

Abstract
Current treatment for late-stage metastatic breast cancer is largely palliative. α-Particles are highly potent, short-range radiation emissions capable of sterilizing individual cells with one to three traversals of the cell nucleus. The α-emitter, 213Bi (T1/2 = 45.6 min), was conjugated to a 100-nm diameter liposomal-CHX-A”-DTPA construct, upon which the rat HER2/neu reactive antibody, 7.16.4, was grafted. A conjugation time of 10 minutes was achieved giving a specific activity corresponding to 0.1 213Bi atom per liposome; stability in vitro and in vivo was confirmed. Efficacy in a rat/neu transgenic mouse model of metastatic mammary carcinoma was investigated. Three days after left cardiac ventricular injection of 105 rat HER-2/neu–expressing syngeneic tumor cells, macrophage-depleted Neu-N mice were treated by i.v. injection with (a) 19.2 MBq (520 μCi) of liposome-CHX-A”-DTPA-213Bi, (b) 19.2 MBq of liposome-CHX-A”-DTPA-213Bi-7.16.4, (c) 4.44 MBq (120 μCi) of 213Bi-7.16.4, and (d) cold (nonradioactive) liposome-CHX-A”-DTPA-7.16.4 as control. Treatment with (a) increased median survival time to 34 days compared with 29 days for the untreated controls (P = 0.013) and 27 days for treated cold controls. Treatment with the radiolabeled antibody–conjugated liposome (b) increased median survival time to 38 days (P = 0.0002 relative to untreated controls). The radiolabeled antibody–treated group (c) gave a median survival of 39 days, which was similar to that for the radiolabeled antibody–conjugated liposome-treated group (P = 0.5). We have shown that the 213Bi radiolabeled immunoliposomes are effective in treating early-stage micrometastases, giving median survival times similar to those obtained with antibody-mediated delivery of 213Bi in this animal model.
Introduction
Disseminated metastatic breast cancer is classified as a stage IV cancer. Treatment at this late stage is generally palliative (1), and the 5-year relative survival rate of patients is 23% (2). α-Particle emitters are being investigated as thera-peutics against metastatic disease. α-Particles travel a very short distance (≈50 to 80 μm) and deposit highly focused energy along their path. The average linear energy transfer of α-particles is ~400 to 800 times greater, on average, than that of β-particles (3). As a result, α-particles can efficiently kill single cells and micrometastases with limited toxicity to surrounding normal tissues. Furthermore, the high prevalence of DNA double-strand breaks caused by α-particle radiation reduces the possibility of repair from sublethal damage, thereby making targeted α-particle therapy less susceptible to the majority of tumor resistance mechanisms. The short half-life of 213Bi is well suited to targeting hematologic malignancies and prevascularized micrometastases. Monoclonal antibody–mediated delivery of the α-emitter 213Bi to target cells expressing the HER-2/neu antigen was effective in treating HER-2/neu–expressing micrometastases in a preclinical model (4-6). The efficacy of targeted α-emitter therapy has been shown to critically depend on antibody-specific activity and the target cell antigen density (7). The number of α-particle–emitting atoms that may be delivered to target cells per carrier might be increased by using nanoscale carrier systems. Several radiolabeled multifunctional nanocarriers have been effective in detecting and treating cancer in animal models (8). Nanoparticles, including liposomes and other nanoscale constructs, could be used to deliver drugs to tumors (9, 10).
Liposomes have been studied for more than 30 years, in particular, as vehicles for drug delivery (11, 12). Their application in drug delivery has been made possible by the development of sterically stabilized structures that use polyethylene glycol (PEG) chains to reduce the uptake and catabolism of i.v. administered liposomes by the reticuloendothelial system, thereby increasing circulatory half-life. Such liposomes are typically 100 to 150 nm in diameter because this size range reduces reticuloendothelial system uptake while retaining adequate aqueous volume for drug delivery. Liposome tumor localization is dependent on the differential permeability of normal versus tumor tissue capillaries (13-15). Liposomes have also been used to deliver radionuclides for tumor diagnosis, therapy, and infectious site imaging (16, 17). The possibility of using liposomes to deliver the α-particle emitter, 225Ac, and its daughters to target cells has been examined (18, 19). In this article, the toxicity and therapeutic efficacy of engineered liposomal vesicles that deliver a greater number of 213Bi atoms per carrier than can be achieved by antibody conjugation are examined.
Materials and Methods
Liposome preparation
Mixtures of 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine (DMPE), cholesterol (1:1 molar ratio; Sigma), and 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] DSPE-PEG–labeled lipids (4 mol% of total lipid) in CHCl3 were dried in a rotary evaporator at 55°C (Avanti Polar Lipids), dried under N2 with a few glass beads, and were hydrated in chelexed PBS (Sigma). The lipid suspension was then annealed to 55°C for 2.5 hours (20). To make unilamellar small liposomes, the lipid suspension was then taken through 21 cycles of extrusion (LiposoFast; Avestin) through two stacked polycarbonate filters (100 nm filter pore diameter) at 55°C and was stored at 4°C until further use.
Preparation of macrophage-depleting liposomes
A mixture of egg phosphatidylcholine, cholesterol, and DSPE-PEG-2000 (4 mol% of total lipid) was dried as explained above and hydrated with a 0.5 mol/L solution of dichloromethylene diphosphonate (DMDP) in PBS (21). Unentrapped DMDP was removed by size exclusion chromatography, passing liposome-DMDP through a Sephadex G-25 PD-10 desalting column (Bio-Rad).
CHX-A”-DTPA conjugation to liposome for 213Bi chelation
To conjugate CHX-A”-DTPA N-[2-amino-3-(p-isothiocyanatophenyl)propyl]-trans-cyclohexane-1,2-diamine-N,N’,N’,N”,N”-pentaacetic acid (Macrocyclics) to the liposome, the NH3 group of DMPE lipid in the liposome was deprotonated with a conjugation buffer that was concentrated 10 times (80.44 g NaHCO3, 4.50 g Na2CO3, 175.32 g NaCl, 2 L double-distilled water, treated with Chelex-100; Sigma). CHX-A”-DTPA solution in DMSO was added to 10× molar excess of DMPE lipid concentration, and pH was adjusted to ~8 to 9 using 1× conjugation buffer and allowed to react at room temperature overnight on a mixer. Unbound CHX-A”-DTPA was removed by size exclusion chromatography on a Sephadex G-50 packed 1 × 10 cm column (Sigma), eluted with an isotonic PBS buffer. The average number of CHX-A”-DTPA on liposome was determined using a standard yttrium arsenazo spectrophotometric method (22), and was found to be ~1,750 ligands per liposome of ~100 nm size.
To prepare DTPA-functionalized liposomes, lipid, 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-DTPA (Avanti Polar Lipids), was included instead of DMPE during the liposome preparation.
Mice, cell line, and antibody
neu-N transgenic mice, at ages 6 to 8 weeks, overexpressing rat HER-2/neu under the mouse mammary tumor virus promoter were maintained and obtained from Harlan. All experiments involving the use of mice were conducted with the approval of the Animal Care and Use Committee of The Johns Hopkins University School of Medicine. The rat HER-2/neu–expressing mouse mammary tumor cell line NT2.5 was established from spontaneous mammary tumors in female neu-N mice (4, 23). The NT lines were grown in RPMI medium containing 20% fetal bovine serum, 0.5% penicillin/streptomycin (Invitrogen), 1% l-glutamine, 1% nonessential amino acids, 1% sodium pyruvate, 0.02% gentamicin, and 0.2% insulin (Sigma) and maintained at 37°C in 5% CO2. The hybridoma cell line for 7.16.4 was kindly provided by Dr. M. Greene (University of Pennsylvania, Philadelphia, PA). 7.16.4 collected from the ascites of athymic mice was purified by a HiTrap protein G column (GE Healthcare Biosciences) using the Biologic LP purification system (Bio-Rad) and dialyzed into PBS using Centricon YM-10 filter units (Millipore).
Anti-HER2 immunoliposomes
Immunoliposome was prepared by covalent conjugation of 7.16.4, a mouse anti-Her2/neu monoclonal antibody to CHX-A”-DTPA liposomes by postinsertion method as described elsewhere (24). As controls, nontargeted CHX-A”-DTPA liposomes were prepared identically except for the omission of monoclonal antibody conjugation. 7.16.4 monoclonal antibodies and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(poly[ethylene glycol])2000] (DSPE-PEG-maleimide lipid; 2 mol% of total lipids) were allowed to react with thiolated 7.16.4 monoclonal antibodies [thiolated using Traut’s reagent (2-iminothiolane); Pierce Biotechnology] overnight at room temperature. 7.16.4 conjugated DSPE-PEG-maleimide lipid (2 mol% of total lipid) in micellar form was later allowed to react with CHX-A”-DTPA liposomes for 1 hour at 55°C for insertion into liposome (postinsertion method). The unbound antibody after postinsertion method was removed by passing the liposomes through a Sepharose 4B (Sigma) column. The liposome concentration was determined using a phosphate assay (25). The antibody concentration was determined by Bio-Rad protein assay. The antibody density turned out to be ~40 antibodies per liposome.
213Bi labeling
Bismuth-213 was generated from 225Ac (10-day half-life; Institute for Transuranium Elements, Karlsruhe, Germany). The 225Ac is bound to an AGMP-50 cation exchange resin (200–400 mesh; Bio-Rad) from which 213Bi may be optimally eluted every 4 to 6 hours (26, 27). The 213Bi was eluted using 1.3 mL of HI (0.1 mol/L HCl/NaI) to a 3 mol/L ammonium acetate (to decrease the pH to ~4) and ascorbic acid solution containing CHX-A”-DTPA immunoliposomes. The binding reaction was carried out for 10 minutes at room temperature with occasional stirring. Upon quenching the reaction by the addition of 50 μL of 10 mmol/L EDTA, unlabeled 213Bi was removed using a PD-10 size exclusion column (Sephadex G-25; Bio-Rad). The 7.16.4 antibody was labeled with 213Bi as described previously (5). Briefly, 7.16.4 was first conjugated to N-[2-amino-3-(p-isothiocyanatophenyl)propyl]-trans-cyclohexane-1,2-diamine-N,N’,N’,N”,N”-pentaacetic acid (SCN-CHX-A”-DTPA). 7.16.4 conjugated to the chelate was incubated with BiI4−/BiI52− (at 10 mCi/mg) for 8 minutes in a reaction buffer (pH 4.5) containing 3 mol/L of ammonium acetate (Fisher Scientific) and 150 mg/mL of l-ascorbic acid (Sigma) preheated to 37°C.
Liposome size distribution determination
Dynamic light scattering of liposome suspensions was studied with a Zetasizer Nano ZS90 (Malvern Instruments), equipped with a 633-nm He-Ne laser (4 mW) light source. To observe the size and external morphology of liposomes, a transmission electron microscope (Hitachi 7600) was used.
Flow cytometry: immunoliposomes
To study the binding of 7.16.4 immunoliposomes to the NT2.5 cell surface, the liposomes were prepared as explained above but with the inclusion of 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(carboxyfluorescein) lipid as a fluorescent label. The BD FACScalibur cytometry system (BD Biosciences) equipped with a 488-nm laser source was used to analyze the cells.
In vitro bismuth stability measurements
To experimentally test the stability of 213Bi-labeled lipo-some-CHX-A”-DTPA, the construct was added to 10% serum (Invitrogen-Life Technologies) and incubated over time at 37°C in 5% CO2. After 7 hours, the sample was quenched with 10 mmol/L of EDTA and run through a PD-10 column with PBS at pH 7.4, and the fractions were collected and counted in a gamma counter (CompuGamma Spectrometer, LKB-Wallac 1282). In vitro stability (in percentage) was calculated as the area under the curve corresponding to liposomal fraction elution divided by the total area under the elution curve times 100.
Immunoreactivity
The immunoreactive fraction of liposome-CHX-A”-DTPA-213Bi(7.16.4) was measured by incubating (2 h on ice) a fixed amount of liposome-CHX-A”-DTPA-111In (7.16.4) with increasing numbers of antigen-expressing cells (NT2.5), and then fitting the expression: to the cell-bound radioactivity (B) over the total activity (T) (ref. 28). The antigen concentration, Ag, was obtained as the product of cell concentration and antigen density (= 1.5 × 105/Avogadro’s number). The fitting software package SAAM II (University of Washington, Seattle, WA) was used to obtain fitted values of the immunoreactive fraction, f, and the effective dissociation constant, KD. Nonspecific binding was evaluated using liposome-CHX-A”-DTPA-111In.
Cell killing
The potency of tumor cell–killing was measured using a colony formation assay. Cells (1 × 104 in 100 μL of medium) plated on a 96-well plate, in their exponential phase of growth, were treated with serial dilutions of liposome-CHX-A”-DTPA-213Bi(7.16.4) or liposome-CHX-A”-DTPA overnight. In vitro toxicity was evaluated by colony formation assay by plating ~1,000 treated cells on separate (60 mm) plates (in duplicate). After 10 days, the colonies (>50 cells) formed were stained using crystal violet (1%; Sigma) and counted. The cell-killing efficacy of each agent was taken in vitro as the slope of the survival curve (plot of surviving fraction against activity).
Maximum tolerated dose
Maximum tolerated dose (MTD) was defined as the highest total activity that allows 100% survival of the mice with no significant (i.e., <15%) body weight loss. Two injections of liposome-CHX-A”-DTPA-213Bi were given to mice (five per group) on 2 consecutive days with total administered activities of 29.6, 22.2, 20.4, 12.3, and 9.3 MBq (800, 600, 550, 350, and 250 μCi). All mice were euthanized, and major organs were collected and examined for histopathology.
The murine model of breast cancer metastases
The murine model for rat HER-2/neu–expressing breast cancer metastasis has been described previously (4). Briefly, neu-N mice, ages 6 to 8 weeks, were injected with 105 NT2.5 cells suspended in 100 μL of cold PBS via the left cardiac ventricle (LCV) after anesthesia with a ketamine (90 mg/kg) and xylazine (10 mg/kg) mixture. Establishment and progression of metastases in multiple organs, including bones, liver, and spleen, was confirmed by histopathology. Necropsy was performed on every mouse to confirm successful LCV injection; mice that were not successfully injected could be identified by tumor confined to the chest wall. Unsuccessfully injected mice were excluded from the analysis.
Biodistribution/in vivo stability
The biodistribution of immunoliposome-CHX-A”-DTPA-213Bi in tumor-bearing mice was obtained at 0.5, 1, 2, and 4 hours after injection [n = 5 (0.5, 1, 2 h) or 4 (4 h)]. To enable identification and collection of metastatic tumor, LCV inoculation was performed 15 days before the biodistribution studies. This was the earliest time that we could reliably identify metastatic tumors. At each time, the mice were sacrificed and the blood, heart, lung, liver, kidney, spleen, intestine, stomach, femur, and tumor were removed, weighed, and counted directly in a gamma counter. Results were corrected for physical decay and presented as a percentage of injected dose per gram (% ID/g). In vivo stability was further evaluated in tumor-free mice by comparing the 4-hour biodistribution of free 213Bi to that of liposome-DTPA-213Bi and liposomeCHX-A”-DTPA-213Bi (n = 3 in each group).
Dosimetry
Absorbed doses were calculated by fitting an exponential expression to each tissue time-activity curve obtained in the biodistribution study. The resulting total number of disintegrations was multiplied by the total α-particle or electron energy emitted per disintegration of 213Bi (29) to give the α-particle or electron-absorbed dose.
Targeted therapy: in vivo
Following LCV injection, radioimmunotherapy was started on the third day with two consecutive daily i.v. injections of (a) liposome-CHX-A”-DTPA-213Bi-7.16.4 with a total administered activity of 19.2 MBq (520 μCi; n = 9); (b) liposome-CHX-A”-DTPA-213Bi with a total administered activity of 19.2 MBq (n = 8); (c) unlabeled immunoliposome-CHX-A”-DTPA (n = 5); (d) no treatment control (n = 8); (e) 213Bi-labeled 7.16.4 antibody. Macrophage depletion in mice was carried out by i.v. injection of liposome-DMDP (0.5–1 h) prior to LCV injection. In LCV-injected mice, weight loss and survival time were monitored.
Histopathology
neu-N mice were treated with 22.2 MBq of liposome-CHX-A”-DTPA-213Bi, 1.85 MBq greater than the MTD. Histopathology analysis was performed to determine the reason for dose-limiting toxicity. Major organs (e.g., heart, lung, liver, kidney, spleen, and femur) were collected, fixed in formalin for 24 hours, and later sectioned and stained with H&E.
Statistical analysis
The statistical significance of differences between two groups was analyzed with a t test and Kaplan-Meier survival analysis using MedCalc (MedCalc Software). Differences with P < 0.05 values were considered statistically significant.
Results
Immunoliposome characterization
Liposomes were prepared by the extrusion method with 100-nm filter pore sizes. Liposome size distribution was measured with dynamic light scattering. The measured average liposome size was ~110 nm. Liposomes were also imaged by transmission electron microscopy using the negative staining method and sizes were confirmed. The reactivity of the immunoliposomes against antigen-expressing cells was confirmed by a shift in cells exposed to fluorescently labeled 7.16.4-immunoliposomes compared with liposomes (Fig. 1).
Figure 1.
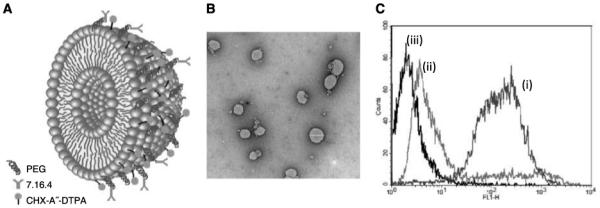
A, schematic illustration of immunoliposome-CHX-A”-DTPA. B, transmission electron microscopy image of liposomes. C, histogramic representation of flow cytometric analysis showing positive shift in fluorescence intensity of (i) cell-bound fluorescent immunoliposomes indicating specific binding with respect to (ii) cell-bound fluorescent liposome without the specific antibody and (iii) untreated cells.
213Bi labeling
The liposome-CHX-A”-DTPA radiolabeling efficiency with 213Bi after 10 minutes of reaction time at room temperature was 83% with a 213Bi generator size of ~111 to 148 MBq. At a pH of 4 to 4.5, 213Bi bound specifically to liposome-CHX-A”-DTPA constructs, whereas at higher pH, 213Bi bound nonspecifically to the phosphate head groups on the liposome surface. Decreasing the pH protonates the head group, and hence, removes the nonspecific binding sites (12). The radiochemical purity of the construct was ~99.8% as assessed by instant TLC. The radiochemical purity of 213Bi-labeled 7.16.4 antibody after purification was 98% and the immunoreactivity was between 80% and 85%.
Stability of 213Bi-labeled liposomes: ex vivo
The 213Bi-labeled immunoliposome-CHX-A”-DTPA construct was stable for 6 hours with a stability of ~96%. Because the half-life was 45 minutes, the radiolabeled constructs only need to be stable for 6 to 7 hours; the liposome-DTPA construct was stable to ~85% over 6 hours in ex vivo serum incubation assays.
Specific activity of 213Bi-labeled liposomes
The specific activity of one 213Bi atom per 10 liposomes with liposome-DTPA and liposome-CHX-A”-DTPA construct was obtained; one 213Bi atom per ~1,900 antibody molecules has been previously reported for antibodies (5), and was also achieved in the current study.
Immunoreactivity/cytotoxicity
The immunoreactive fraction of liposome-CHX-A”-DTPA-213Bi labeled with antibody (7.16.4) was 28 ± 2%, and liposome-CHX-A”-DTPA-213Bi without antibody that nonspecifically adhered to the cell surface gave 3.2 ± 0.4%; the corresponding fitted values for the KD variables were 5 ± 3 and 9 ± 14 nmol/L, respectively (Fig. 2).
Figure 2.
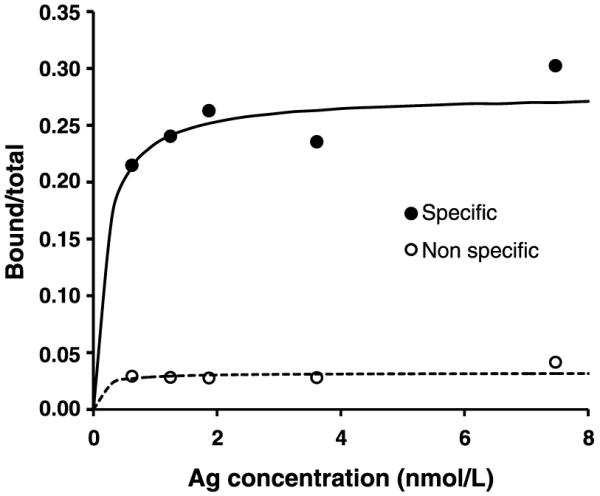
The immunoreactive fraction of liposome-CHX-A”-DTPA with (specific, ●) and without 7.16.4 antibody (nonspecific, ○) was obtained by plotting the bound to total ratio against antigen concentration and fitting a receptor-ligand binding expression that included immunoreactive fraction. The fits are shown as solid or dotted lines. The immunoreactive fraction of liposome-CHX-A”-DTPA with and without antibody was 28 ± 2% and 3.2 ± 0.4%, respectively.
Cell-killing experiments were performed with CHX-A”-DTPA–chelated liposome or immunoliposome 213Bi constructs. Immunoliposomes were more potent at killing cells than liposomes alone but less potent than 213Bi-conjugated 7.16.4 antibody (Fig. 3). The concentrations required to yield 37% cell survival were 0.1, 0.3, and 0.5 MBq/mL (3, 8, and 14 μCi/mL) for antibody, targeted immunoliposomes, and nontargeted liposomes, respectively.
Figure 3.
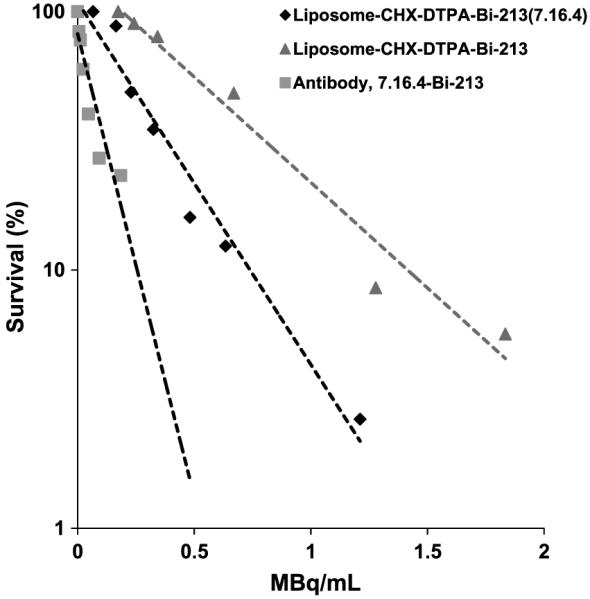
In vitro cell killing assay comparison of antibody (7.16.4) with liposome-CHX-A”-DTPA and immunoliposome-CHX-A”-DTPA. D0 is ~0.12 MBq/mL for antibody, ~0.52 MBq/mL for nontargeted liposome, and the targeted immunoliposome of ~0.3 MBq/mL.
Organ biodistribution
The biodistribution of liposome-CHX-A”-DTPA-213Bi (7.16.4) in tumor-bearing mice is shown in Fig. 4A. At 0.5 h, the construct had already reached its maximum concentration in the blood, heart, lungs, femur, and tumor with mean and SD values of 15 ± 2%, 1.8 ± 0.2%, 4.0 ± 0.4%, 0.7 ± 0.1%, and 3 ± 2% ID/g, respectively. The liver, spleen, kidneys, and intestines reached a maximum 1 hour after injection with mean and SD values of 14 ± 2%, 63 ± 15%, 9 ± 2%, and 0.9 ± 0.4%, respectively. The construct cleared from the blood with a half-life of 1.1 ± 0.4 hours. After a drop from the 1-hour maximum, construct accumulation in the spleen continued to increase over a 4-hour period. At 4 hours after injection, the organ biodistribution of liposome-DTPA-213Bi and free 213Bi was compared with liposome-CHX-A”-DTPA-213Bi. Liposome-DTPA-213Bi and free 213Bi showed a >70% ID/g in the kidneys, whereas <5% ID/g was seen in the liposome-CHX-A”-DTPA-213Bi–injected group, suggesting that the construct was very stable in vivo. Free bismuth is known to accumulate in the kidneys (30); therefore, similar to the observation made in the case of antibody-based chelated constructs (31-33), CHX-A”-DTPA is needed for the in vivo retention of 213Bi on to the liposome surface. The results for in vivo stability after 4 hours are shown in Fig. 4B. The high spleen uptake seen for both the antibody-conjugated and -unconjugated liposome-CHX-A”DTPA-213Bi (Fig. 4A and B, respectively) is consistent with the known splenic uptake of PEGylated liposomes (34).
Figure 4.

A, organ biodistribution at 0.5, 1, 2, and 4 h after i.v. injection of liposome-CHX-A”-DTPA-213Bi(7.16.4). The tumor kinetics shown are for tumors visible 15 d after LCV injection of tumor cells. B, the 4-h biodistribution of liposome-CHX-A”-DTPA-213Bi, liposome-DTPA-213Bi, and 213Bi alone shows the accumulation of free and liposome-DTPA-213Bi in the kidneys, whereas the construct using CHX-A”-DTPA has a high splenic localization consistent with that observed for liposomes.
Dosimetry
Mean absorbed doses are listed on Table 1. The spleen, liver, and intestine received the three largest absorbed doses. The stomach, femur, and heart received the three lowest absorbed doses. The total absorbed dose to tumor was 2.1 ± 0.7 Gy; the total blood-absorbed dose was 9 ± 6 Gy. The high level of uncertainty in the absorbed dose estimates, with SD values ranging from 31% (tumor) to 73% (stomach) of the mean values, respectively, reflect the uncertainty in the fitted variables.
Table 1.
Normal tissue and tumor-absorbed doses ± SD (Gy) from 19.2 MBq (520 μCi) liposome-CHX-A”-DTPA-213Bi(7.16.4)
| α-Particle | Electron | |
|---|---|---|
| Spleen | 28 ± 16 | 2.2 ± 1.3 |
| Intestine | 11 ± 9 | 0.9 ± 0.7 |
| Liver | 9 ± 6 | 0.7 ± 0.5 |
| Blood | 9 ± 5 | 0.7 ± 0.4 |
| Kidneys | 6 ± 2 | 0.5 ± 0.2 |
| Lungs | 3 ± 1 | 0.2 ± 0.1 |
| Tumor* | 1.9 ± 0.6 | 0.15 ± 0.05 |
| Heart | 1.2 ± 0.6 | 0.10 ± 0.05 |
| Femur | 0.5 ± 0.3 | 0.04 ± 0.02 |
| Stomach | 0.4 ± 0.3 | 0.04 ± 0.03 |
Tumor micrometastasis identified 15 d after LCV injection.
Histopathology
The histopathology of liposome-CHX-A”-DTPA-213Bi–treated neu-N mice showed marrow suppression as a probable reason for MTD limitation. Slides of the heart, lung, liver, and kidney clearly show bacterial colonies (septicemia) due to opportunistic infection. Slides of the femur show the depletion of red marrow cells and slides of the spleen show depletion of hematopoietic cells suggesting immune system suppression (Fig. 5).
Figure 5.
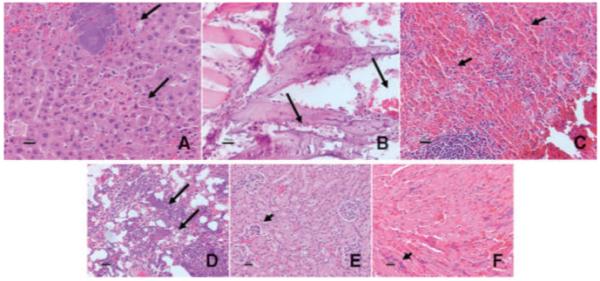
Histopathology staining slides showing (A) bacterial colonies in liver (arrows); (B) bone marrow depletion in the femur (arrows); (C) depletion of hematopoietic cells in the spleen (arrows); (D) bacterial colonies in the lung (arrows); (E) bacterial colonies in the kidney glomerular capillaries (arrow); and (F) multifocal bacterial colonies in capillaries in the heart (arrow) of neu-N mice following treatment with liposome-CHX-A”-DTPA-213Bi at doses greater than the MTD.
Targeted therapy: in vivo
In the metastatic LCV animal model, the median survival of untreated and cold liposome-CHX-A”-DTPA-7.16.4 controls was 29 and 27 days, respectively. Liposome-CHX-A”-DTPA-213Bi-7.16.4–treated mice had a median survival time of 38 days (P = 0.0002 relative to untreated controls). This was not significantly different from the 39-day median survival obtained with 213Bi-labeled 7.16.4 antibody (P = 0.5). The group treated with liposome-CHX-A”-DTPA-213Bi with no antibody had a median survival time of 34 days (P = 0.013) compared with controls (Fig. 6); the 4-day gain in median survival of immunoliposome-treated versus liposome-treated mice was not statistically significant (P = 0.08).
Figure 6.

Survival probability plot (Kaplan-Meier survival curve) for metastatic mouse model in neu-N transgenic mice with treatment groups that include 19.2 MBq of neu-targeted liposome-CHX-A”-DTPA-213Bi (n = 9), 19.2 MBq of nontargeted liposome-CHX-A”-DTPA-213Bi (n = 8), unlabeled targeted liposome-CHX-A”-DTPA (cold; n = 5), and the no-treatment control group (n = 8) and 4.44 MBq 213Bi-labeled 7.16.4 antibody (n = 9).
Discussion
We have developed liposome-CHX-A”-DTPA-213Bi constructs and shown their stability and therapeutic efficacy in vivo. Surface chelation yielded a maximum efficiency of 83% of the initial radioactivity, which translates into 0.1 213Bi atoms per liposome. In this highly aggressive metastatic breast carcinoma model (4), immunoliposomal delivery of the α-particle emitter, 213Bi, led to a significant increase in median survival over untreated controls.
DTPA is the most commonly used chelating agent but has been found to be unsuitable for bismuth (213Bi) conjugation because the 213Bi-DTPA complex is unstable in vivo. The use of CHX-A”-DTPA for bismuth conjugation results in a complex that has been proven to be extremely stable (32, 33) in vitro and in vivo. By surface binding 213Bi onto immunoliposomes via CHX-A”-DTPA, a number of critical advantages are obtained: (a) the chemistry is simpler and faster because, in contrast with 213Bi conjugation of antibodies, only a single purification and buffer exchange step is needed; (b) the number of 213Bi atoms per carrier is substantially increased over that achievable with antibodies; and (c) the liposomal platform makes it possible for surface coating of liposomes with antibodies against more than one target antigen, thereby reducing the possibility of treatment failure due to the escape of a subpopulation of low antigen–expressing tumor cells. Also, receptor-mediated endocytosis of immunoliposomes has been previously reported (35, 36). Endocytosis of such radiolabeled immunoliposomes would deliver a much greater effective number of radioactive atoms to a tumor cell than would be possible with radiolabeled antibodies, given currently achievable specific activities and typical cell surface antigen densities.
The increase in survival using the higher specific activity immunoliposomal construct was not better than that obtained by antibody-mediated delivery of 213Bi. The higher specific activity is counterbalanced by the low, 28% immunoreactive fraction of the immunoliposome-CHX-A”-DTPA-213Bi construct compared with the 80% to 85% achievable with 7.16.4 antibody. This is reflected in the 3-fold higher activity concentration required to yield 37% cell survival in the cytotoxicity assay. The MTD of the liposomal constructs was ~4.5 times greater than the 4.44 MBq (120 μCi) value reported for 213Bi-7.16.4 (5). This may be explained by the larger size and, therefore, reduced mobility of liposomes compared with antibodies. The immunoreactive and cell-killing experiments would favor antibody-mediated 213Bi delivery. The comparable in vivo efficacy of the 213Bi-labeled immunoliposomal construct, compared with the 213Bi-labeled antibody, might be explained by a combination of the greater MTD of the labeled immunoliposomes and the high specific activity of the fraction that is reactive. Because of their larger size, the liposomes showed a number of disadvantages as target vehicles. In this study mice had to be macrophage-depleted prior to treatment, suggesting that the constructs were recognized by the reticuloendothelial system and that additional liposomal engineering is needed to reduce such uptake. Possible modifications would include conjugating Fab antibody fragments, instead of intact antibody, to reduce Fc-mediated recognition of the constructs as well as the use of longer PEG chains to assure that the CHX-A”-DTPA-Bi-213 groups do not extend beyond the reticuloendothelial system–repelling PEG brush border.
The estimated absorbed doses resulting from the near-MTD administered activity level are generally higher than corresponding values previously reported for antibody-mediated delivery of 213Bi (5). In the antibody-targeting studies, blood received the greatest absorbed dose at the MTD administered activity level. In this study, the spleen and intestine received higher absorbed doses than the blood but the blood α-particle–absorbed dose of 9 ± 6 Gy is within the 6.2 Gy value reported previously. In contrast, the tumor-absorbed dose of 1.9 ± 0.6 Gy is substantially lower than the 4.43 Gy reported for micrometastases. The assumption of a uniform α-particle energy distribution within tumor and normal tissue for both constructs is known to not accurately represent the true distribution; the latter is essential in predicting the likely biological effects of α-emitters. More detailed, microscopic distributions of α-emissions in each organ and in the tumor are required to explain these discrepancies.
In conclusion, the high–specific activity 213Bi-conjugated immunoliposome constructs that were produced were stable in vitro and in vivo. Specific cell–killing and a significant increase in median survival over untreated mice was shown in vitro. The median survival was similar to that obtained for antibody-mediated targeting of 213Bi in the same highly aggressive metastatic breast cancer model. Immunoliposomal delivery of α-emitters to target rapidly accessible metastatic cancer merits further consideration.
Acknowledgments
Grant Support
NIH/NCI grant R01 CA113797, DOD Concept Award BC052595 and DOD Fellowship BC062968 (M. Lingappa).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.NCI Breast Cancer PDQ [Accessed November 5, 2009]; http://www.cancer.gov/cancertopics/pdq/treatment/breast/HealthProfessional/page8#Reference8.15.
- 2.American Cancer Society Cancer facts and figures 2006. 2006;2007 [Google Scholar]
- 3.Sgouros G, Roeske JC, McDevitt MR, et al. MIRD pamphlet no. 22—(abridged): Radiobiology and dosimetry of α-particle emitters for targeted radionuclide therapy. J Nucl Med. 2010;51:311–28. doi: 10.2967/jnumed.108.058651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Song H, Shahverdi K, Huso DL, et al. An immunotolerant HER-2/neu transgenic mouse model of metastatic breast cancer. Clin Cancer Res. 2008;14:6116–24. doi: 10.1158/1078-0432.CCR-07-4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Song H, Shahverdi K, Huso DL, et al. 213Bi (α-emitter)-antibody targeting of breast cancer metastases in the neu-N transgenic mouse model. Cancer Res. 2008;68:3873–80. doi: 10.1158/0008-5472.CAN-07-6308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Song H, Hobbs RF, Vajravelu R, et al. Radioimmunotherapy of breast cancer metastases with α-particle-emitter 225Ac: comparing efficacy with 213Bi and 90Y. Cancer Res. 2009;69:8941–8. doi: 10.1158/0008-5472.CAN-09-1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sgouros G, Song H. Cancer stem cell targeting using the α-particle emitter, 213Bi: mathematical modeling and feasibility analysis. Cancer Biother Radiopharm. 2008;23:74–81. doi: 10.1089/cbr.2007.0408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mitra A, Nan A, Line BR, Ghandehari H. Nanocarriers for nuclear imaging and radiotherapy of cancer. Curr Pharm Des. 2006;12:4729–49. doi: 10.2174/138161206779026317. [DOI] [PubMed] [Google Scholar]
- 9.Noble CO, Kirpotin DB, Hayes ME, et al. Development of ligand-targeted liposomes for cancer therapy. Expert Opin Ther Targets. 2004;8:335–53. doi: 10.1517/14728222.8.4.335. [DOI] [PubMed] [Google Scholar]
- 10.Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science. 2004;303:1818–22. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 11.Lasic DD. Liposomes from physics to applications. Elsevier; Amsterdam: 1993. [Google Scholar]
- 12.Drummond DC, Meyer O, Hong K, Kirpotin DB, Papahadjopoulos D. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol Rev. 1999;51:691–743. [PubMed] [Google Scholar]
- 13.Litzinger DC, Buiting AM, van Rooijen N, Huang L. Effect of liposome size on the circulation time and intraorgan distribution of amphipathic poly(ethylene glycol)-containing liposomes. Biochim Biophys Acta. 1994;1190:99–107. doi: 10.1016/0005-2736(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 14.Wu NZ, Da D, Rudoll TL, Needham D, Whorton AR, Dewhirst MW. Increased microvascular permeability contributes to preferential accumulation of Stealth liposomes in tumor tissue. Cancer Res. 1993;53:3765–70. [PubMed] [Google Scholar]
- 15.Yuan F, Leunig M, Huang SK, Berk DA, Papahadjopoulos D, Jain RK. Microvascular permeability and interstitial penetration of sterically stabilized (stealth) liposomes in a human tumor xenograft. Cancer Res. 1994;54:3352–6. [PubMed] [Google Scholar]
- 16.Boerman OC, Storm G, Oyen WJ, et al. Sterically stabilized liposomes labeled with indium-111 to image focal infection. J Nucl Med. 1995;36:1639–44. [PubMed] [Google Scholar]
- 17.Sofou S, Sgouros G. Antibody-targeted liposomes in cancer therapy and imaging. Expert Opin Drug Deliv. 2008;5:189–204. doi: 10.1517/17425247.5.2.189. [DOI] [PubMed] [Google Scholar]
- 18.Sofou S, Thomas JL, Lin HY, McDevitt MR, Scheinberg DA, Sgouros G. Engineered liposomes for potential {alpha}-particle therapy of metastatic cancer. J Nucl Med. 2004;45:253–60. [PubMed] [Google Scholar]
- 19.Sofou S, Kappel BJ, Jaggi JS, McDevitt MR, Scheinberg DA, Sgouros G. Enhanced retention of the α-particle-emitting daughters of Actinium-225 by liposome carriers. Bioconjug Chem. 2007;18:2061–7. doi: 10.1021/bc070075t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castile JD, Taylor KMG. Factors affecting the size distribution of liposomes produced by freeze-thaw extrusion. Int J Pharm. 1999;188:87–95. doi: 10.1016/s0378-5173(99)00207-0. [DOI] [PubMed] [Google Scholar]
- 21.Naito M, Nagai H, Kawano S, et al. Liposome-encapsulated dichloromethylene diphosphonate induces macrophage apoptosis in vivo and in vitro. J Leukoc Biol. 1996;60:337–44. doi: 10.1002/jlb.60.3.337. [DOI] [PubMed] [Google Scholar]
- 22.Pippin CG, Parker TA, McMurry TJ, Brechbiel MW. Spectrophotometric method for the determination of a bifunctional DTPA ligand in DTPA-monoclonal antibody conjugates. Bioconjug Chem. 1992;3:342–5. doi: 10.1021/bc00016a014. [DOI] [PubMed] [Google Scholar]
- 23.Reilly RT, Gottlieb MBC, Ercolini AM, et al. HER-2/neu is a tumor rejection target in tolerized HER-2/neu transgenic mice. Cancer Res. 2000;60:3569–76. [PubMed] [Google Scholar]
- 24.Moreira JN, Ishida T, Gaspar R, Allen TM. Use of the post-insertion technique to insert peptide ligands into pre-formed stealth liposomes with retention of binding activity and cytotoxicity. Pharm Res. 2002;19:265–9. doi: 10.1023/a:1014434732752. [DOI] [PubMed] [Google Scholar]
- 25.Bartlett GR. Phosphorus assay in column chromatography. J Biol Chem. 1959;234:466–8. [PubMed] [Google Scholar]
- 26.McDevitt MR, Finn RD, Sgouros G, Ma D, Scheinberg DA. An 225Ac/213Bi generator system for therapeutic clinical applications: construction and operation. Appl Radiat Isot. 1999;50:895–904. doi: 10.1016/s0969-8043(98)00151-1. [DOI] [PubMed] [Google Scholar]
- 27.Apostolidis C, Molinet R, Rasmussen G, Morgenstern A. Production of Ac-225 from Th-229 for targeted α therapy. Anal Chem. 2005;77:6288–91. doi: 10.1021/ac0580114. [DOI] [PubMed] [Google Scholar]
- 28.Konishi S, Hamacher K, Vallabhajosula S, et al. Determination of immunoreactive fraction of radiolabeled monoclonal antibodies: what is an appropriate method? Cancer Biother Radiopharm. 2004;19:706–15. doi: 10.1089/cbr.2004.19.706. [DOI] [PubMed] [Google Scholar]
- 29.Sgouros G, Ballangrud AM, Jurcic JG, et al. Pharmacokinetics and dosimetry of an α-particle emitter labeled antibody: 213Bi-HuM195 (anti-CD33) in patients with leukemia. J Nucl Med. 1999;40:1935–46. [PubMed] [Google Scholar]
- 30.Durbin PW. Metabolic characteristics within a chemical family. Health Phys. 1960;2:225–38. doi: 10.1097/00004032-195907000-00001. [DOI] [PubMed] [Google Scholar]
- 31.Brechbiel MW, Gansow OA, Atcher RW, et al. Synthesis of 1-(p-isothiocyanatobenzyl) derivatives of DTPA and EDTA. Antibody labeling and tumor-imaging studies. Inorg Chem. 2002;25:2772–81. [Google Scholar]
- 32.Brechbiel MW, Pippin CG, McMury TJ, et al. An effective chelating agent for labeling of monoclonal antibody with 212Bi for α-particle mediated radioimmunotherapy. J Chem Soc Chem Commun. 1991;17:1169–70. [Google Scholar]
- 33.Nikula TK, Curcio MJ, Brechbiel MW, Gansow OA, Finn RD, Scheinberg DA. A rapid, single-vessel method for preparation of clinical grade ligand conjugated monoclonal-antibodies. Nucl Med Biol. 1995;22:387–90. doi: 10.1016/0969-8051(94)00126-5. [DOI] [PubMed] [Google Scholar]
- 34.Boerman OC, Oyen WJ, van Bloois L, et al. Optimization of technetium-99m-labeled PEG liposomes to image focal infection: effects of particle size and circulation time. J Nuclei Med. 1997;38:489–93. [PubMed] [Google Scholar]
- 35.Mamot C, Drummond DC, Noble CO, et al. Epidermal growth factor receptor-targeted immunoliposomes significantly enhance the efficacy of multiple anticancer drugs in vivo. Cancer Res. 2005;65:11631–8. doi: 10.1158/0008-5472.CAN-05-1093. [DOI] [PubMed] [Google Scholar]
- 36.Kirpotin DB, Park JW, Hong K, et al. Sterically stabilized anti-HER2 immunoliposomes: design and targeting to human breast cancer cells in vitro. Biochemistry. 1997;36:66–75. doi: 10.1021/bi962148u. [DOI] [PubMed] [Google Scholar]