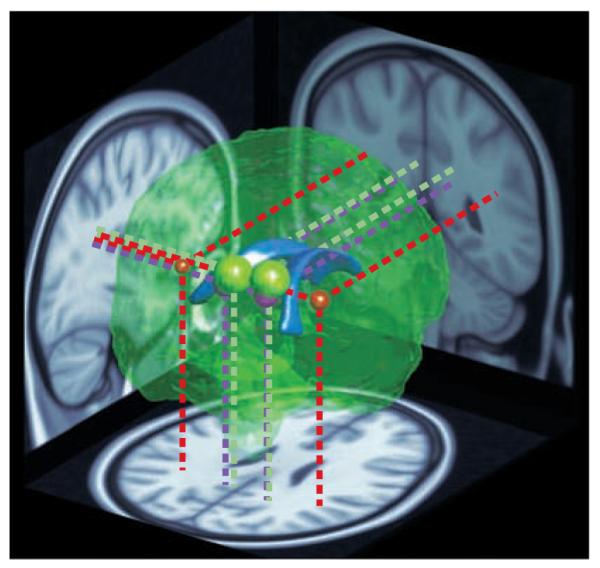
A reconstruction of the brain viewed from the posterior right side. The lateral ventricles (shown in blue) are depicted in the centre as a point of reference. The coloured spheres indicate the location and approximate extent of major clusters of reduced fractional anisotropy (FA) — a measure of neural connectivity — in children with 22q11.2 deletion syndrome (22q11.2DS), as reported by REF. 32 (red), REF. 36 (green) and REF. 87 (purple). The data were obtained using diffusion tensor imaging, an MRI technique that measures the directionality of water diffusion in the brain as an indicator of the organization and integrity of neuronal tracts wrapped in myelin (white matter). The location and size of the coloured spheres depict the sites of peak FA differences and their extent. Projections onto brain slices (dotted lines) indicate the positions of the clusters in major white matter tracts. The red spheres represent the left and right extremities in a wedge-shaped cluster reported by REF. 32. The entire area between them showed higher FA in children with 22q11.2DS compared with typical developing controls, but because this large cluster would completely envelope the other regions, it is not represented here in its entirety. In summary, three separate studies from two different teams have reported similar, and in one case directly overlapping, differences in neural connectivity in children and young adults with 22q11.2DS.
There have been a few fMRI studies on children with 22q11.2DS, which revealed atypical parietal lobe activation during an arithmetic task90, and atypical parietal activation (rather than prefrontal activation) in adolescents with 22q11.2DS in a non-spatial WM task51 and an inhibitory Go–NoGo task91.
This pattern of cortical and subcortical changes is generally consistent with the cognitive impairments observed in 22q11.2DS and with well-established links between particular neuroanatomical structures and cognitive functions in typical individuals. These differences thus provide evidence for atypical development of particular brain circuits in 22q11.2DS. Although the mechanisms that drive this atypical development are not well understood, neuroimaging studies in adolescent and adult carriers of the 22q11.2 microdeletion suggest the presence of a progressive, abnormal maturation process. In individuals with 22q11.2DS, the rostrocaudal gradient of structural alterations described in childhood seems to disappear with age, giving way to more widespread loss of brain tissue, particularly in the frontal and temporal lobes.
The structural anomalies that distinguish those 22q11.2 microdeletion carriers who develop schizophrenia from those who do not are unknown. Preliminary results point to some distinguishing features that are consistent with changes reported for general schizophrenia. These include larger lateral and third ventricles and a decrease in grey and especially white matter volumes in frontal and temporal lobes92,93. In addition, carriers who develop schizophrenia exhibit cognitive impairments that may reflect differences in the development and function of frontal brain regions52,94. One longitudinal study also suggests that decreases in frontal grey matter volume contribute to the susceptibility of adolescents with 22q11.2DS to schizophrenia95.
The findings described indicate a pattern of cognitive dysfunction characterized by an impairment of attentional functioning, accompanied by a very high risk of developing schizophrenia. These symptoms seem to be correlated with aberrant structure, connectivity patterns or activity in several cortical and subcortical areas. Given that a large number of genes are affected by the 22q11.2 microdeletion, these aberrations are likely to be due to combined anomalies in neuronal development, connectivity and synaptic plasticity.
Genetic influences on 22q11.2 deletion syndrome
There are at least three ways in which the removal of genes from the region deleted in 22q11.2DS could cause the cognitive and psychiatric phenotypes of the disorder: firstly, the microdeletion may unmask one or more recessive mutations; secondly, a single dosage-sensitive gene may exert a major effect; and lastly, underexpression of a number of dosage-sensitive genes may have a cumulative effect. According to the cumulative under expression model, although some genes may have a greater impact on phenotype, the imbalance of several deleted genes determines the overall phenotype. The presence of additional trans- or cis-acting genetic modifiers (including DNA variation in genes in the intact chromosome) may also contribute to the variability of the cognitive and psychiatric phenotypes95,96-98.
As outlined below, existing data from animal models also seem to support the underexpression model, and this suggests that there is an oligogenic basis for the neural, behavioural and cognitive 22q11.2DS phenotypes and that the sets of genes responsible for specific deficits may only partially overlap. Indeed, systematic and candidate genetic association approaches also seem to implicate more than one 22q11.2 gene in the psychiatric and cognitive phenotypes of 22q11.2DS99,100. Studies that have linked specific 22q11.2 genes to schizophrenia are indicated in Supplementary information S1 (table), and a more comprehensive account can be found on the Schizophrenia Research Forum website. A detailed discussion of each study is beyond the scope of this Review. These studies typically assume that common variants in the 22q11.2 genes contribute to the disease risk in karyo-typically normal individuals, and they have provided important initial insights into the neurobiology of the 22q11.2DS101-107. Given the well-established difficulties in determining genetic causality in psychiatric disorders using genetic association studies of common variants108, these findings remain for the most part only suggestive due to the small sample sizes used, especially in the absence of functional data. Until these issues are resolved using novel next-generation sequencing approaches, genetically engineered animal models offer a complementary and powerful approach for dissecting the genetic and neural influences that shape the cognitive and behavioural manifestations of the 22q11.2DS.
Animal models of 22q11.2 deletion syndrome
Several questions arise from the human studies described above. These studies suggest that function and connectivity of several brain areas are altered in 22q11.2DS. However, the studies could not address how abnormal function and connectivity are manifested in neural circuits at the molecular, cellular and synaptic levels. Moreover, given the aforementioned limitations of current human genetic approaches, it remains unclear how human findings relate to individual genes from this locus. Model organisms that mimic the human condition are likely to have an important role in clarifying these issues because they allow direct assessment of the impact of genetic factors on neural activity across multiple brain areas.
Cognitive and psychiatric disorders can be deconstructed into individual phenotypic components, which in turn can be analysed in animal models109. The mouse equivalent of the human 1.5 Mb 22q11.2 locus harbours orthologues of all the human functional genes except clathrin, heavy polypeptide-like 1 (CLTCL1). Another difference is that humans (but not mice) carry two functional genes (DiGeorge syndrome critical region 6 (DGCR6) and DGCR6-like (DGCR6L)) in the deleted locus, as a result of intralocus duplication. Overall, there is a high degree of conservation with the 22q11.2 region, which provides an opportunity to create etiologically valid mutant animal models. Indeed, several studies have reported the presence of neuronal and behavioural abnormalities resulting from deletions of this locus (FIG. 2). In addition, several groups have inactivated the mouse orthologues of individual genes from the 22q11.2 region and tested the mutant mice for neuronal and behavioural abnormalities.
Figure 2. Mouse models of the 22q11.2 microdeletion.

The chromosomal location and genetic organization of the 22q11.2 locus in humans is shown at the top. Each light green circle represents one gene. This 1.5 megabase (Mb) region is flanked by low-copy-repeat sequences (represented by grey boxes), making it prone to non-homologous recombination. The syntenic region of mouse chromosome 16 (MMU 16qA13) and the genetic organization of the corresponding orthologues are shown. Single-gene deletion models that have been characterized as heterozygotes for neuronal and behavioural abnormalities are indicated by dark green circles. Also shown are the various multigene deletion models that have been characterized for neuronal and behavioural abnormalities labelled using their Mouse Genome Database (MGD) allele symbols and commonly used synonyms. They include mice deficient for seven genes spanning ~150 kilobases (kb) of the 1.5 Mb deletion syntenic region (Del(16Zpf520–Slc25a1)1Awb mice (also known as DelAwb mice))149; a 22q11.2 model spanning ~1 Mb and containing 18 orthologues of the human genes in the 1.5 Mb deletion (Del(16Es2el–Ufd1l)217Bld mice (also known as Df1 mice))148,154; mice with a hemizygous deletion spanning ~1.3 Mb and containing all but one of the orthologous genes in the 1.5 Mb deletion (Del(Dgcr2–Hira)1Rak mice (also known as Del1Rak or LgDel mice))153 and mice with a hemizygous deletion syntenic to the human 1.5 Mb deletion (Del(Dgcr2–Hira)2Aam mice)114. ARVCF, armadillo repeat gene deleted in velocardiofacial syndrome; CDC45L, cell division cycle 45-like; CLDN5, claudin 5; CLTCL1, clathrin, heavy chain-like 1; COMT, catechol-O-methyltransferase; DGCR, DiGeorge syndrome critical region; DGCR6L, DiGeorge syndrome critical region 6-like; GNB1L, guanine-nucleotide-binding protein (G protein) β-polypeptide 1-like; GP1BB, glycoprotein Ib, β-polypeptide; GSCL, goosecoid-like; HIRA, histone cell cycle regulation defective A; HTF9C, HpaII tiny fragments locus 9C (also known as TRMT2A); MRPL40, mitochondrial ribosomal protein L40; PRODHP, proline dehydrogenase pseudogene; RANBP1, RAN binding protein 1; RTN4R, reticulon 4 receptor; SEPT5, septin 5; SLC25A1, solute carrier family 25, member 1; Stk22a, serine/threonine kinase 22A (also known as Tssk1); STK22B, serine/threonine kinase 22B (also known as TSSK2); TBX1, T-box 1; TXNRD2, thioredoxin reductase 2; UFD1L, ubiquitin fusion degradation 1-like; Vpreb2, pre-B lymphocyte gene 2; ZDHHC8, zinc finger, DHHC-type containing 8.
Analysis of mouse models of 22q11.2DS has focused primarily on hippocampal and frontal circuitry — for example, the mouse frontal cortex region that is functionally analogous to the human dorsolateral PFC based on pharmacological and lesions studies110 — because many cognitive impairments described in individuals with 22q11.2DS probably depend on these brain areas. Furthermore, several studies suggest that functional connectivity between the PFC and temporal lobe is abnormal in schizophrenia111-113. Specific genetic pathways have been identified that affect neuronal connectivity and neuromodulation and may be crucial for the cognitive dysfunction and high risk of schizophrenia seen in 22q11.2DS.
Alterations in microRNA processing
Recently, insights have emerged from the Del(Dgcr2–Hira)2Aam mouse strain (in which Aam refers to the initials of the genetic engineer who created the strain) (FIG. 2). Studies in this strain have provided compelling evidence that the 22q11.2 microdeletion results in abnormal processing of brain microRNAs (miRNAs)114, a class of small, non-coding RNAs that regulate the stability and translation of mRNA115 (Supplementary information S3 (figure)). One gene disrupted by the 22q11.2 microdeletion is Dgcr8, a component of the ‘microprocessor’ complex that is essential for miRNA production116. Dgcr8 haploinsufficiency results in the downregulation (by ~20–70%) of a specific subset of mature miRNAs (Supplementary information S3 (figure)). Two additional miRNA genes (mir-185 and mir-649) are removed by the 22q11.2 microdeletion. A synchronous, modest decrease in several miRNAs may have considerable impact on target transcript stability114 but direct targets for these miRNAs have not been reported so far. Nevertheless, the potential of miRNAs to contribute to the epigenetic regulation of expression of multiple genes in the brain could be an important component of the genetically complex architecture of psychiatric and cognitive phenotypes associated with the 22q11.2DS and may also contribute to the phenotypic variability117. This discovery highlights emerging novel connections between miRNAs and psychiatric and neurodevelopmental disorders.
Alterations in neuronal architecture
Evidence has been found for abnormalities in the formation of dendritic spines and dendritic complexity in cultured hippocampal neurons and in CA1 pyramidal neurons in Del(Dgcr2–Hira)2Aam mice118. This is consistent with the dysregulation of a number of neuronal and synaptic genes (see below). Cultured neurons showed a reduction in the density of mushroom spines (the larger, mature and most stable spines), as well as a decrease in spine head-width and length. Furthermore, electrophysiological recordings and quantitative immunocytochemistry indicated a reduction in the density of functional glutamatergic synapses. Finally, there was a decrease in the number of primary dendrites, the extent of dendrite branching and the total length of dendrites. The concordance of the analysis in dissociated cultures with that in the intact animal was extensive; however, several differences were observed. The reduction in the number of glutamatergic synapses and the density of mushroom spines was less pronounced in vivo than in vitro and the length of mushroom spines seemed to be normal in vivo118. This may be due to long-range network effects that cannot be recapitulated in culture and to compensatory changes in gene expression during development. Nevertheless, even modest alterations in dendritic and spine formation may result in a suboptimal number of synaptic connections, an increased number of inappropriate connections or changes in the integration of synaptic inputs, all of which will ultimately lead to considerable changes in information processing119,120.
It is unknown whether similar alterations in dendritic morphology are also present in the cortex. However, using mice from the Del(Dgcr2–Hira)1Rak strain (FIG. 2), it was shown that diminished dosage of 22q11.2 genes compromises neurogenesis and subsequent neuronal differentiation in the cerebral cortex121.
Future experiments need to address the developmental course of all of these changes. Unfortunately, it is unknown whether similar cytoarchitectural alterations are present in the brains of humans with 22q11.2DS. As such, it remains unclear whether the severity and extent of these alterations correlates with cognitive and behavioural dysfunction. Nevertheless, it is tempting to speculate that changes in dendritic complexity and neuronal density may account, at least in part, for the reduction in cortical thickness or regional decreases in grey matter volumes observed in some individuals with 22q11DS. Changes in the distribution of cortical inhibitory neurons may contribute to higher-order cognitive dysfunctions, whereas alterations in spine densities and size may contribute to the observed intellectual dysfunction.
Genetic contributions to changes in neuronal architecture
To understand how the abnormalities in neural structure in mouse models of 22q11.2DS emerge, it is important to consider the contribution of individual genes and genetic pathways. Hemizygosity for DGCR8 and the consequent changes in miRNA abundance may contribute to deficits in synaptic development and maturation122-124. In addition, other genes in the 22q11.2 locus might contribute to these changes (Supplementary information S1 (table)) — for example, zinc finger, DHHC-type containing 8 (ZDHHC8) encodes a palmitoyl-transferase (PAT) and is a member of a multigene family of PATs101,118,125,126. Evidence from several studies suggests that ZDHHC8 is localized in the Golgi apparatus and vesicular compartment of neurons118,127 and that protein palmitoylation is its central function. One report has suggested that ZDHHC8 is localized to the mitochondria, where it has been proposed to have other functions128. However, this finding needs to be confirmed, as it is based on the overexpression of potentially mistargeted carboxy-terminal green fluorescent protein (GFP) fusion protein in non-neuronal cell lines and the use of antibodies of unknown specificity. Palmitoylated proteins can regulate the formation and function of dendritic spines and glutamatergic synapses129,130. ZDHHC8 seems to be involved in the palmitoylation of a specific subset of neuronal proteins, including the postsynaptic density protein PSD95 (also known as DLG4), an adaptor molecule that was previously shown to modulate the number of dendritic spines and possibly of dendritic branches118. Recent studies show that the complementary effects of impaired miRNA biogenesis and impaired palmitoylation contribute to the impaired dendritic growth and spine development observed in Del(Dgcr2–Hira)2Aam mice. Decreases in spine density could be accounted for by deficiency of Zdhhc8, whereas decreases in spine size could be accounted for by deficiency of Dgcr8. Similar complementary effects seem to influence dendritic growth. Hemizygosity of other genes in the 1.5 Mb region (Supplementary information S1 (table)) may also contribute to the observed neural abnormalities, although evidence from animals lacking some candidate genes has been mostly negative131,132.
Genes located outside the 1.5 Mb region, but within the 3 Mb region of the 22q11.2 deletion (Supplementary information S1 (table)), may also contribute to the neuronal manifestations of 22q11.2DS. A ubiquitously expressed syntaxin-binding protein (synaptosomal-associated protein 29 (SNAP29)) modulates the function of the machinery that mediates membrane fusion, a crucial step in synaptic vesicle recycling. Low SNAP29 levels in primary neurons affect synaptic transmission133, whereas homozygous truncating mutations of the gene cause cortical dysplasia134 and abnormalities in the corpus callosum. Deficiency of another gene (CRK-like (CRKL)) in the 3 Mb region may affect signalling mediated by reelin, an extracellular matrix protein that is crucial for cell positioning and neuronal migration during brain development135. Unfortunately, it remains unknown whether any one of these genes is haploinsufficient in 22q11.2DS.
In addition to the reduction in dosage of genes in the 22q11.2 region, microarray analysis of the Del(Dgcr2–Hira)2Aam mouse strain revealed more widespread, genome-wide alterations of transcriptional programs114. These reflect either downstream effects of gene deletions in the 22q11.2 region (for example, of DGCR8) or compensatory changes. Large-scale changes in expression of genes flanking the deletion have not been reported. In the hippocampus (HPC), the genes affected were primarily related to synaptic transmission. In the PFC, expression changes involved mitochondrial genes; it remains to be determined whether this was directly related to inadequate levels of known mitochondrial genes (such as proline dehydrogenase (Prodh), mitochondrial ribosomal protein L40 (Mrpl40), thioredoxin reductase 2 (Txnrd2) and solute carrier family 25, member 1 (Slc25a1)) or putative mitochondrial genes (such as T10 (also known as D16H22S680E))128 (Supplementary information S1 (table)) in the 22q11.2 region or to adaptive energetic changes caused by altered PFC function. Interestingly, changes in mitochondrial genes in the dentate gyrus of the Del(16Es2el–Ufd1l)217Bld strain (FIG. 2) were also reported136.
Abnormal dopamine transmission
Dopamine modulates cognition and psychosis but its role in 22q11.2DS has not been assessed directly in mouse models. However, accumulating evidence shows that catechol-O-methyltransferase (COMT), a gene deleted in 22q11.2DS that encodes an intracellular, postsynaptic enzyme, modulates dopamine clearance in the PFC. Removal of dopamine from the extracellular space following evoked release was twofold slower in Comt (also known as Comt1)-homozygous knockout mice137. Notably, baseline dopamine levels seemed to be normal in both heterozygous and homozygous Comt-deficient mice138,139. This suggests that the partial reduction in CoMT levels that occurs in 22q11.2DS may have an effect primarily under conditions of increased dopamine release. Such an increase might result from deficiencies in other genes in or outside the microdeletion, from environmental factors or from both of these.
This scenario is supported by analysis of mice lacking Prodh, another gene located in the 22q11.2 microdeletion that has been linked by human genetic studies to schizophrenia103. Prodh encodes an enzyme that metabolizes l-proline140 — a putative neuromodulatory amino acid that may affect glutamatergic or GABA (γ-aminobutyric acid)-ergic transmission96,141. Approximately half of the individuals with 22q11.2DS have elevated serum levels of l-proline, which can predispose to learning impairments, epilepsy142 and schizoaffective disorder143,144. Mice carrying a missense mutation in Prodh have high serum levels of l-proline that are comparable to those observed in 22q11.2DS145. Although no anomalies in basal cortical dopamine levels were detected in these mice, in vivo microdialysis revealed increased cortical dopamine release in the PFC following acute, systemic administration of the psychostimulant amphetamine146. Prodh-deficient mice also show hypersensitivity to the locomotor effects of amphetamine. Notably, this effect is potentiated by pharmacological inhibition of COMT activity and levels of COMT transcript and protein are upregulated in the PFC of Prodh-deficient mice, probably indicating a negative-feedback loop that restrains increased local hyperdopaminergia. Thus, there is an epistatic interaction between the two genes, and the effect of the Prodh gene is modified by Comt146.
Behavioural changes in mouse models
The neural circuitry and synaptic plasticity subserving perception, attention and memory can be readily characterized in mice through objective tests. The most important criterion in choosing behavioural tests to model the human deficit is the conservation of neural circuitry underlying a given behaviour109. However, the underlying neural circuitry for the behaviours and the psychological capacities that are impaired in individuals with 22q11.2DS has not yet been fully characterized. Nevertheless, studies suggesting functional and structural pathology in the hippocampal and frontal circuitry of individuals with 22q11.2DS have prompted assessment of behavioural and cognitive modalities that typically depend on these areas.
Behavioural analysis of animal models of the 22q11.2DS has provided evidence that one or more genes from this locus modulate sensorimotor gating, fear memory and WM (FIG. 3a–c). Although other types of memory (such as reference or recognition memory) have not been studied, future studies comparing performance across various tasks will help to disentangle the mechanisms underlying the observed learning impairments. Unfortunately, considering the central importance of attentional dysfunction in 22q11.2DS and schizophrenia, there is a dearth of studies measuring attention in mutant models of 22q11.2DS. However, several rodent paradigms for measuring attentional control exist147 and can be used in future studies designed to model and dissect attentional deficits.
Figure 3. Changes in behaviour and brain connectivity exhibited by mouse models of the 22q11.2 microdeletion.

Representative results from different behavioural studies are shown from the Del(Dgcr2–Hira)2Aam mouse strain (also known as Del2Aam or Df(16)A mice) (FIG. 2). a ∣ Del(Dgcr2–Hira)2Aam mice (dark green bars) showed decreased prepulse inhibition (PPI) of the acoustic startle when compared with controls (light green bars; the asterisks represent significant differences between mutant and wild-type (WT) response). PPI is a reduction in the magnitude of the startle reflex that occurs when mice are presented with a non-startling stimulus (a prepulse) before being presented with the startling stimulus. b ∣ Del(Dgcr2–Hira)2Aam mice (dark green bars) exhibit robust deficits in both cued and contextual fear memory compared with controls (light green bars), as indicated by a decrease in the time spent exhibiting freezing behaviour. The cued and contextual fear conditioning test quantifies the ability to associate a neutral conditioned stimulus (CS; either a light or a tone) with an unconditioned stimulus (an electric shock); the cued version of the test requires the amygdala, and the context version of the test typically requires both the hippocampus (HPC) and the amygdala172. c ∣ Del(Dgcr2–Hira)2Aam mice (dark green bar) are impaired in working memory-dependent cognitive performance, as shown by decreased accuracy in the delayed non-match to place (DNMP) task compared with controls (light green bar). For the DNMP task, mice are trained to enter the appropriate arm of a two-arm T-maze to obtain a food reward, the location of which varies across trials. In both the training and testing phases, short delays of various lengths are introduced, which require frontal regions of the mouse neocortex173 and their interaction with the HPC158,174 for the active maintenance of information. d ∣ Del(Dgcr2–Hira)2Aam mice (dark green circles) showed reduced connectivity and synchrony in the HPC–prefrontal cortex (HPC–PFC) compared with controls (light green circles) as demonstrated by recordings from the medial PFC (mPFC) and HPC of mice while they performed a task that required working memory. An example of a field potential recording from the HPC (grey trace) showing θ-oscillations (blue trace) and spikes recorded simultaneously from a PFC neuron (red marks) is also shown. Note the robust modulation of prefrontal neuron spiking by hippocampal θ-oscillations (synchrony), which is disrupted in Del(Dgcr2–Hira)2Aam mice.
Cognitive impairments are core features of schizophrenia and may be mediated by the same abnormal neural processes as other human symptoms, such as hallucinations (which are less amenable to study in animal models)147. Therefore, analysis of cognition in animal models of 22q11.2DS may also have important clinical implications for understanding the increased schizophrenia risk associated with this locus147.
Mice deficient for 18 of the orthologues of human genes in the 1.5 Mb region (Del(16Es2el–Ufd1l)217Bld mice) (FIG. 2) have impaired conditioned fear memory148, whereas those hemizygous for just 7 of the orthologues (Del(16Zpf520–Slc25a1)1Awb mice (FIG. 2) do not149. Similarly, mice with a hemizygous deletion spanning all of the orthologous genes in the 1.5 Mb region (Del(Dgcr2–Hira)2Aam mice) (FIG. 2) exhibit robust deficits in both cued and contextual fear memory114. One simple interpretation of these findings is that one or more of the genes between Slc25a1 and Hira (FIG. 2) are important for the formation of fear memory. However, as explained below, it may be difficult to infer such simple genotype–phenotype relationships in 22q11.2DS based on phenotypic characterization of variable size deficiencies.
Del(Dgcr2–Hira)2Aam mice also show deficits in spatial WM tasks, consistent with studies in children and adolescents with the 22q11.2DS27,48,51. These mice are impaired in the acquisition of a delayed non-match to place (DNMP) task114 that assesses spatial WM-dependent performance. Interestingly, this deficit arises in part from deficiency of Dgcr8 (and presumably miRNAs), as heterozygous deletion of Dgcr8 (Dgcr8Gt(XH158)Byg) alone also affects acquisition of this task114 without affecting associative memory. By contrast, hemizygosity of other genes in the 1.5 Mb deletion does not seem to cause robust WM deficits: mice deficient for the NOGO receptor (reticulon 4 receptor (Rtn4r)tm1Gogo), Comt150-152 or Prodh146 perform normally in DNMP. However, interfering pharmacologically with the epistatic interaction between Comt and Prodh unmasks an underlying dopamine dysfunction and reveals WM deficits in Prodh mutant mice146. Notably, inhibition of COMT also exacerbates other behaviours that are influenced by cortical dopamine (such as sensitivity to amphetamine and PPI)146.
These observations indicate that microdeletion carriers, who carry only one COMT copy, may be particularly disadvantaged by their inability to restrain emergent dopamine dysregulation. Indeed, subsequent studies of human carriers confirmed an epistatic interaction between l-proline levels and low COMT activity. These studies provided additional evidence that individuals with 22q11.2DS who carry a low-activity COMT variant (Met-COMT) in the intact chromosome, especially those with the most elevated serum levels of l-proline, are at higher risk for psychosis or related cognitive and neurophysiological impairments95-97. These studies collectively show the power of iterative human genetic and animal model studies for understanding the neural substrates and genetic influences of the 22q11.2DS.
Studies addressing individual genetic contributions to PPI have been particularly informative in addressing the genetic architecture of 22q11.2-linked neurobehavioural impairments. Like children with 22q11.2DS, animal models carrying large deficiencies at the 22q11.2 locus114,148,153 have consistently shown abnormalities in PPI, although the pattern of alterations differs among studies. A number of genes in the deleted region differentially affect PPI. Heterozygous deficiency of guanine-nucleotide-binding protein (G protein) β-polypeptide 1-like (Gnb1l), Dgcr8 and possibly T-box 1 (Tbx1), as well as elevations in l-proline levels (especially in conjunction with inhibition of COMT activity) decrease PPI114,146,153,154, whereas heterozygous deficiency of Rtn4r, Zdhhc8, Comt and septin 5 (Sept5) has no effect101,131,150,155. It is interesting that one set of 22q11.2 deletion mice (Del(16Zpf520–Slc25a1)1Awb mice) (FIG. 2) were shown to have increased PPI149. This suggests that alterations in PPI are the result of interplay between both positive and negative contributions from genes in this locus. Similar patterns of genetic contributions may underlie other behavioural impairments, underscoring the difficulty in using traditional deletion-mapping approaches to dissect genotype–phenotype relationships from models of large CNVs.
These behavioural studies provided new and unexpected insights into the genetic architecture of the human syndrome, including the contribution of distinct or partially overlapping sets of genes to specific behavioural deficits, the identification of positive and negative influences of local genes and the existence of local negative-genetic-feedback loops. Behavioural studies also greatly facilitated the identification of neural substrates — for example, observation of deficits in fear memory indicates a previously largely unappreciated amygdala circuit dysfunction in 22q11.2DS. Although fear memory has not been assessed in humans with 22q11.2DS, amygdala dysfunction may have a crucial role in the pathophysiology of 22q11.2-associated anxiety disorders. Finally, the fact that a number of behavioural alterations described in humans (such as deficits in PPI or WM) could be recapitulated in mice supports the view that 22q11.2 mouse models have strong validity and can be used to dissect the underlying abnormalities in genes and neural circuits.
Brain connectivity and synchrony changes
Macroscopic measurements of brain structure and activity in 22q11.2DS individuals suggest that structural and functional long-range connectivity between brain areas may be altered, an anomaly that may also be related to the onset of psychosis. Recent work capitalizing on the observation of impaired WM performance in 22q11.2 animal models has helped to provide important new insights into the nature of altered brain connectivity that emerges as a result of the 22q11.2 microdeletion. The coordinated, rhythmic activity of neuronal populations in the brain gives rise to oscillations at a broad range of frequencies156,157, which are often used to synchronize neural firing of interconnected cells in and between brain areas — for example, PFC neuron spiking is modulated by HPC θ-frequency (4–12 Hz) oscillations158. In a recent study159, HPC–PFC synchrony was recorded in Del(Dgcr2–Hira)2Aam mice while they performed a task requiring WM (FIG. 3d). In wild-type mice, this synchrony was increased during WM performance, whereas Del(Dgcr2–Hira)2Aam mice showed dramatically reduced HPC–PFC synchrony159 (FIG. 3d). Furthermore, the magnitude of HPC–PFC synchrony at the onset of training predicted the time it took the mutant mice to learn the WM task. Notably, synchrony in HPC or PFC was not impaired, suggesting that θ-oscillations can organize local neural activity. The results show that the 22q11.2 microdeletion can disrupt functional connectivity and synchrony among brain areas. Abnormal PFC–HPC coupling has been observed in schizophrenia patients111-113 as well as in healthy individuals carrying candidate schizophreniarisk variants160,161, and may affect not only cognition but also other aspects of the clinical diagnosis. Therefore, the results suggest that 22q11.2 deletion may contribute to the development of schizophrenia by altering prefrontal connectivity.
Conclusions and future directions
FIG. 4 attempts to conceptualize the pathogenesis and pathophysiology of 22q11.2DS and to guide future studies of this locus, as well as those of other pathogenic CNV loci. According to this framework, reduction in dosage of a subset of neighbouring genes has a cumulative effect on the properties of neural networks. This results in locus-specific neuronal dysconnectivity, aberrant synaptic plasticity and neuromodulation, as well as altered functional integration in and across brain regions. These effects lead to cognitive dysfunction and schizophrenia when a critical threshold is surpassed. Although one or a few genes may have a greater impact, it is the cumulative effect of the imbalance of several genes in the deletion that determines the overall phenotype. Genes responsible for specific deficits are only partially overlapping. The presence of additional genetic (trans- or cis-acting) and environmental modifiers may also contribute to the variability of the cognitive and psychiatric phenotypes of the syndrome.
Figure 4. A framework for the pathogenesis and pathophysiology of 22q11.2 deletion syndrome.

Reduced gene-dosage of more than one gene affects brain function at multiple levels in 22q11.2 deletion syndrome (22q11.2DS). For simplicity, only the 1.5 megabase (Mb) region is shown. In the upper row, red circles indicate genes (proline dehydrogenase (PRODH), catechol-O-methyltransferase (COMT), DiGeorge syndrome critical region 8 (DGCR8) and zinc finger, DHHC-type containing 8 (ZDHHC8)) that have been characterized at the molecular, cellular, neural circuit and behavioural levels. Blue circles indicate genes (septin 5 (SEPT5), T-box 1 (TBX1) and guanine-nucleotide-binding protein (G protein) β-polypeptide 1-like (GNB1L)) that were studied only at the behavioural level and were found to cause changes in heterozygous mutant mice. The lower rows show how mutations in these genes disrupt the molecular and cellular properties of neurons, and how these disruptions interact with each other across different levels and may ultimately lead to a psychiatric dysfunction. Black arrows represent experimentally determined effects of 22q11.2 genes and grey arrows represent potential links inferred from existing knowledge. The cumulative effect of the imbalance of several genes in the deletion determines the overall phenotype. Genes responsible for specific phenotypic deficits are only partially overlapping, and additional genetic and environmental modifiers may also contribute to the expression and variability of these deficits. The grey boxes represent low-copy-repeat sequences that flank the 22q11.2 locus. ARVCF, armadillo repeat gene deleted in velocardiofacial syndrome; CDC45L, cell division cycle 45-like; CLDN5, claudin 5; CLTCL1, clathrin, heavy chain-like 1; DGCR6L, DiGeorge syndrome critical region 6-like; GP1BB, glycoprotein Ib, β-polypeptide; GSCL, goosecoid-like; HIRA, histone cell cycle regulation defective A; HTF9C, HpaII tiny fragments locus 9C (also known as TRMT2A); MRPL40, mitochondrial ribosomal protein L40; PRODHP, proline dehydrogenase pseudogene; RANBP1, RAN binding protein 1; RTN4R, reticulon 4 receptor; SLC25A1, solute carrier family 25, member 1; STK22B, serine/threonine kinase 22B (also known as TSSK2); TXNRD2, thioredoxin reductase 2; UFD1L, ubiquitin fusion degradation 1-like.
What determines the specificity of the phenotype? Similar neuronal abnormalities in dendrites or spines, for instance, have been observed in other neurogenetic syndromes, such as fragile X syndrome, that do not share the pattern of cognitive and psychiatric impairments observed in 22q11.2DS. There is not yet a satisfactory answer to this question; however, as analyses become more detailed, differences at the cellular level as well as in the spatial distribution pattern across the whole brain may reveal distinct anomalies in local and long-range connectivity and synaptic plasticity that may account for the phenotypic differences among syndromes. In addition, abnormal neuromodulatory function in dopamine neurotransmission brought on by combined deficiency in genes specific to this locus could further alter network properties, modify the cognitive phenotypes and possibly predispose a subset of microdeletion carriers to psychiatric symptoms.
A number of details at the circuit and neuronal level remain to be elucidated. The relationship among brain structure, function, connectivity and cognitive impairments in children and adults with 22q11.2DS is an important but challenging75 area of future research and will be essential to compare anomalies in mouse models and humans. In addition, there is a need to undertake longitudinal prospective studies — including experimental and standardized cognitive assessments alongside neuroimaging — to identify one or more endophenotypes of schizophrenia, as well as a predictive prodrome that can be preventively treated during childhood and adolescence.
It will also be interesting to determine how the affected genetic pathways (such as miRNAs) modulate the input and output function of specific brain areas and what accounts for the variable degree of penetrance of the genetic effects. However, the complexities of the genetic architecture of the disease risk associated with this locus suggest that approaches that focus on the mutation as an entity rather than deconstructing it into individual genetic components may prove equally fruitful. A number of questions remain to be addressed. What is the effect of the microdeletion on inhibitory neurotransmission or on both short- and long-term synaptic plasticity? How do such deficits correlate with the cognitive impairment? How does brain dysconnectivity relate to the etiology of the psychiatric and cognitive symptoms and how does it manifest in the activity of neural circuits or at the level of single neurons? How widespread are the recently identified deficits in brain synchrony? And are anatomical connections affected in terms of axonal extension, branching, myelination or synaptic properties? Finally, there is also a need to identify which of the molecular, cellular and circuit level deficits can serve as the best targets for the development of treatments for the cognitive and psychiatric symptoms.
Supplementary Material
Acknowledgements
The authors wish to acknowledge grant support for their work from the US National Institutes of Health (grant R01MH67068 to M.K. and J.A.G. and grant R01HD42974 to T.J.S.). J.A.G. also gratefully acknowledges support from the Simons Foundation and M.K. from the McKnight and March of Dimes Foundations. The authors wish to thank A. Arguello for help with creating figures 2,3 and 4, B. Xu for help with Supplementary information S1 (table) and Supplementary information S3 (figure) and S. Srivastava for help with box 3 and figure 1. The authors also thank A. Arguello, L. Drew, B. Xu and other members of their laboratories for comments and critical feedback.
Glossary
- Microdeletion
A submicroscopic loss of a segment of DNA of varying size, typically several kilobases long.
- Breakpoint
A specific site of chromosomal breakage associated with a chromosomal abnormality.
- Full scale IQ
A standardized composite measure of global intellectual functioning generated from scores in specific domains, such as verbal, perceptual, memory and speeded functions. Typically the median age-adjusted score is 100 ± 15 points.
- Attention
A cognitive process that is mainly thought to be involved in selectively processing or focusing on one aspect of the environment at the expense of others. Several types of attention are thought to exist, including focused, sustained and divided attention, each of which seems to depend on different cognitive, neural and neurotransmitter systems.
- Executive function
Also referred to as ‘cognitive control’, this is a broad category of cognitive functions that are generally associated in typical humans with the prefrontal cortex. Executive functions are thought to modulate or control the use of other cognitive resources and include planning, problem solving, error monitoring, decision making and the use of working memory.
- Prepulse inhibition
A reduction in the magnitude of the startle reflex that occurs when an organism is presented with a non-startling stimulus (a prepulse) before being presented with the startling stimulus. Deficits in prepulse inhibition have been observed in patients with schizophrenia as well as in patients with other psychiatric and neurological disorders.
- Mismatch negativity
A component of the electro-encephalographic (EEG) brain response that is typically generated 150–250 ms after an unusual stimulus is detected in a sequence of similar stimuli.
- Diffusion tensor imaging
An MRI imaging technique that takes advantage of the restricted diffusion of water through myelinated nerve fibres in the brain to map the anatomical connectivity among brain areas.
- Genetic modifiers
Genetic variation in (cis) or outside (trans) a gene or genetic locus that alters the phenotypic expression of the gene.
- Oligogenic
A phenotypic trait produced by two or more (but only a few) genes working together.
- Next-generation sequencing
High-throughput parallel sequencing of several megabases of DNA.
- Haploinsufficiency
The situation in which one copy of a gene is incapable of providing sufficient protein production to ensure normal function.
- Endophenotype
A state-independent biomarker or cognitive marker of an illness (present whether or not the illness is active) that is heritable and present in unaffected relatives of subjects that have the illness.
Footnotes
Competing interests statement The authors declare no competing financial interests.
DATABASES
Entrez Gene: http://www.ncbi.nlm.nih.gov/geneCOMT|CLTCL1|DGCR6|DGCR6L|Dgcr8|Hira|Mrpl40|Prodh|Rtn4r|Sept5|Slc25a1|Txnrd2|ZDHHC8
miRBase: http://www.mirbase.orgmir-185|mir-649
OMIM: http://www.ncbi.nlm.nih.gov/omim22q11.2DS
UniProtKB: http://www.uniprot.org SNAP29
FURTHER INFORMATION
Maria Karayiorgou and Joseph A. Gogos’ homepage: http://schizophreniaresearch.columbia.edu
Tony J. Simon’s homepage: http://cabil.mindinstitute.org
Schizophrenia Research Forum: http://www.szgene.org
SUPPLEMENTARY INFORMATION
See online article: S1 (table)|S2 (table)|S3 (figure)
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
References
- 1.Shprintzen RJ, et al. A new syndrome involving cleft palate, cardiac anomalies, typical facies, and learning disabilities: velo-cardio-facial syndrome. Cleft Palate J. 1978;15:56–62. [PubMed] [Google Scholar]
- 2.DiGeorge A. A new concept of the cellular basis of immunity. Disabil. Rehabil. 1965;67:907–908. [Google Scholar]
- 3.Robin NH, Shprintzen RJ. Defining the clinical spectrum of deletion. 22q11.2. J. Pediatr. 2005;147:90–96. doi: 10.1016/j.jpeds.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 4.Kobrynski LJ, Sullivan KE. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet. 2007;370:1443–1452. doi: 10.1016/S0140-6736(07)61601-8. [DOI] [PubMed] [Google Scholar]
- 5.Botto LD, et al. A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics. 2003;112:101–107. doi: 10.1542/peds.112.1.101. [DOI] [PubMed] [Google Scholar]
- 6.Urban AE, et al. High-resolution mapping of DNA copy alterations in human chromosome 22 using high-density tiling oligonucleotide arrays. Proc. Natl Acad. Sci. USA. 2006;103:4534–4539. doi: 10.1073/pnas.0511340103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edelmann L, Pandita RK, Morrow BE. Low-copy repeats mediate the common 3-Mb deletion in patients with velo-cardio-facial syndrome. Am. J. Hum. Genet. 1999;64:1076–1086. doi: 10.1086/302343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shaikh TH, et al. Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: genomic organization and deletion endpoint analysis. Hum. Mol. Genet. 2000;9:489–501. doi: 10.1093/hmg/9.4.489. [DOI] [PubMed] [Google Scholar]
- 9.Maynard TM, et al. A comprehensive analysis of 22q11 gene expression in the developing and adult brain. Proc. Natl Acad. Sci. USA. 2003;24:14433–14438. doi: 10.1073/pnas.2235651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ryan AK, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J. Med. Genet. 1997;34:798–804. doi: 10.1136/jmg.34.10.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carlson C, et al. Molecular definition of 22q11 deletions in 151 velo-cardio-facial syndrome patients. Am. J. Hum. Genet. 1997;61:620–629. doi: 10.1086/515508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sullivan KE. The clinical, immunological, and molecular spectrum of chromosome 22q11.2 deletion syndrome and DiGeorge syndrome. Curr. Opin. Allergy Clin. Immunol. 2004;4:505–512. doi: 10.1097/00130832-200412000-00006. [DOI] [PubMed] [Google Scholar]
- 13.Cook EH, Jr, Scherer SW. Copy-number variations associated with neuropsychiatric conditions. Nature. 2008;455:919–923. doi: 10.1038/nature07458. [DOI] [PubMed] [Google Scholar]
- 14.De Smedt B, et al. Mathematical disabilities in children with velo-cardio-facial syndrome. Neuropsychologia. 2007;45:885–895. doi: 10.1016/j.neuropsychologia.2006.08.024. [DOI] [PubMed] [Google Scholar]
- 15.Campbell L, Swillen A. In: Velo-Cardio-Facial Syndrome: A Model for Understanding Microdeletion Disorders. Murphy KC, Scambler PJ, editors. Cambridge Univ. Press; 2005. pp. 147–164. [Google Scholar]
- 16.Wang PP, et al. Research on behavioral phenotypes: velocardiofacial syndrome (deletion 22q11.2) Dev. Med. Child Neurol. 2000;42:422–427. doi: 10.1017/s0012162200000785. [DOI] [PubMed] [Google Scholar]
- 17.Moss EM, et al. Psychoeducational profile of the 22q11.2 microdeletion: a complex pattern. J. Pediatrics. 1999;134:193–198. doi: 10.1016/s0022-3476(99)70415-4. [DOI] [PubMed] [Google Scholar]
- 18.Gerdes M, et al. Cognitive and behavior profile of preschool children with chromosome 22q11.2 deletion. Am. J. Med. Genet. 1999;85:127–133. [PubMed] [Google Scholar]
- 19.Scherer NJ, D’Antonio LL, Kalbfleisch JH. Early speech and language development in children with velocardiofacial syndrome. Am. J. Med. Genet. 1999;88:714–723. [PubMed] [Google Scholar]
- 20.Solot CB, et al. Communication disorders in the 22Q11.2 microdeletion syndrome. J. Commun. Disord. 2000;33:187–203. doi: 10.1016/s0021-9924(00)00018-6. [DOI] [PubMed] [Google Scholar]
- 21.Solot CB, et al. Communication issues in 22q11.2 deletion syndrome: children at risk. Genet. Med. 2001;3:67–71. doi: 10.1097/00125817-200101000-00015. [DOI] [PubMed] [Google Scholar]
- 22.Swillen A, et al. Neuropsychological, learning and psychosocial profile of primary school aged children with the velo-cardio-facial syndrome (22q11 deletion): evidence for a nonverbal learning disability? Child Neuropsychol. 1999;5:230–241. doi: 10.1076/0929-7049(199912)05:04;1-R;FT230. [DOI] [PubMed] [Google Scholar]
- 23.Woodin M, et al. Neuropsychological profile of children and adolescents with the 22q11.2 microdeletion. Genet. Med. 2001;3:34–39. doi: 10.1097/00125817-200101000-00008. An early comprehensive review of the neuropsychological profile of school-aged children with 22q11.2DS.
- 24.Bearden CE, et al. The neurocognitive phenotype of the 22q11.2 deletion syndrome: selective deficit in visual-spatial memory. J. Clin. Exper. Neuropsychol. 2001;23:447–464. doi: 10.1076/jcen.23.4.447.1228. [DOI] [PubMed] [Google Scholar]
- 25.Lajiness-O’Neill RR, et al. Memory and learning in children with 22q11.2 deletion syndrome: evidence for ventral and dorsal stream disruption? Neuropsychol. Dev. Cogn. C Child Neuropsychol. 2005;11:55–71. doi: 10.1080/09297040590911202. [DOI] [PubMed] [Google Scholar]
- 26.Sobin C, et al. Neuropsychological characteristics of children with the 22q11 deletion syndrome: a descriptive analysis. Neuropsychol. Dev. Cogn. C Child Neuropsychol. 2005;11:39–53. doi: 10.1080/09297040590911167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sobin C, et al. Networks of attention in children with the 22q11 deletion syndrome. Dev. Neuropsychol. 2004;26:611–626. doi: 10.1207/s15326942dn2602_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simon TJ, et al. Overlapping numerical cognition impairments in children with chromosome 22q11.2 deletion or Turner syndromes. Neuropsychologia. 2008;46:82–94. doi: 10.1016/j.neuropsychologia.2007.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cavanagh P. In: Cognitive Neuroscience of Attention. Posner MI, editor. Guilford Press; New York: 2004. pp. 13–28. [Google Scholar]
- 30.Fan J, McCandliss BD, Sommer T, Raz A, Posner MI. Testing the efficiency and independence of attentional networks. J. Cogn. Neurosci. 2002;14:340–347. doi: 10.1162/089892902317361886. [DOI] [PubMed] [Google Scholar]
- 31.Posner MI, Petersen SE. The attention system of human brain. Ann. Rev. Neurosci. 1990;13:25–42. doi: 10.1146/annurev.ne.13.030190.000325. [DOI] [PubMed] [Google Scholar]
- 32.Simon TJ, et al. Volumetric, connective, and morphologic changes in the brains of children with chromosome 22q11.2 deletion syndrome: an integrative study. Neuroimage. 2005;25:169–180. doi: 10.1016/j.neuroimage.2004.11.018. [DOI] [PubMed] [Google Scholar]
- 33.Simon TJ, et al. A multiple levels analysis of cognitive dysfunction and psychopathology associated with chromosome 22q11.2 deletion syndrome in children. Dev. Psychopathol. 2005;17:753–784. doi: 10.1017/S0954579405050364. An integrative review of evidence from cognitive experimental studies, standardized measures, neuroimaging and genetics concerning children with 22q11.2DS.
- 34.Bish JP, et al. Domain specific attentional impairments in children with chromosome 22q11.2 deletion syndrome. Brain Cogn. 2007;64:265–273. doi: 10.1016/j.bandc.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Corbetta M, Shulman GL. Control of goal-directed and stimulus-driven attention in the brain. Nature Rev. Neurosci. 2002;3:201–215. doi: 10.1038/nrn755. [DOI] [PubMed] [Google Scholar]
- 36.Simon TJ, et al. Atypical cortical connectivity and visuospatial cognitive impairments are related in children with chromosome 22q11.2 deletion syndrome. Behav. Brain Funct. 2008;4:25. doi: 10.1186/1744-9081-4-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Piazza M, Giacomini E, Le Bihan D, Dehaene S. Single-trial classification of parallel pre-attentive and serial attentive processes using functional magnetic resonance imaging. Proc. Biol. Sci. 2003;270:1237–1245. doi: 10.1098/rspb.2003.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sathian K, et al. Neural evidence linking visual object enumeration and attention. J. Cogn. Neurosci. 1999;11:36–51. doi: 10.1162/089892999563238. [DOI] [PubMed] [Google Scholar]
- 39.Simon TJ, Vaishnavi S. Subitizing and counting depend on different attentional mechanisms: evidence from visual enumeration in afterimages. Percept. Psychophys. 1996;58:915–926. doi: 10.3758/bf03205493. [DOI] [PubMed] [Google Scholar]
- 40.Debbané M, Glaser B, Gex-Fabry M, Eliez S. Temporal perception in velo-cardio-facial syndrome. Neuropsychologia. 2005;43:1754–1762. doi: 10.1016/j.neuropsychologia.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 41.Ansari D, Lyons IM, van Eimeren L, Xu F. Linking visual attention and number processing in the brain: the role of the temporo-parietal junction in small and large symbolic and nonsymbolic number comparison. J. Cogn. Neurosci. 2007;19:1845–1853. doi: 10.1162/jocn.2007.19.11.1845. [DOI] [PubMed] [Google Scholar]
- 42.Molko N, et al. Functional and structural alterations of the intraparietal sulcus in a developmental dyscalculia of genetic origin. Neuron. 2003;40:847–858. doi: 10.1016/s0896-6273(03)00670-6. [DOI] [PubMed] [Google Scholar]
- 43.Shuman M, Kanwisher N. Numerical magnitude in the human parietal lobe; tests of representational generality and domain specificity. Neuron. 2004;44:557–569. doi: 10.1016/j.neuron.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 44.Zorzi M, Priftis K, Meneghello F, Marenzi R, Umiltà C. The spatial representation of numerical and non-numerical sequences: evidence from neglect. Neuropsychologia. 2006;44:1061–1067. doi: 10.1016/j.neuropsychologia.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 45.Bish JP, et al. Maladaptive conflict monitoring as evidence for executive dysfunction in children with chromosome 22q11.2 deletion syndrome. Dev. Sci. 2005;8:36–43. doi: 10.1111/j.1467-7687.2005.00391.x. [DOI] [PubMed] [Google Scholar]
- 46.Gratton G, Coles MG, Donchin E. Optimizing the use of information: strategic control of activation of responses. J. Exp. Psychol. 1992;121:480–506. doi: 10.1037//0096-3445.121.4.480. [DOI] [PubMed] [Google Scholar]
- 47.Takarae Y, Schmidt L, Tassone F, Simon TJ. Catechol-O-methyltransferase polymorphism modulates cognitive control in children with chromosome 22q11.2 deletion syndrome. Cogn. Affect. Behav. Neurosci. 2009;9:83–90. doi: 10.3758/CABN.9.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sobin C, Kiley-Brabeck K, Karayiorgou M. Lower prepulse inhibition in children with the 22q11 deletion syndrome. Am. J. Psychiatry. 2005;162:1090–1099. doi: 10.1176/appi.ajp.162.6.1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baker K, et al. COMT Val108/158Met modifies mismatch negativity and cognitive function in 22q11 deletion syndrome. Biol. Psychiatry. 2005;58:23–31. doi: 10.1016/j.biopsych.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 50.Cheour M, et al. The first neurophysiological evidence for cognitive brain dysfunctions in children with CATCH. Neuroreport. 1997;8:1785–1787. doi: 10.1097/00001756-199705060-00043. [DOI] [PubMed] [Google Scholar]
- 51.Kates W, et al. The neural correlates of non-spatial working memory in velocardiofacial syndrome (22q11.2 deletion syndrome) Neuropsychologia. 2007;45:2863–2873. doi: 10.1016/j.neuropsychologia.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Amelsvoort T, et al. Cognitive deficits associated with schizophrenia in velo-cardio-facial syndrome. Schizophr. Res. 2004;70:223–232. doi: 10.1016/j.schres.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 53.Karayiorgou M, et al. Schizophrenia susceptibility associated with interstitial deletions of chromosome 22q11. Proc. Natl Acad. Sci. USA. 1995;92:7612–7616. doi: 10.1073/pnas.92.17.7612. A seminal paper that provided the first evidence to support the importance of CNVs in schizophrenia vulnerability.
- 54.Xu B, et al. Strong association of de novo copy number mutations with sporadic schizophrenia. Nature Genet. 2008;40:880–885. doi: 10.1038/ng.162. A systematic study that confirms the extensive contribution of de novo CNVs, such as the 22q11.2 microdeletions, in schizophrenia vulnerability.
- 55.Arnold PD, et al. Velo-cardio-facial syndrome: implications of microdeletion 22q11 for schizophrenia and mood disorders. Am. J. Med. Genet. 2001;105:354–362. doi: 10.1002/ajmg.1359. [DOI] [PubMed] [Google Scholar]
- 56.Feinstein C, et al. Psychiatric disorders and behavioral problems in children with velocardiofacial syndrome: usefulness as phenotypic indicators of schizophrenia risk. Biol. Psychiatry. 2002;51:312–318. doi: 10.1016/s0006-3223(01)01231-8. [DOI] [PubMed] [Google Scholar]
- 57.Antshel KM, et al. ADHD, major depressive disorder, and simple phobias are prevalent psychiatric conditions in youth with velocardiofacial syndrome. J. Am. Acad. Child Adolesc. Psychiatry. 2006;45:596–603. doi: 10.1097/01.chi.0000205703.25453.5a. [DOI] [PubMed] [Google Scholar]
- 58.Antshel KM, et al. Autistic spectrum disorders in velo-cardiofacial syndrome (22q11.2 deletion) J. Autism Dev. Disord. 2007;37:1776–1786. doi: 10.1007/s10803-006-0308-6. [DOI] [PubMed] [Google Scholar]
- 59.Vorstman JA, et al. The 22q11.2 deletion in children: high rate of autistic disorders and early onset of psychotic symptoms. J. Am. Acad. Child Adolesc. Psychiatry. 2006;45:1104–1113. doi: 10.1097/01.chi.0000228131.56956.c1. [DOI] [PubMed] [Google Scholar]
- 60.Flint J, Yule W. In: Behavioural Phenotypes, Child and Adolescent Psychiatry. 3rd edn Rutter M, Taylor E, Hersov L, editors. Blackwell Scientific; Oxford: 1994. pp. 666–687. [Google Scholar]
- 61.Flint J. Behavioral phenotypes: conceptual and methodological issues. Am. J. Med. Genet. 1998;81:235–240. [PubMed] [Google Scholar]
- 62.Ogilvie CM, Moore J, Daker M, Palferman S, Docherty Z. Chromosome 22q11 deletions are not found in autistic patients identified using strict diagnostic criteria. IMGSAC. International Molecular Genetics Study of Autism Consortium. Am. J. Med. Genet. 2000;96:15–17. [PubMed] [Google Scholar]
- 63.Elia J, et al. Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol. Psychiatry. 2009 Jun;23 doi: 10.1038/mp.2009.57. (doi:10.1038/mp.2009.57) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Glessner JT, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eliez S. Autism in children with 22Q11.2 deletion syndrome. J. Am. Acad. Child Adolesc. Psychiatry. 2007;46:433–434. doi: 10.1097/CHI.0b013e31802f5490. [DOI] [PubMed] [Google Scholar]
- 66.Pulver AE, et al. Psychotic illness in patients diagnosed with velo-cardio-facial syndrome and their relatives. J. Nerv. Ment. Dis. 1994;182:476–478. doi: 10.1097/00005053-199408000-00010. [DOI] [PubMed] [Google Scholar]
- 67.Murphy KC, Jones LA, Owen MJ. High rates of schizophrenia in adults with velocardio-facial syndrome. Arch. Gen. Psychiatry. 1999;56:940–945. doi: 10.1001/archpsyc.56.10.940. [DOI] [PubMed] [Google Scholar]
- 68.Gothelf D, et al. Risk factors for the emergence of psychotic disorders in adolescents with 22q11.2 deletion syndrome. Am. J. Psychiatry. 2007;164:663–669. doi: 10.1176/ajp.2007.164.4.663. [DOI] [PubMed] [Google Scholar]
- 69.Green T, et al. Psychiatric disorders and intellectual functioning throughout development in velocardiofacial (22q11.2 deletion) syndrome. J. Am. Acad. Child Adolesc. Psychiatry. 2009;48:1060–1068. doi: 10.1097/CHI.0b013e3181b76683. [DOI] [PubMed] [Google Scholar]
- 70.American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders. 4th edn American Psychiatric Publishing; Washington, DC: 1994. [Google Scholar]
- 71.International Schizophrenia Consortium Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. A comprehensive large-scale study that confirmed the role of 22q11.2 microdeletions in schizophrenia and identified additional candidate pathogenic CNVs.
- 72.Stefansson H, et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bassett AS, et al. The schizophrenia phenotype in 22q11 deletion syndrome. Am. J. Psychiatry. 2003;160:1580–1586. doi: 10.1176/appi.ajp.160.9.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bassett AS, et al. 22q11 deletion syndrome in adults with schizophrenia. Am. J. Med. Genet. 1998;81:328–337. [PMC free article] [PubMed] [Google Scholar]
- 75.Johnson MH, Halit H, Grice SJ, Karmiloff-Smith A. Neuroimaging of typical and atypical development: a perspective from multiple levels of analysis. Dev. Psychopathol. 2002;14:521–536. doi: 10.1017/s0954579402003073. An important review of special interpretive assumptions as they relate to neuroimaging studies of typically and, more importantly, atypically developing children.
- 76.Campbell LE, et al. Brain and behaviour in children with 22q11.2 deletion syndrome: a volumetric and voxel-based morphometry MRI study. Brain. 2006;129:1218–1228. doi: 10.1093/brain/awl066. [DOI] [PubMed] [Google Scholar]
- 77.Eliez S, et al. Children and adolescents with velocardiofacial syndrome: a volumetric study. Am. J. Psychiatry. 2000;3:409–415. doi: 10.1176/appi.ajp.157.3.409. [DOI] [PubMed] [Google Scholar]
- 78.Kates WR, et al. Regional cortical white matter reductions in velocardiofacial syndrome: a volumetric MRI analysis. Biol. Psychiatry. 2001;49:677–684. doi: 10.1016/s0006-3223(00)01002-7. [DOI] [PubMed] [Google Scholar]
- 79.Kates WR, et al. Frontal and caudate alterations in velocardiofacial syndrome (deletion at chromosome 22q11.2) J. Child Neurol. 2004;5:337–342. doi: 10.1177/088307380401900506. [DOI] [PubMed] [Google Scholar]
- 80.Simon TJ, et al. Visuospatial and numerical cognitive deficits in children with chromosome 22q11.2 deletion syndrome. Cortex. 2005;2:145–155. doi: 10.1016/s0010-9452(08)70889-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tan G, et al. Meta-analysis of magnetic resonance imaging studies in chromosome 22q11.2 deletion syndrome (velocardiofacial syndrome) Schizophr. Res. 2009;115:173–181. doi: 10.1016/j.schres.2009.09.010. An extensive review of structural neuroimaging findings in 22q11.2DS.
- 82.Bearden C, et al. Mapping cortical thickness in children with 22q11.2 deletions. Cereb. Cortex. 2006;8:1889–1898. doi: 10.1093/cercor/bhl097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bearden CE, et al. Alterations in midline cortical thickness and gyrification patterns mapped in children with 22q11.2 deletions. Cereb. Cortex. 2009;19:115–126. doi: 10.1093/cercor/bhn064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schaer M, et al. Abnormal patterns of cortical gyrification in velo-cardio-facial syndrome (deletion 22q11.2): an MRI study. Psychiatry Res. 2006;146:1–11. doi: 10.1016/j.pscychresns.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 85.Schaer M, et al. Congenital heart disease affects local gyrification in 22q11.2 deletion syndrome. Dev. Med. Child Neurol. 2009;51:746–753. doi: 10.1111/j.1469-8749.2009.03281.x. [DOI] [PubMed] [Google Scholar]
- 86.Van Essen DC, et al. Symmetry of cortical folding abnormalities in Williams syndrome revealed by surface-based analyses. J. Neurosci. 2006;26:5470–5483. doi: 10.1523/JNEUROSCI.4154-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Barnea-Goraly N, et al. Investigation of white matter structure in velocardiofacial syndrome: a diffusion tensor imaging study. Am. J. Psychiatry. 2003;160:1863–1869. doi: 10.1176/appi.ajp.160.10.1863. [DOI] [PubMed] [Google Scholar]
- 88.Machado AM, et al. Corpus callosum morphology and ventricular size in chromosome 22q11.2 deletion syndrome. Brain Res. 2007;1131:197–210. doi: 10.1016/j.brainres.2006.10.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Barnea-Goraly N, et al. Arithmetic ability and parietal alterations: a diffusion tensor imaging study in velocardiofacial syndrome. Brain Res. Cogn. Brain Res. 2005;25:735–740. doi: 10.1016/j.cogbrainres.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 90.Eliez S, et al. Functional brain imaging study of mathematical reasoning abilities in velocardiofacial syndrome (del22q11.2) Genet. Med. 2001;3:49–55. doi: 10.1097/00125817-200101000-00011. [DOI] [PubMed] [Google Scholar]
- 91.Gothelf D, et al. Abnormal cortical activation during response inhibition in 22q11.2 deletion syndrome. Hum. Brain Mapp. 2007;28:533–542. doi: 10.1002/hbm.20405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.van Amelsvoort T, et al. Brain anatomy in adults with velocardiofacial syndrome with and without schizophrenia: preliminary results of a structural magnetic resonance imaging study. Arch. Gen. Psychiatry. 2004;61:1085–1096. doi: 10.1001/archpsyc.61.11.1085. [DOI] [PubMed] [Google Scholar]
- 93.Chow EW, et al. Structural brain abnormalities in patients with schizophrenia and 22q11 deletion syndrome. Biol. Psychiatry. 2002;51:208–215. doi: 10.1016/s0006-3223(01)01246-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chow EW, Watson M, Young DA, Bassett AS. Neurocognitive profile in 22q11 deletion syndrome and schizophrenia. Schizophr. Res. 2006;87:270–278. doi: 10.1016/j.schres.2006.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gothelf D, et al. COMT genotype predicts longitudinal cognitive decline and psychosis in 22q11.2 deletion syndrome. Nature Neurosci. 2005;8:1500–1502. doi: 10.1038/nn1572. [DOI] [PubMed] [Google Scholar]
- 96.Raux G, et al. Involvement of hyperprolinemia in cognitive and psychiatric features of the 22q11 deletion syndrome. Hum. Mol. Genet. 2007;16:83–91. doi: 10.1093/hmg/ddl443. [DOI] [PubMed] [Google Scholar]
- 97.Vorstman JA, et al. Proline affects brain function in 22q11DS children with the low activity COMT158 allele. Neuropsychopharmacology. 2009;34:739–746. doi: 10.1038/npp.2008.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Taddei I, et al. Genetic factors are major determinants of phenotypic variability in a mouse model of the DiGeorge/del22q11 syndromes. Proc. Natl Acad. Sci. USA. 2001;98:11428–11431. doi: 10.1073/pnas.201127298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Karayiorgou M, Gogos JA. The molecular genetics of the 22q11-associated schizophrenia. Brain Res. Mol. Brain Res. 2004;132:95–104. doi: 10.1016/j.molbrainres.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 100.Arinami T. Analyses of the associations between the genes of 22q11 deletion syndrome and schizophrenia. J. Hum. Genet. 2006;51:1037–1045. doi: 10.1007/s10038-006-0058-5. [DOI] [PubMed] [Google Scholar]
- 101.Mukai J, et al. Evidence that the gene encoding ZDHHC8 contributes to the risk of schizophrenia. Nature Genet. 2004;36:725–731. doi: 10.1038/ng1375. [DOI] [PubMed] [Google Scholar]
- 102.Liu H, et al. Genetic variation in the 22q11 locus and susceptibility to schizophrenia. Proc. Natl Acad. Sci. USA. 2002;26:16859–16864. doi: 10.1073/pnas.232186099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liu H, et al. Genetic variation at the 22q11 PRODH2/DGCR6 locus presents an unusual pattern and increases susceptibility to schizophrenia. Proc. Natl Acad. Sci. USA. 2002;99:3717–3722. doi: 10.1073/pnas.042700699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Karayiorgou M, et al. Genotype determining low catechol-O-methyltransferase activity as a risk factor for obsessive-compulsive disorder. Proc. Natl Acad. Sci. USA. 1997;94:4572–4575. doi: 10.1073/pnas.94.9.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Karayiorgou M, et al. Family-based association studies support a sexually dimorphic effect of COMT and MAOA on genetic susceptibility to obsessive-compulsive disorder. Biol. Psychiatry. 1999;45:1178–1189. doi: 10.1016/s0006-3223(98)00319-9. [DOI] [PubMed] [Google Scholar]
- 106.Pooley EC, Fineberg N, Harrison PJ. The met allele of catechol-O-methyltransferase (COMT) is associated with obsessive-compulsive disorder in men: case–control study and meta-analysis. Mol. Psychiatry. 2007;12:556–561. doi: 10.1038/sj.mp.4001951. [DOI] [PubMed] [Google Scholar]
- 107.Williams NM, et al. Strong evidence that GNB1L is associated with schizophrenia. Hum. Mol. Genet. 2008;17:555–566. doi: 10.1093/hmg/ddm330. [DOI] [PubMed] [Google Scholar]
- 108.Goldstein DB. Common genetic variation and human traits. N. Engl. J. Med. 2009;360:1696–1698. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- 109.Arguello PA, Gogos JA. Modeling madness in mice: one piece at a time. Neuron. 2006;52:179–196. doi: 10.1016/j.neuron.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 110.Wise SP. Forward frontal fields: phylogeny and fundamental function. Trends Neurosci. 2008;31:599–608. doi: 10.1016/j.tins.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Meyer-Lindenberg AS, et al. Regionally specific disturbance of dorsolateral prefrontal-hippocampal functional connectivity in schizophrenia. Arch. Gen. Psychiatry. 2005;62:379–386. doi: 10.1001/archpsyc.62.4.379. [DOI] [PubMed] [Google Scholar]
- 112.Lawrie SM, et al. Reduced frontotemporal functional connectivity in schizophrenia associated with auditory hallucinations. Biol. Psychiatry. 2002;51:1008–1011. doi: 10.1016/s0006-3223(02)01316-1. [DOI] [PubMed] [Google Scholar]
- 113.Ford JM, Mathalon DH, Whitfield S, Faustman WO, Roth WT. Reduced communication between frontal and temporal lobes during talking in schizophrenia. Biol. Psychiatry. 2002;51:485–492. doi: 10.1016/s0006-3223(01)01335-x. [DOI] [PubMed] [Google Scholar]
- 114.Stark KL, et al. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nature Genet. 2008;40:751–760. doi: 10.1038/ng.138. This paper provided compelling evidence that the 22q11.2 microdeletion results in abnormal processing of brain miRNAs, which results in altered neuronal connectivity, behaviour and cognition in mice, including deficits in WM.
- 115.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 116.Tomari Y, Zamore PD. MicroRNA biogenesis: drosha can’t cut it without a partner. Curr. Biol. 2005;15:R61–R64. doi: 10.1016/j.cub.2004.12.057. [DOI] [PubMed] [Google Scholar]
- 117.Hornstein E, Shomron N. Canalization of development by microRNAs. Nature Genet. 2006;38:S20–S24. doi: 10.1038/ng1803. [DOI] [PubMed] [Google Scholar]
- 118.Mukai J, et al. Palmitoylation-dependent neurodevelopmental deficits in a mouse model of 22q11 microdeletion. Nature Neurosci. 2008;11:1302–1310. doi: 10.1038/nn.2204. This paper provided evidence that the 22q11.2 microdeletion results in impaired development of dendrites, dendritic spines and excitatory synapses, in part due to abnormal palmitoylation of neuronal proteins.
- 119.Yuste R, Tank DW. Dendritic integration in mammalian neurons, a century after Cajal. Neuron. 1996;16:701–716. doi: 10.1016/s0896-6273(00)80091-4. [DOI] [PubMed] [Google Scholar]
- 120.Mainen ZF, Sejnowski TJ. Influence of dendritic structure on firing pattern in model neocortical neurons. Nature. 1996;382:363–366. doi: 10.1038/382363a0. [DOI] [PubMed] [Google Scholar]
- 121.Meechan DW, Tucker ES, Maynard TM, LaMantia AS. Diminished dosage of 22q11 genes disrupts neurogenesis and cortical development in a mouse model of 22q11 deletion/DiGeorge syndrome. Proc. Natl Acad. Sci. USA. 2009;106:16434–16445. doi: 10.1073/pnas.0905696106. This paper provided evidence that diminished dosage of 22q11.2 genes subtly compromises neurogenesis and subsequent neuronal differentiation in the cerebral cortex.
- 122.Kosik KS. The neuronal microRNA system. Nature Rev. Neurosci. 2006;7:911–920. doi: 10.1038/nrn2037. [DOI] [PubMed] [Google Scholar]
- 123.Fiore R, et al. Mef2-mediated transcription of the miR379–410 cluster regulates activity-dependent dendritogenesis by fine-tuning Pumilio2 protein levels. EMBO J. 2009;28:697–710. doi: 10.1038/emboj.2009.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Schratt GM, et al. A brain-specific microRNA regulates dendritic spine development. Nature. 2006;439:283–289. doi: 10.1038/nature04367. [DOI] [PubMed] [Google Scholar]; 2006;441:902. erratum in. [Google Scholar]
- 125.Fukata M, Fukata Y, Adesnik H, Nicoll RA, Bredt DS. Identification of PSD-95 palmitoylating enzymes. Neuron. 2004;44:987–996. doi: 10.1016/j.neuron.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 126.Mitchell DA, Vasudevan A, Linder ME, Deschenes RJ. Protein palmitoylation by a family of DHHC protein S-acyltransferases. J. Lipid Res. 2006;47:1118–1127. doi: 10.1194/jlr.R600007-JLR200. [DOI] [PubMed] [Google Scholar]
- 127.Ohno Y, Kihara A, Sano T, Igarashi Y. Intracellular localization and tissue-specific distribution of human and yeast DHHC cysteine-rich domain-containing proteins. Biochim. Biophys. Acta. 2006;1761:474–483. doi: 10.1016/j.bbalip.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 128.Maynard TM, et al. Mitochondrial localization and function of a subset of 22q11 deletion syndrome candidate genes. Mol. Cell. Neurosci. 2008;39:439–451. doi: 10.1016/j.mcn.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.El-Husseini AED, Bredt DS. Protein palmitoylation: a regulator of neuronal development and function. Nature Rev. Neurosci. 2002;3:791–802. doi: 10.1038/nrn940. [DOI] [PubMed] [Google Scholar]
- 130.Kang R, et al. Neural palmitoyl-proteomics reveals dynamic synaptic palmitoylation. Nature. 2008;456:904–909. doi: 10.1038/nature07605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hsu R, et al. Nogo receptor 1 (RTN4R) as a candidate gene for schizophrenia: analysis using human and mouse genetic approaches. PLoS ONE. 2007;2:e1234. doi: 10.1371/journal.pone.0001234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tsang CW, et al. Superfluous role of mammalian septins 3 and 5 in neuronal development and synaptic transmission. Mol. Cell. Biol. 2008;28:7012–7029. doi: 10.1128/MCB.00035-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Su Q, Mochida S, Tian JH, Mehta R, Shen ZH. SNAP-29: a general SNARE protein that inhibits SNARE disassembly and is implicated in synaptic transmission. Proc. Natl Acad. Sci. USA. 2001;98:14038–14043. doi: 10.1073/pnas.251532398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Park TJ, Curran T. Crk and Crk-like play essential overlapping roles downstream of disabled-1 in the Reelin pathway. J. Neurosci. 2008;28:13551–13562. doi: 10.1523/JNEUROSCI.4323-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sprecher E, et al. A mutation in SNAP29, coding for a SNARE protein involved in intracellular trafficking, causes a novel neurocutaneous syndrome characterized by cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma. Am. J. Hum. Genet. 2005;77:242–251. doi: 10.1086/432556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jurata LW, et al. Altered expression of hippocampal dentate granule neuron genes in a mouse model of human 22q11 deletion syndrome. Schizophr. Res. 2006;88:251–259. doi: 10.1016/j.schres.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 137.Yavich L, Forsberg MM, Karayiorgou M, Gogos JA, Männistö PT. Site-specific role of catechol-O-methyltransferase in dopamine overflow within prefrontal cortex and dorsal striatum. J. Neurosci. 2007;27:10196–10209. doi: 10.1523/JNEUROSCI.0665-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Huotari M, et al. Brain catecholamine metabolism in catechol-O-methyltransferase (COMT)-deficient mice. Eur. J. Neurosci. 2002;15:246–256. doi: 10.1046/j.0953-816x.2001.01856.x. [DOI] [PubMed] [Google Scholar]
- 139.Huotari M, García-Horsman JA, Karayiorgou M, Gogos JA, Männistö PT. d-Amphetamine responses in catechol-O-methyltransferase (COMT) disrupted mice. Psychopharmacology (Berlin) 2004;172:1–10. doi: 10.1007/s00213-003-1627-3. [DOI] [PubMed] [Google Scholar]
- 140.Hayward DC, et al. The sluggish-A gene of Drosophila melanogaster is expressed in the nervous system and encodes proline oxidase, a mitochondrial enzyme involved in glutamate biosynthesis. Proc. Natl Acad. Sci. USA. 1993;90:2979–2983. doi: 10.1073/pnas.90.7.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Goodman BK, Rutberg J, Lin WW, Pulver AE, Thomas GH. Hyperprolinaemia in patients with deletion (22)(q11.2) syndrome. J. Inherit. Metab. Dis. 2000;23:847–848. doi: 10.1023/a:1026773005303. [DOI] [PubMed] [Google Scholar]
- 142.Jacquet H, et al. The severe form of type I hyperprolinaemia results from homozygous inactivation of the PRODH gene. J. Med. Genet. 2003;40:e7. doi: 10.1136/jmg.40.1.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jacquet H, et al. PRODH mutations and hyperprolinemia in a subset of schizophrenic patients. Hum. Mol. Genet. 2002;11:2243–2249. doi: 10.1093/hmg/11.19.2243. [DOI] [PubMed] [Google Scholar]
- 144.Jacquet H, et al. Hyperprolinemia is a risk factor for schizoaffective disorder. Mol. Psychiatry. 2005;10:479–485. doi: 10.1038/sj.mp.4001597. [DOI] [PubMed] [Google Scholar]
- 145.Gogos JA, et al. The gene encoding proline dehydrogenase modulates sensorimotor gating in mice. Nature Genet. 1999;21:434–439. doi: 10.1038/7777. [DOI] [PubMed] [Google Scholar]
- 146.Paterlini M, et al. Transcriptional and behavioral interaction between 22q11.2 orthologs modulates schizophrenia-related phenotypes in mice. Nature Neurosci. 2005;8:1586–1594. doi: 10.1038/nn1562. This paper and reference 96 provided the first demonstration of an epistatic interaction between two candidate schizophrenia risk genes in the 22q11.2 locus and showed the power of iterative human genetic and animal model studies for dissecting the genetic and neural substrates of the 22q11.2DS.
- 147.Arguello PA, Gogos JA. Cognition in mouse models of schizophrenia susceptibility genes. Schizophr. Bull. 2010;36:289–300. doi: 10.1093/schbul/sbp153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Paylor R, et al. Mice deleted for the DiGeorge/velocardiofacial syndrome region show abnormal sensorimotor gating and learning and memory impairments. Hum. Mol. Genet. 2001;10:2645–2650. doi: 10.1093/hmg/10.23.2645. [DOI] [PubMed] [Google Scholar]
- 149.Kimber WL, et al. Deletion of 150 kb in the minimal DiGeorge/velocardiofacial syndrome critical region in mouse. Hum. Mol. Genet. 1999;8:2229–2237. doi: 10.1093/hmg/8.12.2229. [DOI] [PubMed] [Google Scholar]
- 150.Gogos JA, et al. Catechol-O-methyltransferase-deficient mice exhibit sexually dimorphic changes in catecholamine levels and behavior. Proc. Natl Acad. Sci. USA. 1998;95:9991–9996. doi: 10.1073/pnas.95.17.9991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Papaleo F, et al. Genetic dissection of the role of catechol-O-methyltransferase in cognition and stress reactivity in mice. J. Neurosci. 2008;28:8709–8723. doi: 10.1523/JNEUROSCI.2077-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Babovic D, et al. Phenotypic characterization of cognition and social behavior in mice with heterozygous versus homozygous deletion of catechol-O-methyltransferase. Neuroscience. 2008;155:1021–1029. doi: 10.1016/j.neuroscience.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 153.Long JM, et al. Behavior of mice with mutations in the conserved region deleted in velocardiofacial/DiGeorge syndrome. Neurogenetics. 2006;7:247–257. doi: 10.1007/s10048-006-0054-0. [DOI] [PubMed] [Google Scholar]
- 154.Paylor R, et al. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: implications for 22q11 deletion syndrome. Proc. Natl Acad. Sci. USA. 2006;103:7729–7734. doi: 10.1073/pnas.0600206103. This paper described the first traditional deletion-mapping approach to dissect genotype–phenotype relationships in the 22q11.2 locus.
- 155.Nitta T, et al. Size-selective loosening of the blood–brain barrier in claudin-5-deficient mice. J. Cell Biol. 2003;161:653–660. doi: 10.1083/jcb.200302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Steriade M, Gloor P, Llinas RR, de Silva F. H. Lopes, Mesulam MM. Report of IFCN Committee on Basic Mechanisms. Basic mechanisms of cerebral rhythmic activities. Electroencephalogr. Clin. Neurophysiol. 1990;76:481–508. doi: 10.1016/0013-4694(90)90001-z. [DOI] [PubMed] [Google Scholar]
- 157.Singer W. Neuronal synchrony: a versatile code for the definition of relations? Neuron. 1999;24:49–65. doi: 10.1016/s0896-6273(00)80821-1. [DOI] [PubMed] [Google Scholar]
- 158.Jones MW, Wilson MA. Theta rhythms coordinate hippocampal–prefrontal interactions in a spatial memory task. PLoS Biol. 2005;3:e402. doi: 10.1371/journal.pbio.0030402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Sigurdsson T, Stark KL, Karayiorgou M, Gogos JA, Gordon JA. Impaired hippocampal–prefrontal synchrony in a genetic mouse model of schizophrenia. Nature. 2010;464:763–767. doi: 10.1038/nature08855. Building on the findings of reference 114, this paper provides compelling evidence that impaired long-range connectivity and synchrony of neural activity is one consequence of the 22q11.2 deletion and could be a fundamental component of the pathophysiology underlying schizophrenia.
- 160.Bertolino A, et al. Prefrontal–hippocampal coupling during memory processing is modulated by COMT val158met genotype. Biol. Psychiatry. 2006;60:1250–1258. doi: 10.1016/j.biopsych.2006.03.078. [DOI] [PubMed] [Google Scholar]
- 161.Esslinger C, et al. Neural mechanisms of a genome-wide supported psychosis variant. Science. 2009;324:605. doi: 10.1126/science.1167768. [DOI] [PubMed] [Google Scholar]
- 162.Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nature Genet. 2008;40:695–701. doi: 10.1038/ng.f.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Van Veen V, Carter CS. The anterior cingulated as a conflict monitor: fMRI and ERP studies. Physiol. Behav. 2002;77:477–482. doi: 10.1016/s0031-9384(02)00930-7. [DOI] [PubMed] [Google Scholar]
- 164.Devrim-Uçok M, Keskin-Ergen HY, Uçok A. Mismatch negativity at acute and post-acute phases of first-episode schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 2008;258:179–185. doi: 10.1007/s00406-007-0772-9. [DOI] [PubMed] [Google Scholar]
- 165.Carroll CA, Boggs J, O’Donnell BF, Shekhar A, Hetrick WP. Temporal processing dysfunction in schizophrenia. Brain Cogn. 2008;67:150–161. doi: 10.1016/j.bandc.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Elvevåg B, Goldberg TE. Cognitive impairment in schizophrenia is the core of the disorder. Crit. Rev. Neurobiol. 2000;14:1–21. [PubMed] [Google Scholar]
- 167.Debbané M, Glaser B, David MK, Feinstein C, Eliez S. Psychotic symptoms in children and adolescents with 22q11.2 deletion syndrome: neuropsychological and behavioral implications. Schizophr. Res. 2006;84:187–193. doi: 10.1016/j.schres.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 168.Sugama S, et al. Morphometry of the head of the caudate nucleus in patients with velocardiofacial syndrome (del 22q11.2) Acta Paediatr. 2000;89:546–549. doi: 10.1080/080352500750027826. [DOI] [PubMed] [Google Scholar]
- 169.Eliez S, et al. Increased basal ganglia volumes in velo-cardio-facial syndrome (deletion 22q11.2) Biol. Psychiatry. 2002;52:68–70. doi: 10.1016/s0006-3223(02)01361-6. [DOI] [PubMed] [Google Scholar]
- 170.Eliez S, et al. A quantitative MRI study of posterior fossa development in velocardiofacial syndrome. Biol. Psychiatry. 2001;49:540–546. doi: 10.1016/s0006-3223(00)01005-2. [DOI] [PubMed] [Google Scholar]
- 171.Bish JP, et al. Specific cerebellar reductions in children with chromosome 22q11.2 deletion syndrome. Neurosci. Lett. 2006;399:245–248. doi: 10.1016/j.neulet.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 172.Fanselow MS, Poulos AM. The neuroscience of mammalian associative learning. Annu Rev. Psychol. 2005;56:207–234. doi: 10.1146/annurev.psych.56.091103.070213. [DOI] [PubMed] [Google Scholar]
- 173.Aultman JM, Moghaddam B. Distinct contributions of glutamate and dopamine receptors to temporal aspects of rodent working memory using a clinically relevant task. Psychopharmacology (Berlin) 2001;153:353–364. doi: 10.1007/s002130000590. [DOI] [PubMed] [Google Scholar]
- 174.Lee I, Kesner RP. Time-dependent relationship between the dorsal hippocampus and the prefrontal cortex in spatial memory. J. Neurosci. 2003;23:1517–1523. doi: 10.1523/JNEUROSCI.23-04-01517.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Goldberg MC, Maurer D, Lewis TL. Developmental changes in attention: the effects of endogenous cueing and of distractors. Dev. Sci. 2001;4:209–219. [Google Scholar]
- 176.Posner MI, et al. Effects of parietal injury on covert orienting of attention. J. Neurosci. 1984;4:1863–1874. doi: 10.1523/JNEUROSCI.04-07-01863.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Rueda MR, et al. Development of attentional networks in childhood. Neuropsychologia. 2004;42:1029–1040. doi: 10.1016/j.neuropsychologia.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 178.Egly R, Driver J, Rafal RD. Shifting visual attention between objects and locations: evidence from normal and parietal lesion subjects. J. Exp. Psychol. Gen. 1994;123:161–177. doi: 10.1037//0096-3445.123.2.161. [DOI] [PubMed] [Google Scholar]
- 179.Klein RM. Inhibition of return. Trends Cogn. Sci. 2000;4:138–147. doi: 10.1016/s1364-6613(00)01452-2. [DOI] [PubMed] [Google Scholar]
- 180.MacPherson AC, Klein RM, Moore C. Inhibition of return in children and adolescents. J. Exp. Child Psychol. 2003;85:337–351. doi: 10.1016/s0022-0965(03)00104-8. [DOI] [PubMed] [Google Scholar]
- 181.Chi MT, Klahr D. Span and rate of apprehension in children and adults. J. Exp. Child Psychol. 1975;19:434–439. doi: 10.1016/0022-0965(75)90072-7. [DOI] [PubMed] [Google Scholar]
- 182.Trick LM, Pylyshyn ZW. Why are small and large numbers enumerated differently? A limited-capacity preattentive stage in vision. Psychol. Rev. 1994;101:80–102. doi: 10.1037/0033-295x.101.1.80. [DOI] [PubMed] [Google Scholar]
- 183.Ansari D, Karmiloff-Smith A. Atypical trajectories of number development: a neuroconstructivist perspective. Trends Cogn. Sci. 2002;6:511–516. doi: 10.1016/s1364-6613(02)02040-5. [DOI] [PubMed] [Google Scholar]
- 184.Dehaene S, Cohen L. Towards an anatomical and functional model of number processing. Math. Cogn. 1995;1:83–120. [Google Scholar]
- 185.Ornitz EM, Guthrie D, Kaplan AR, Lane SJ, Norman RJ. Maturation of startle modulation. Psychophysiology. 1986;23:624–634. doi: 10.1111/j.1469-8986.1986.tb00681.x. [DOI] [PubMed] [Google Scholar]
- 186.Ahmmed AU, Clarke EM, Adams C. Mismatch negativity and frequency representational width in children with specific language impairment. Dev. Med. Child Neurol. 2008;50:938–944. doi: 10.1111/j.1469-8749.2008.03093.x. [DOI] [PubMed] [Google Scholar]
- 187.Näätänen R. Mismatch negativity (MMN): perspectives for application. Int. J. Psychophysiol. 2000;37:3–10. doi: 10.1016/s0167-8760(00)00091-x. [DOI] [PubMed] [Google Scholar]
- 188.Casey BJ, et al. Activation of prefrontal cortex in children during a nonspatial working memory task with functional MRI. Neuroimage. 1995;2:221–229. doi: 10.1006/nimg.1995.1029. [DOI] [PubMed] [Google Scholar]
- 189.Durston S, Thomas KM, Worden MS, Yang Y, Casey BJ. The effect of preceding context on inhibition: an event-related fMRI study. Neuroimage. 2002;16:449–453. doi: 10.1006/nimg.2002.1074. [DOI] [PubMed] [Google Scholar]
- 190.Schulz KP, et al. Response inhibition in adolescents diagnosed with attention deficit hyperactivity disorder during childhood: an event-related FMRI study. Am. J. Psychiatry. 2004;161:1650–1657. doi: 10.1176/appi.ajp.161.9.1650. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.